
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A lanthanide tag for a complementary set of pseudocontact shifts†
Lydia
Topping
a,
Adarshi P.
Welegedara
b,
Martyna
Judd
c,
Elwy H.
Abdelkader
 b,
Nicholas
Cox
c,
Gottfried
Otting
b and
Stephen J.
Butler
*a
b,
Nicholas
Cox
c,
Gottfried
Otting
b and
Stephen J.
Butler
*a
aDepartment of Chemistry, Loughborough University, Epinal Way, Loughborough, LE11 3TU, UK. E-mail: s.j.butler@lboro.ac.uk
bARC Centre of Excellence for Innovations in Peptide & Protein Science, Research School of Chemistry, Australian National University, Canberra, ACT 2601, Australia
cResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia
First published on 16th July 2024
Abstract
Pseudocontact shifts (PCS) generated by paramagnetic lanthanide ions deliver powerful restraints for protein structure analysis by NMR spectroscopy. We present a new lanthanide tag that generates different PCSs than that of a related tag, which differs in structure by a single oxygen atom. It is highly reactive towards cysteine and performs well in turn-on luminescence and in EPR spectroscopy.
Lanthanide complexes enable structural characterisation of biological macromolecules by different types of spectroscopy.1 For example, in nuclear magnetic resonance (NMR), paramagnetic lanthanide tags elicit large pseudocontact shifts (PCS), which form the basis of long-range structure restraints for 3D structure determination.2 In electron paramagnetic resonance (EPR), the interaction of pairs of gadolinium tags introduced into proteins, can measure distances ranging between 25 and 75 Å.3 In addition, terbium and europium tags feature long-lived luminescence,4–7 which can be engineered to ‘switch on’ upon conjugation of the tag with the target biomolecule.8,9
Recently, we showed that labelling a single solvent-accessible cysteine residue of a target protein with three to four different lanthanide tags delivers PCSs that allow the coordinates of the detected nuclear spins to be determined with high accuracy.10 Most remarkably, the information content provided by the PCSs in this single-site-multiple-tag experiment is almost equivalent to that of the previously established multiple-site-single-tag approach,11–13 where different protein constructs must be produced for site-specific tag attachment. The ability to work with a single site is important, as mutations of some sites can result in unforeseen structural perturbations. Notably, the single-site-multiple-tag experiment is only effective if the set of tags, relative to the protein, display different orientations of their magnetic susceptibility anisotropy (Δχ) tensor. This Δχ tensor is an attribute of the chelated paramagnetic lanthanide ion.10
Unfortunately, a set of tags with a different Δχ tensor cannot be generated by simply changing the lanthanide ion; for a given ligand field, different lanthanides generally have very similar Δχ tensor orientations.14 However, different Δχ tensor orientations can be achieved by changing the chirality of the tag10 or the nature of the linker between the lanthanide tag and the protein. Ideally, the linker is conformationally rigid, as flexible reorientation of the lanthanide complex relative to the protein averages the PCSs and leads to smaller effective Δχ tensors.15
Here we show that a small structural modification to the previously published C12 tag8 creates a new tag that is more reactive towards cysteine and produces a significantly different Δχ tensor orientation. Furthermore, we demonstrate that it performs like the C12 tag in double-electron–electron resonance (DEER) experiments and possesses switch-on luminescence properties.
We refer to this new tag as C14 as it is based on the cyclen macrocycle like its predecessor C12 (Fig. 1).8 Cyclen-based lanthanide complexes are attractive for their high thermodynamic and kinetic stability and ease of use, but their synthesis requires multiple steps.1 The C14 tag differs from the C12 tag only by the oxidation of the nitrogen of the pyridyl ring to an N-oxide, simplifying the synthetic protocol, which is otherwise identical to that of the C12 tag (see Scheme S1 and ESI† for experimental details). The C14 tag reacts spontaneously with the thiol group of a cysteine residue in aqueous solution, forming a stable thioether bond.16 The tag is stable to storage and shipping.‡
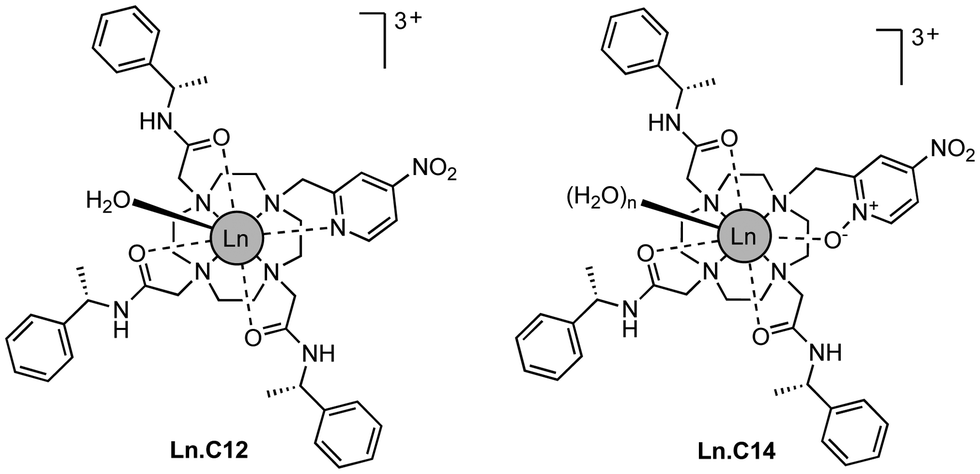 | ||
| Fig. 1 Chemical structures of Ln.C12 and Ln.C14 tags. | ||
We initially investigated the photophysical properties of the Tb3+ complex Tb.C14 and its reactivity towards cysteine. Fig. 2a shows the change in UV-Vis absorption and emission spectra of Tb.C14 upon reaction with cysteine in buffered aqueous solution at pH 8.0 and 37 °C. Tb.C14 alone exhibits a broad featureless absorption band centred at 292 nm (ε = 1800 M−1 cm1). Indirect excitation of the 4-nitropyridine N-oxide group of Tb.C14 produces weak Tb(III)-centred luminescence with four characteristic bands in the green region of the visible spectrum (475–630 nm). Upon reacting with cysteine, Tb.C14 shows a pronounced 4-fold enhancement in Tb(III)-centred luminescence, attributed to displacement of the electron withdrawing nitro group and subsequent formation of an electron donating thioether bond. The absorption spectrum was slightly red-shifted to 302 nm upon cysteine ligation, and the extinction coefficient increased to 4600 M−1 cm−1, approximately 2.5-fold higher than the untagged complex. The increase in both the extinction coefficient and quantum yield means that the overall brightness of the cysteine-tagged Tb(III) complex, defined as the product of ε and Φ, increases approximately 10-fold. This increase in brightness, although less pronounced than that observed for the previously reported 4-nitropyridine complex Tb.C12,8 is still highly effective for monitoring the cysteine labelling reaction using UV-Vis and luminescence spectroscopy, discussed further below.
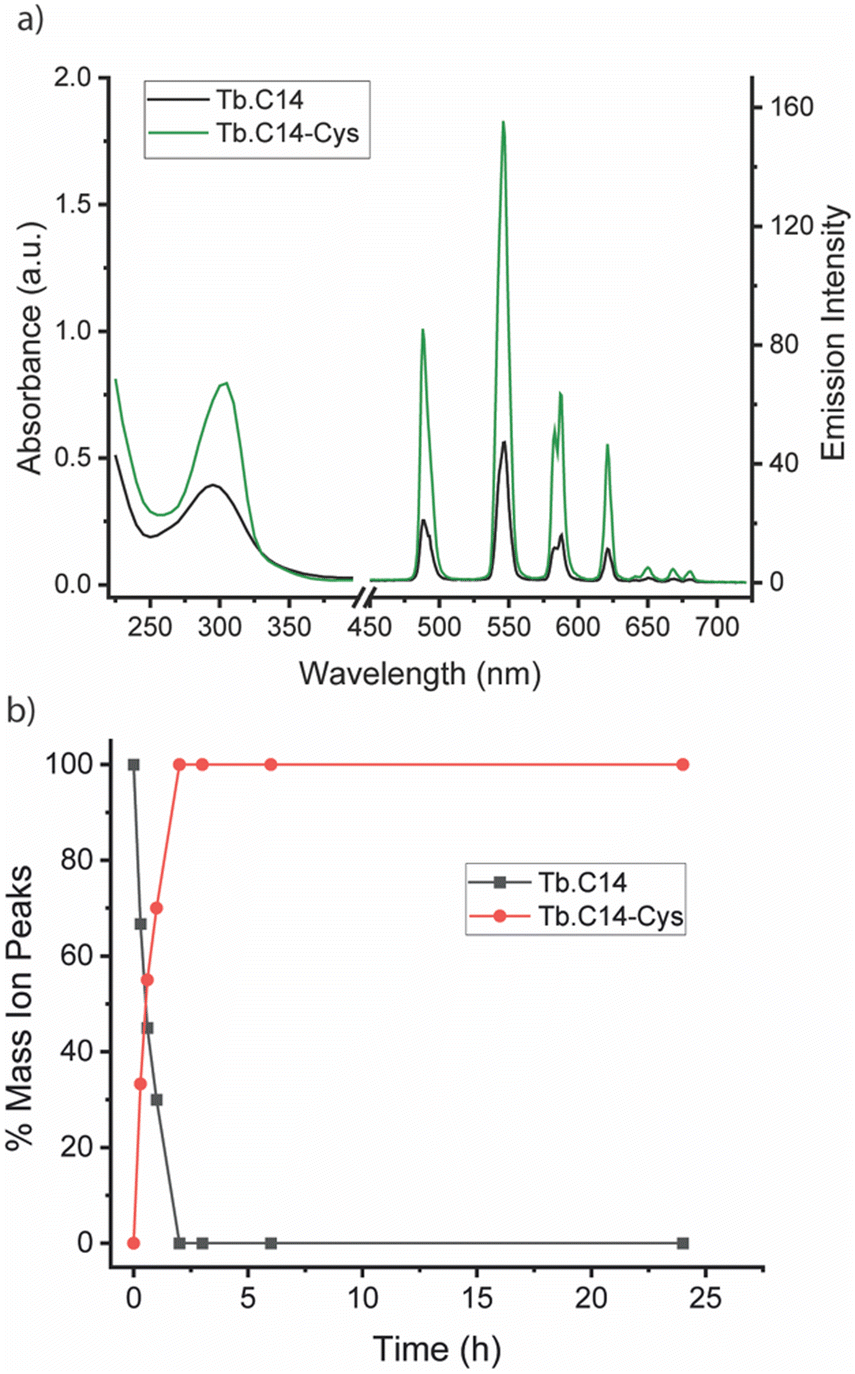 | ||
| Fig. 2 (a) Absorption and emission spectra of Tb.C14 (black) and its cysteine-tagged derivative Tb.C14-Cys (green), measured in 50 mM ammonium bicarbonate buffer at pH 8.0. To confirm that the absorbance and luminescence increase was due to the successful reaction of Tb.C14 with cysteine, Tb.C14-Cys was purified by preparative reverse-phase HPLC. (b) Monitoring the reaction between Tb.C14 and cysteine by LCMS analysis, showing quantitative reaction after 2 hours. The percentage mass ion peaks of Tb.C14 (m/z = 322.1) and Tb.C14-Cys (m/z = 346.8) are plotted as a function of time, following incubation with 4 equivalents of cysteine in water at pH 8.0 and 37 °C. | ||
The rate of reaction between Tb.C14 and cysteine was evaluated by LCMS analysis (Fig. 2b and Fig. S1, S2, ESI†). Incubation of Tb.C14 (250 μM) with four equivalents of cysteine at 37 °C resulted in quantitative labelling of the Tb(III) complex within approximately 2 hours. This is a significantly faster rate of reaction compared with the previously studied Tb.C12, which achieved approximately 95% conversion at 37 °C after 16 hours under similar conditions.8 The increased reactivity of Tb.C14 can be attributed to the ability of the N-oxide to stabilise the developing negative charge in the transition state of the rate-determining step, which is the nucleophilic addition of the thiol to the pyridyl ring. We demonstrated the ability to track the cysteine labelling reaction using UV-Vis and luminescence spectroscopy (Fig. S3, ESI†), taking advantage of the increase in both extinction coefficient and luminescence quantum yield. The spectra revealed steep increases in both the UV-Vis absorption and Tb(III) emission intensity within the first hour of the reaction, followed by stable signals after 2.5 hours, indicating quantitative reaction consistent with the LCMS data.
To explore the performance of the C14 tag for PCS measurements, we loaded the tag with Y3+, Tm3+ and Tb3+ (named Y.C14, Tm.C14 and Tb.C14, respectively) and reacted the tags with the S57C mutant of uniformly 15N-labelled ubiquitin. Full details of the tagging reaction conditions are provided in the ESI.† Mass spectra confirming tagging of the ubiquitin S57C mutant are given in Fig. S4 (ESI†). Fig. 3 shows the 15N-HSQC spectra obtained.
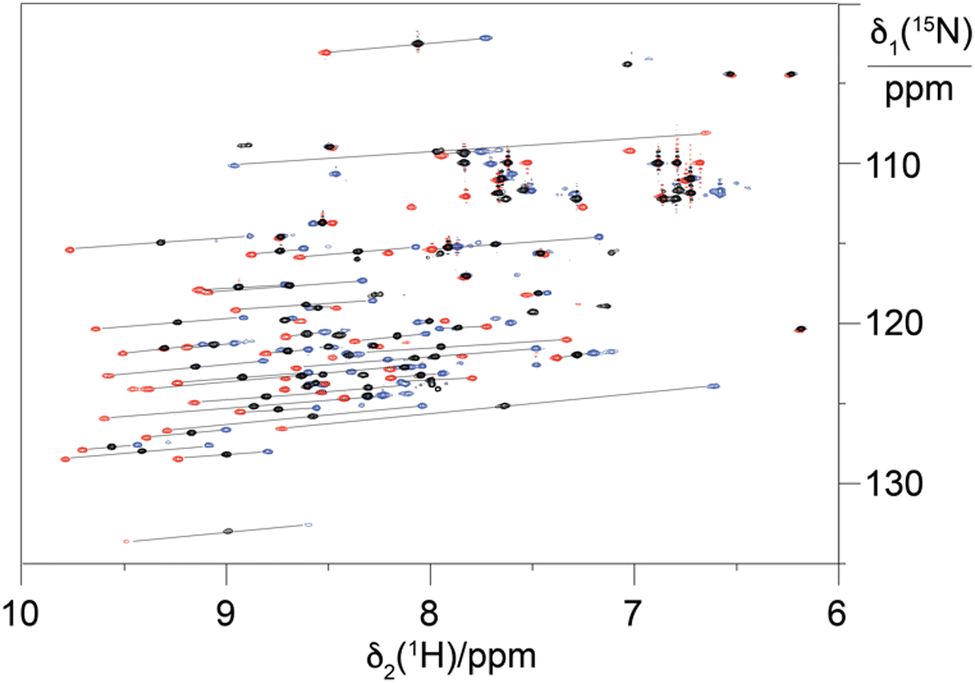 | ||
| Fig. 3 Superimposition of 15N-HSQC spectra of ubiquitin S57C ligated with the C14 tag loaded with different metal ions. The spectra recorded with diamagnetic Y.C14 tag and paramagnetic Tm.C14 and Tb.C14 are plotted in black, blue and red, respectively. Lines link paramagnetic with diamagnetic cross-peaks, identifying the PCSs. | ||
PCSs could be measured for most amide protons of the protein, allowing the accurate fitting of Δχ tensors. Table 1 compares the fitted Δχ tensors to those obtained previously for ubiquitin S57C tagged with Tm.C12 and Tb.C12. The C14 tags gave slightly smaller tensors than the C12 tags with comparable qualities of fit (Fig. S5, ESI†). Importantly, the tensor orientations are very different from those of the C12 tags (Fig. 4) and the fitted metal positions differ by about 4.5 Å (Table S2, ESI†). This shows that the single additional oxygen in the C14 tag changes the conformation of the linker between lanthanide ion and protein. In contrast, the tensor orientations obtained with Tm.C14 and Tb.C14 tags were closely similar, as expected for similar metal coordination geometries. Performing measurements with both metal ions thus adds little structural information but facilitates assignment of the paramagnetic cross-peaks, as they shift the peaks in opposite directions (Fig. 3).
| Tag | Δχax/10−23 m3 | Δχrh/10−23 m3 | Q |
|---|---|---|---|
| a The program Paramagpy17 was used to fit the PCSs of amide protons to the structure coordinates 1UBQ.18 The quality factor of each fit was calculated as the root-mean-square (RMS) of the differences between experimental and back-calculated PCSs divided by the MRS of the experimental PCSs. The full set of fitted tensor parameters is given in Table S2 (ESI). | |||
| Tm.C14 | 8.9 | 0.5 | 0.05 |
| Tb.C14 | −10.9 | −1.6 | 0.04 |
| Tm.C12 | 5.8 | 3.0 | 0.06 |
| Tb.C12 | 8.3 | 3.3 | 0.05 |
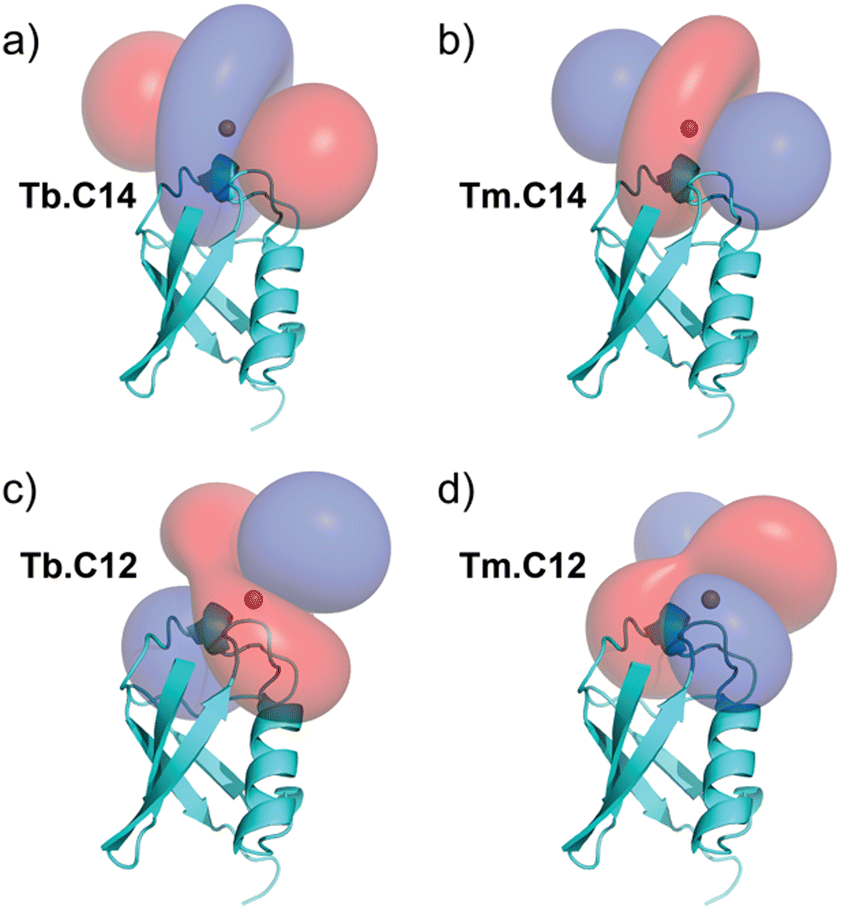 | ||
| Fig. 4 PCS isosurfaces illustrating the orientations of the Δχ tensors obtained with the (a) Tb.C14 and (b) Tm.C14 tags. (c) and (d) show the corresponding isosurfaces obtained with the previously published Tb.C12 and Tm.C12 tags, respectively, for comparison. The isosurfaces correspond to PCSs of ±1 ppm (blue and red, respectively). | ||
1D 1H-NMR spectra recorded of the free Tm.C14 tag showed chemical shifts ranging between about 270 and −160 ppm (Fig. S6, ESI†), which is comparable to the range observed for the Tm.C12 tag (250 to −150 ppm). This suggests that the intrinsic Δχ-tensor magnitudes are comparable for both tags. A q value of 2 obtained from the Tb luminescence lifetimes of Tb.C14 (Table S1, ESI†) contrasts with a value of 1 determined previously for the Tb.C12 tag, indicating that the N-oxide renders the lanthanide ion more solvent accessible.
Gadolinium tags are also of great utility for distance measurements between Gd3+ ions by EPR spectroscopy. To test the performance of the Gd.C14 tag in DEER experiments, we tagged the single-cysteine mutant S114C of the homodimeric protein ERp29.11
The EPR spectrum is very similar to that of the same protein tagged with the C12 homologue, with a slightly broader line width of the central −½ to ½ transition (Fig. S7, ESI†). This suggests that the zero-field splitting is the same or only slightly larger than that of the C12 tag. Also, the relaxation properties of the Gd.C14 tag are similar to those of the Gd.C12 tag (Fig. S8 and Table S4, ESI†) and therefore should perform equally well as an EPR spin tag.
Fig. 5 shows the DEER dipolar evolution trace and distance distribution calculated from it. The distance distribution has a Gaussian shape with mean distance of 5.7 nm. The modulation depth of the Gd.C14 DEER trace was only 0.975, compared to the 0.91 modulation depth of the Gd.C12 DEER. Subsequent analysis by non-reducing SDS-PAGE revealed a significant fraction of disulfide-bonded dimer in the ERp29 S114C sample used for Gd.C14 labelling, which would have been undetectable in the DEER experiment. Consequently, only a fraction of the protein formed the native dimer with a Gd.C14 tag in each monomer as required, explaining the relatively low modulation depth compared with ERp29 S114C tagged with Gd.C12 (Fig. 5).8 Importantly, a high-quality DEER trace was obtained despite incomplete tagging, and the relatively narrow width of the distance distribution obtained with the Gd.C14 tag indicates a smaller variation of the Gd3+–Gd3+ distance.
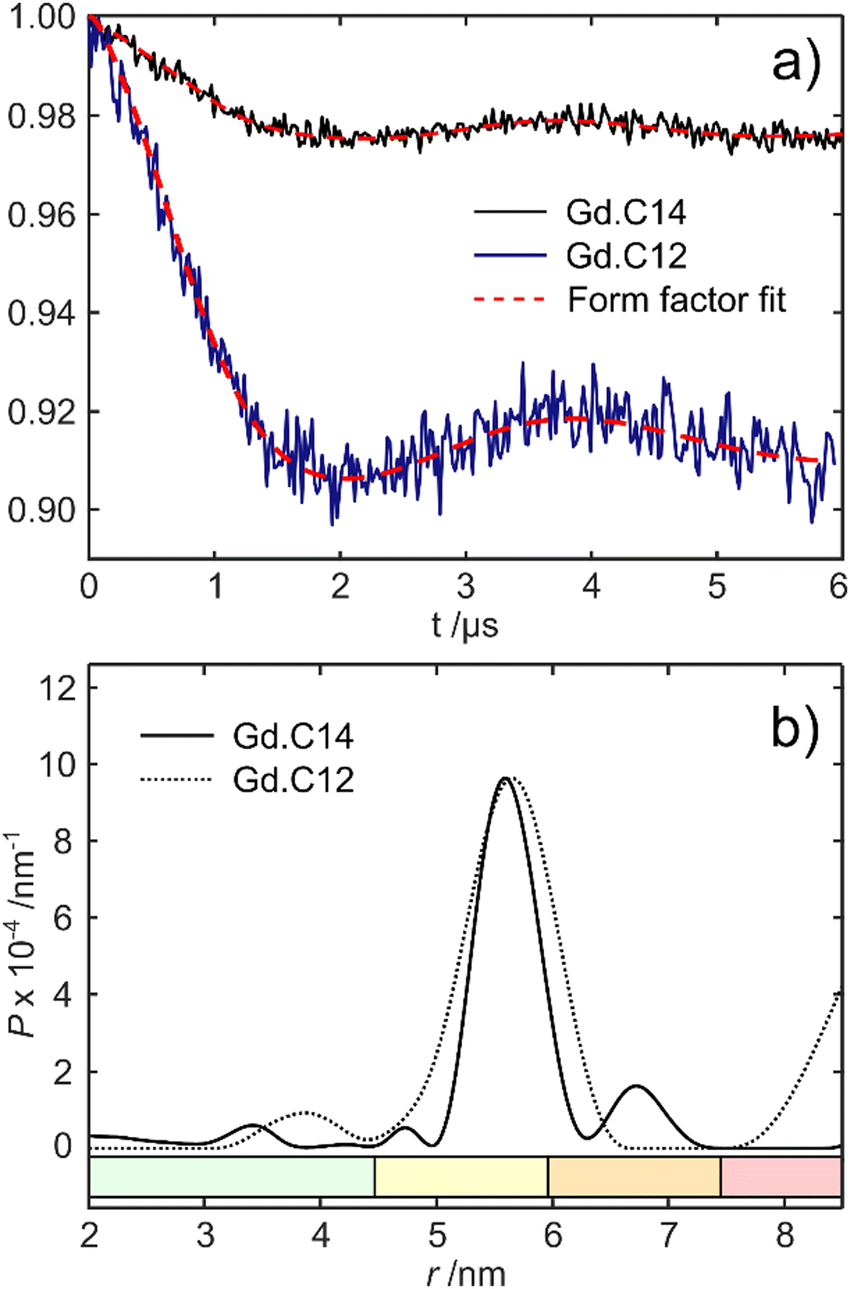 | ||
| Fig. 5 Comparison of DEER data obtained with ERp29 S114C tagged with either Gd.C14 or Gd.C12. (a) Background-subtracted dipolar evolution time trace of the protein with Gd.C14 tag (black trace) and the Gd.C12 tag (blue trace) showing the form factor fits calculated in DeerAnalysis2019 (red traces). The noise amplitudes differ because of different number of scans used. (b) Distance distributions obtained with the Gd.C14 (solid line) and Gd.C12 (dotted line) tags. The data in (a) were processed by DeerAnalysis2019 with Tikhonov regularisation top obtain the distance distributions in (b). Reliability ranges are identified by different colours: green – the shape of the distance distribution is reliable, yellow – the maximum of the distribution and its width are reliable, orange – the mean distance is reliable, red – long-range distances may be observable but cannot be quantified. | ||
In conclusion, the C14 tag proves to be as versatile as the related C12 tag for NMR, luminescence and EPR studies. Despite differing by only a single oxygen atom, the C14 tag generates different and highly complementary Δχ tensors. This behaviour can be attributed to different linker conformations between the tag and protein.
We have previously reported that the enantiomer of the C12 tag, C13, also produces different Δχ tensors,10 although the differences are smaller than those reported here. We envisage the same will be true for the enantiomer of the C14 tag. This set of four tags is thus perfectly suited for protein structure determinations from PCSs using a single tagging site.
L. T. synthesized the C14 tags and evaluated the NMR data, E. A. produced the protein, A. W. optimized tagging conditions together with L. T., M. J. and N. C. measured and interpreted the EPR data, G. O. wrote the first draft of the manuscript, and S. J. B. designed the C14 tag, coordinated the project and wrote the manuscript with G. O.
This work was supported by the Loughborough University Doctoral College for international research activity. Financial support by the Australian Research Council (grants CE200100012 and DP230100079) is gratefully acknowledged.
Data availability
The data supporting this article have been included as part of the ESI,† including synthesis and characterisation of Ln.C14, photophysical analysis of Tb.C14 and Tb.C14-Cys, NMR and EPR spectroscopic data of protein tagging experiments.Conflicts of interest
There are no conflicts to declare.Notes and references
- Q. Miao, C. Nitsche, H. Orton, M. Overhand, G. Otting and M. Ubbink, Chem. Rev., 2022, 122, 9571–9642 CrossRef CAS.
- H. Yagi, K. B. Pilla, A. Maleckis, B. Graham, T. Huber and G. Otting, Structure, 2013, 21, 883–890 CrossRef CAS PubMed.
- A. Feintuch, G. Otting and D. Goldfarb, in Methods in Enzymology, ed. P. Z. Qin and K. Warncke, Academic Press, 2015, vol. 563, pp. 415–457 Search PubMed.
- E. G. Moore, J. Xu, C. J. Jocher, T. M. Corneillie and K. N. Raymond, Inorg. Chem., 2010, 49, 9928–9939 CrossRef CAS.
- J. M. Zwier, H. Bazin, L. Lamarque and G. Mathis, Inorg. Chem., 2014, 53, 1854–1866 CrossRef CAS PubMed.
- S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, D. Parker, J. W. Walton and J. M. Zwier, Dalton Trans., 2015, 44, 4791–4803 RSC.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef.
- I. D. Herath, C. Breen, S. H. Hewitt, T. R. Berki, A. F. Kassir, C. Dodson, M. Judd, S. Jabar, N. Cox, G. Otting and S. J. Butler, Chem. – Eur. J., 2021, 27, 13009–13023 CrossRef CAS PubMed.
- S. Wheeler and S. J. Butler, Anal. Sens., 2022, 3, e202200036 Search PubMed.
- H. W. Orton, E. H. Abdelkader, L. Topping, S. J. Butler and G. Otting, Magn. Reson., 2022, 3, 65–76 CrossRef CAS.
- H. Yagi, D. Banerjee, B. Graham, T. Huber, D. Goldfarb and G. Otting, J. Am. Chem. Soc., 2011, 133, 10418–10421 CrossRef CAS.
- K. B. Pilla, G. Otting and T. Huber, J. Mol. Biol., 2016, 428, 522–532 CrossRef CAS.
- F.-J. Wu, P. S. Rieder, L. A. Abiko, P. Rößler, A. D. Gossert, D. Häussinger and S. Grzesiek, J. Am. Chem. Soc., 2022, 144, 21728–21740 CrossRef CAS.
- I. Bertini, M. B. L. Janik, Y.-M. Lee, C. Luchinat and A. Rosato, J. Am. Chem. Soc., 2001, 123, 4181–4188 CrossRef CAS.
- D. Shishmarev and G. Otting, J. Biomol. NMR, 2013, 56, 203–216 CrossRef CAS PubMed.
- K. L. Gempf, S. J. Butler, A. M. Funk and D. Parker, Chem. Commun., 2013, 49, 9104–9106 RSC.
- H. W. Orton, T. Huber and G. Otting, Magn. Reson., 2020, 1, 1–12 CrossRef.
- S. Vijay-Kumar, C. E. Bugg and W. J. Cook, J. Mol. Biol., 1987, 194, 531–544 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03007a |
| ‡ The tag demonstrated excellent stability during shipment between UK and Australian laboratories, maintaining its structural and functional integrity throughout the journey in solid form. Moreover, it can be stored at room temperature for several months without any degradation, ensuring reliability and ease of use in various protein tagging applications. |
| This journal is © The Royal Society of Chemistry 2024 |