DOI:
10.1039/D4CC02165G
(Communication)
Chem. Commun., 2024,
60, 8462-8465
An anchor for help: a cross-linking moiety for block copolymer membrane stabilization for ultrafiltration applications in water†
Received
4th May 2024
, Accepted 16th July 2024
First published on 17th July 2024
Abstract
Block copolymer membranes are an exciting class of materials used to separate small contaminants from water. Covalent cross-linking of the membrane matrix is one approach to alleviate stability issues, which limit their application nowadays. In the current work, membranes from amphiphilic block copolymers are manufactured and cross-linked using a UV-active radical initiator moiety.
Purifying mixtures without using heat would lower global energy use, emissions, and pollution – and open up new routes to resources.1 With these words opens a comment by Sholl and Lively recently published. One possibility to facilitate purification steps is separation via membrane technologies. Currently, there is already a diverse portfolio of interesting materials available, ranging from inorganic ceramics to composites to polymers and combinations thereof. Ultrafiltration membranes can be manufactured via different approaches, where the self-assembly and non-solvent induced phase separation (SNIPS) process, pioneered by Peinemann et al.,2 produces tailored isoporous integral asymmetric membranes. Within this strategy, an amphiphilic block copolymer (BCP) is dissolved in a solvent mixture of a selective solvent for one block segment, and a non-selective solvent. During the membrane formation process, the amphiphilic BCP undergoes self-assembly into isoporous membranes with pore sizes usually in the range of 20–70 nm, according to the microphase-separated morphologies of the underlying BCP.3,4 For more information on this process the authors refer to some recent and seminal reviews in the field.5–7 Negatively, these membranes suffer from poor oxidative/chlorine-, pH-, thermal- and compacting resistance.8 Within the present study, we aim to increase the compacting resistance of these polymeric membranes. On the one hand, the fabrication of composite membranes can result in more stable membranes as shown by various groups.9,10 These composite membranes feature improved antifouling and mechanical- and chemical resistance, although the applicability of these composites in combination with the SNIPS process has not yet been shown. Another way to enhance the compacting resistance is the cross-linking strategy of the membrane matrix, as shown by Luo et al., who used metal ions to cross-link their carboxyl group functionalized polyimide membranes for organic solvent filtration.11 Another method was used by Yang et al. who used UV-light to cross-link the polystyrene matrix of their membrane,12 or Sierke et al. who used thiol–ene click reactions to achieve cross-linking of their dehydrofluorinated PVDF membrane.13 Another interesting approach is utilizing cycloaddition reactions, like the Diels–Alder Reaction, as shown by Rangou et al., who elegantly used a postmodified polymer to prepare ordered BCP membranes.14 Recently, we described a BCP with an incorporated UV-activated radical generator and its application in ultrafiltration membranes focusing on its enhanced solvent resistance.15 In the present work, the membrane matrix further consists of a statistical copolymer of methyl methacrylate (MMA), n-butyl methacrylate (BMA) and allyl methacrylate (ALMA). The allylic double bonds in ALMA are later utilized as a feasible anchoring group to enable a higher and more uniform cross-linking density by a radical grafting-through polymerization approach throughout this matrix, by enabling the reaction between the generated radical and saturated hydrocarbons as well as other remaining double bonds, compared to earlier work.16 A two-step atom transfer radical polymerization (ATRP) and two different initiators were investigated to tailor the polymer architectures on the molecular level. On the one hand, the initiator tert-butyl 2-bromo-2-methylpropionate (tBib) is investigated because of its structural similarity to methacrylate monomers. On the other hand, ethyl α-bromo phenylacetate (EPBA) as a more reactive ATRP initiator is compared.17 The reaction temperature was chosen with a maximum of 65 °C, as side reactions stemming from the allylic double bond of ALMA during the polymerization reaction can be suppressed, as reported by Mennicken et al.18 Additionally, a low conversion is also favorable for the methacrylic double bond reaction over the allylic moiety reactions.19 The hydrophilic block was chosen to be poly(2-hydroxy ethyl methacrylate) (P(HEMA)), because of its desirable antifouling, water flux and biocompatibility features as well as its better suitability for radical reactions compared to its acrylate derivate.20–23 Generally, the formation of well-defined BCPs through ATRP can be accomplished, when there is a halogen exchange during the reaction of the second block segment.24 Here, the halogen terminating the polymer chain is exchanged from a more reactive one like bromine to a less reactive one like chlorine. Therefore, it is ensured, that the initiation of the second monomer is faster than its chain propagation. Several BCPs reported utilizing ALMA as a cross-linkable component,25–27 but to the best of our knowledge this monomer has not been used to form a cross-linkable BCP membrane. For this purpose, the macroinitiator P(ALMA-co-BMA-co-MMA) (MI) was synthesized using the two different initiators tBib and EPBA, followed by the preparation of a hydrophilic block consisting of P(HEMA) in a second ATRP step, as shown in Scheme 1. The reaction conditions for the synthesis of P(ALMA-co-BMA-co-MMA)-b-P(HEMA) (BCP) were varied concerning monomer, solvent, and catalyst system to find the best polymerization conditions and to account for the ALMA units in the macroinitiator. The polymerization of the macroinitiator proceeded with high control over molecular weight and distribution with both investigated initiators, as can be concluded from the dispersity index values shown in Table 1. The EPBA initiator, however, allowed for a narrower distribution compared to the tBib initiator, presumably because of the higher initiation rate. Furthermore, the comparison between 60 °C and 65 °C showed a lower dispersity index for the lower temperature (Fig. S1, ESI†). This is attributed to the increased control over the ATRP. All polymers MI 1 to MI 5 exhibited an increased incorporation rate for ALMA, and a decreased MMA content. Comparison of the corresponding NMR spectra and the integral of the –CH2 group of ALMA-sidechain to the double-bond protons revealed a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 ratio (Fig. S2, ESI†), which indicated no reaction of the allylic double bond. The ratios between the different monomers were kept constant in MI 1 to MI 5 to make a comparison between the different BCPs possible. First, during the reaction in a polar solvent mixture of 1-propanol and methyl ethyl ketone (MEK), only a low amount of hydrophilic component (1–3 mol%, Table 1) could be achieved with both HEMA and HEMA-TMS (4%, Table 1). Although the usage of HEMA-TMS over HEMA (BCP 2 vs. BCP 3) afforded a higher amount of P(HEMA), a formation of a shoulder at a higher molar mass (Fig. S3, ESI†) was observed. The problems for this synthesis were attributed to several factors: (i) the macroinitiator only had a limited solubility in the chosen solvent mixture, which is necessary for the polymerization of the HEMA monomer. (ii) the reaction mixture had a high viscosity, which was more pronounced at higher molecular weight for the macroinitiator. To solve this issue, the catalyst system was changed from the 2,2′-bypridine (Bpy) to N,N,N′,N′′,N′′-pentamethyl diethylenetriamine (PMDETA) ligand to gain higher molar masses for the same reaction time due to the higher reactivity of the latter ligand.28 As can be concluded from the results for BCP 4 and 5, a higher amount of P(HEMA) was obtained. Although, the formation of the shoulder at higher MW was more pronounced (Fig. S3, ESI†), than for polymer BCP 3. Lastly, the solvent was exchanged for the less polar anisole, combined with the more active catalyst system of PMDETA. This change alleviated the solubility problems of the macroinitiator in the more polar solvent mixture. As can be concluded from SEC traces of BCP 6 and BCP 7 (Fig. S3, ESI†), the usage of the bromine salt again led to the formation of a more pronounced shoulder, which was absent in the case of the reaction with chlorine salt. Since the propagation of bromine-terminated chain ends is reported to be a magnitude faster than chlorine-terminated ones,29 the inhibition of chain growth speed successfully leads to a uniform initiation of the polymerization of HEMA from the macroinitiator. From the SEC traces of the BCPs 1 and 2 with a small amount of P(HEMA) as second block an apparent reduction in Mn can be observed before the two distributions start to match maxima at higher contents of P(HEMA) again. This is attributed to the changes in hydrodynamic volume due to the hydroxy groups of the PHEMA block segment. These hydroxy groups furthermore lead to an increase
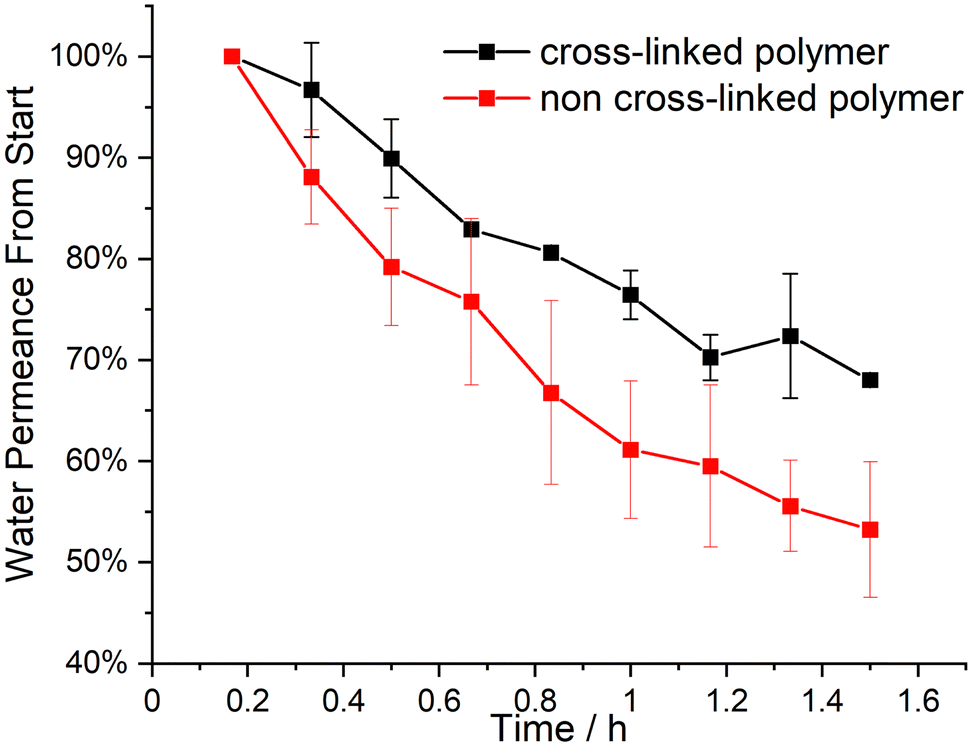
in dispersity due to interactions during SEC measurements. To give proof of BCP formation, the hydroxy group of the incorporated HEMA monomer was protected by a benzoyl-protecting group. This protection also eliminates the interactions of the –OH groups and decreases dispersity (BCP 6 vs. BCP 6-benz). The theoretical molecular weight, calculated via1H-NMR measurements, and the molecular weight of the macroinitiator agreed with the measured value through SEC. In Table 1, all analytical data for the discussed polymers are given. Using the macroinitiator MI 5, the cross-linking reaction for the polymer films was examined. As radical initiator benzophenone (BP) was added with 1, 2 and 5 wt% concerning the amount of polymer, to enable the cross-linking reaction through the allylic double bonds of ALMA. UV-light was applied through a broadband UV source operating at 1000 W. As can be concluded from Fig. 1e, after an induction period of 2 min, the amount of THF-soluble polymer decreased sharply. For the samples with 1 and 2% BP, around 50% of the polymer became insoluble at 5 min and around 80% after 10 min. Compared to this finding, with 5% BP the insoluble polymer fraction reached 100% after the full duration time, with around 80% after 5 min. This effect can be observed in the photographs in Fig. (1a)–(d), as the polymer/THF mixture transitions from a solution to a gel. As conclusion from these tests, the macroinitiator with 5 wt% of BP was the most efficient strategy for enabling cross-linking. Subsequently, the solubility of BP was tested in THF, DMF and 1,4-dioxane, where good solubility was found for all three solvents. BCP 6 was chosen to investigate the applicability of the SNIPS process because of the amount of P(HEMA), i.e. the hydrophilic pore-forming segment, and its monomodal molar mass distribution. For HEMA-containing polymer systems, mole fractions of around 10% have been found to enable porous cylinder formation.15,30 For the SNIPS process, the polymer was dissolved in a mixture of THF, DMF and 1,4-dioxane (2:1:1 by weight) in a semi-concentrated solution featuring 0.5% of CuCl2 to enhance micellization and processability through coordination, similar to previous work published22 and 5% BP was added. This polymer solution was cast on a PE/PP nonwoven with a doctor blade with a blade gap of 200 μm. In Fig. 2, the topography of the resulting BCP membrane is shown, together with a cross-section, for both prior to the UV cross-linking reaction (Fig. 2(a) and (c)) and after (Fig. 2(b) and (d)). From the SEM micrographs, an integral asymmetric membrane structure could be observed, i.e., an isoporous top layer followed by a sponge-like support layer as expected for SNIPS membranes. Pore diameters of 33 ± 6 nm before the UV exposure and 31 ± 5 nm after UV exposure could be determined. To test the SNIPS membranes’ compacting resistance, water permeance measurements were carried out. For these measurements, membranes without exposure to UV-light, and membranes with exposure for 5 and 10 min were used. The membranes with 10 min exposure did not exhibit water permeance and are therefore not shown in Fig. 3 (SEM micrographs in ESI,† Fig. S4). The high amount of energy introduced into the BCP membranes during UV exposure has likely caused the porous structure to collapse after 10 min. The permeance was measured in 10 min intervals over 1.5 h, according to a modified standard procedure in our group (cf. Material section). The reduction in permeance with increasing time under pressure is a well-known phenomenon for polymer membranes, as the applied mechanical pressure is thought to squeeze the membrane and therefore reduce the free volume inside of the porous membrane.31 In Fig. 3 the red line indicates the behavior of the non-cross-linked membrane. During the measurements, a reduction in water permeance of around 50% compared to the initial value could be observed. Comparatively, after cross-linking the reduction is only about 30%, signifying the enhanced compacting resistance through the cross-linking process and the associated stiffening of the polymer matrix of the membrane. In conclusion, the amphiphilic BCP P(ALMA-co-BMA-co-MMA)-b-P(HEMA) featuring cross-linkable ALMA sites were successfully synthesized via a two-step ATRP. The importance of the fast initiation was highlighted in the synthesis of the macroinitiator, followed by the feasibility of a halogen exchange from bromine to chlorine for the polymerization of HEMA monomer. The prepared BCPs were used to fabricate ultrafiltration membranes featuring a pore size of approximately 30 to 50 nm. The UV-mediated cross-linking reaction of these membranes, using benzophenone as a radical initiator and ALMA as a cross-linkable unit, enhanced the compacting resistance significantly. The present work paves the way for a universally applicable synthesis strategy to enhance the performance of porous BCP membranes in the field of water filtration processes.
:1:1:1 ratio (Fig. S2, ESI†), which indicated no reaction of the allylic double bond. The ratios between the different monomers were kept constant in MI 1 to MI 5 to make a comparison between the different BCPs possible. First, during the reaction in a polar solvent mixture of 1-propanol and methyl ethyl ketone (MEK), only a low amount of hydrophilic component (1–3 mol%, Table 1) could be achieved with both HEMA and HEMA-TMS (4%, Table 1). Although the usage of HEMA-TMS over HEMA (BCP 2 vs. BCP 3) afforded a higher amount of P(HEMA), a formation of a shoulder at a higher molar mass (Fig. S3, ESI†) was observed. The problems for this synthesis were attributed to several factors: (i) the macroinitiator only had a limited solubility in the chosen solvent mixture, which is necessary for the polymerization of the HEMA monomer. (ii) the reaction mixture had a high viscosity, which was more pronounced at higher molecular weight for the macroinitiator. To solve this issue, the catalyst system was changed from the 2,2′-bypridine (Bpy) to N,N,N′,N′′,N′′-pentamethyl diethylenetriamine (PMDETA) ligand to gain higher molar masses for the same reaction time due to the higher reactivity of the latter ligand.28 As can be concluded from the results for BCP 4 and 5, a higher amount of P(HEMA) was obtained. Although, the formation of the shoulder at higher MW was more pronounced (Fig. S3, ESI†), than for polymer BCP 3. Lastly, the solvent was exchanged for the less polar anisole, combined with the more active catalyst system of PMDETA. This change alleviated the solubility problems of the macroinitiator in the more polar solvent mixture. As can be concluded from SEC traces of BCP 6 and BCP 7 (Fig. S3, ESI†), the usage of the bromine salt again led to the formation of a more pronounced shoulder, which was absent in the case of the reaction with chlorine salt. Since the propagation of bromine-terminated chain ends is reported to be a magnitude faster than chlorine-terminated ones,29 the inhibition of chain growth speed successfully leads to a uniform initiation of the polymerization of HEMA from the macroinitiator. From the SEC traces of the BCPs 1 and 2 with a small amount of P(HEMA) as second block an apparent reduction in Mn can be observed before the two distributions start to match maxima at higher contents of P(HEMA) again. This is attributed to the changes in hydrodynamic volume due to the hydroxy groups of the PHEMA block segment. These hydroxy groups furthermore lead to an increase
in dispersity due to interactions during SEC measurements. To give proof of BCP formation, the hydroxy group of the incorporated HEMA monomer was protected by a benzoyl-protecting group. This protection also eliminates the interactions of the –OH groups and decreases dispersity (BCP 6 vs. BCP 6-benz). The theoretical molecular weight, calculated via1H-NMR measurements, and the molecular weight of the macroinitiator agreed with the measured value through SEC. In Table 1, all analytical data for the discussed polymers are given. Using the macroinitiator MI 5, the cross-linking reaction for the polymer films was examined. As radical initiator benzophenone (BP) was added with 1, 2 and 5 wt% concerning the amount of polymer, to enable the cross-linking reaction through the allylic double bonds of ALMA. UV-light was applied through a broadband UV source operating at 1000 W. As can be concluded from Fig. 1e, after an induction period of 2 min, the amount of THF-soluble polymer decreased sharply. For the samples with 1 and 2% BP, around 50% of the polymer became insoluble at 5 min and around 80% after 10 min. Compared to this finding, with 5% BP the insoluble polymer fraction reached 100% after the full duration time, with around 80% after 5 min. This effect can be observed in the photographs in Fig. (1a)–(d), as the polymer/THF mixture transitions from a solution to a gel. As conclusion from these tests, the macroinitiator with 5 wt% of BP was the most efficient strategy for enabling cross-linking. Subsequently, the solubility of BP was tested in THF, DMF and 1,4-dioxane, where good solubility was found for all three solvents. BCP 6 was chosen to investigate the applicability of the SNIPS process because of the amount of P(HEMA), i.e. the hydrophilic pore-forming segment, and its monomodal molar mass distribution. For HEMA-containing polymer systems, mole fractions of around 10% have been found to enable porous cylinder formation.15,30 For the SNIPS process, the polymer was dissolved in a mixture of THF, DMF and 1,4-dioxane (2:1:1 by weight) in a semi-concentrated solution featuring 0.5% of CuCl2 to enhance micellization and processability through coordination, similar to previous work published22 and 5% BP was added. This polymer solution was cast on a PE/PP nonwoven with a doctor blade with a blade gap of 200 μm. In Fig. 2, the topography of the resulting BCP membrane is shown, together with a cross-section, for both prior to the UV cross-linking reaction (Fig. 2(a) and (c)) and after (Fig. 2(b) and (d)). From the SEM micrographs, an integral asymmetric membrane structure could be observed, i.e., an isoporous top layer followed by a sponge-like support layer as expected for SNIPS membranes. Pore diameters of 33 ± 6 nm before the UV exposure and 31 ± 5 nm after UV exposure could be determined. To test the SNIPS membranes’ compacting resistance, water permeance measurements were carried out. For these measurements, membranes without exposure to UV-light, and membranes with exposure for 5 and 10 min were used. The membranes with 10 min exposure did not exhibit water permeance and are therefore not shown in Fig. 3 (SEM micrographs in ESI,† Fig. S4). The high amount of energy introduced into the BCP membranes during UV exposure has likely caused the porous structure to collapse after 10 min. The permeance was measured in 10 min intervals over 1.5 h, according to a modified standard procedure in our group (cf. Material section). The reduction in permeance with increasing time under pressure is a well-known phenomenon for polymer membranes, as the applied mechanical pressure is thought to squeeze the membrane and therefore reduce the free volume inside of the porous membrane.31 In Fig. 3 the red line indicates the behavior of the non-cross-linked membrane. During the measurements, a reduction in water permeance of around 50% compared to the initial value could be observed. Comparatively, after cross-linking the reduction is only about 30%, signifying the enhanced compacting resistance through the cross-linking process and the associated stiffening of the polymer matrix of the membrane. In conclusion, the amphiphilic BCP P(ALMA-co-BMA-co-MMA)-b-P(HEMA) featuring cross-linkable ALMA sites were successfully synthesized via a two-step ATRP. The importance of the fast initiation was highlighted in the synthesis of the macroinitiator, followed by the feasibility of a halogen exchange from bromine to chlorine for the polymerization of HEMA monomer. The prepared BCPs were used to fabricate ultrafiltration membranes featuring a pore size of approximately 30 to 50 nm. The UV-mediated cross-linking reaction of these membranes, using benzophenone as a radical initiator and ALMA as a cross-linkable unit, enhanced the compacting resistance significantly. The present work paves the way for a universally applicable synthesis strategy to enhance the performance of porous BCP membranes in the field of water filtration processes.
M. G. expresses his gratitude for the partial financial support provided by the European Union through the European Regional Development Fund (EFRE) and the State of Saarland, Germany, in the SWIMEMSYS project.
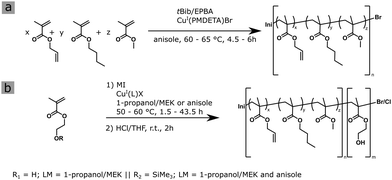 |
| | Scheme 1 Synthesis of the amphiphilic BCP P(ALMA-co-BMA-co-MMA)-b-P(HEMA) in a two-step ATRP. (a) Synthesis for the macroinitiator MI. (b) General procedure for the synthesis of the hydrophilic block segment with HEMA or HEMA-TMS, respectively. | |
Table 1 Overview of the analytical data for the macroinitiators and BCPs. The amount of substances was calculated from the corresponding 1H-NMR spectra. Molecular weights, as well as dispersity index values, were determined from SEC measurements in THF (MI) and DMF (BCP), respectively
| Polymer |
x(ALMA)/% |
x(BMA)/% |
x(MMA)/% |
M
n/g mol−1 |
Đ
|
|
Measured in THF vs. PS standard.
|
| MI 1 |
6 |
44 |
50 |
62900 |
1.12 |
| MI 2 |
6 |
44 |
50 |
59300 |
1.10 |
| MI 3 |
6 |
44 |
50 |
39600 |
1.05 |
| MI 4 |
6 |
44 |
50 |
46900 |
1.09 |
| MI 5 |
6 |
44 |
50 |
62400 |
1.24 |
| Polymer |
MI |
x(HEMA)/% |
M
n,SEC/g mol−1 |
M
n,calc./g mol−1 |
Đ
|
| BCP 1 |
1 |
3 |
52900 |
65000 |
1.33 |
| BCP 2 |
1 |
1 |
50700 |
63900 |
1.34 |
| BCP 3 |
3 |
4 |
42600 |
41400 |
1.25 |
| BCP 4 |
4 |
9 |
54500 |
51800 |
1.93 |
| BCP 5 |
4 |
11 |
57600 |
53200 |
1.43 |
| BCP 6 |
5 |
11 |
70400 |
71000 |
1.70 |
| BCP 6_benz |
5 |
11 |
80200a |
79800 |
1.46 |
| BCP 7 |
2 |
7 |
67500 |
64000 |
2.53 |
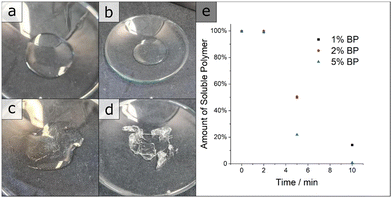 |
| | Fig. 1 Photographs of the polymer MI 5, featuring 5% benzophenone after UV exposure and subsequent treatment with THF. (a) 0 min (b) 2 min (c) 5 min and (d) 10 min (e) Graph of the amount of insoluble polymer determined gravimetrically after the cross-linking procedure. | |
 |
| | Fig. 2 SEM photographs of the SNIPS membrane of BCP 6. (a) and (c) Are photographs of the neat membrane, and (b) and (d) are photographs of the same membrane after 5 min of UV exposure. | |
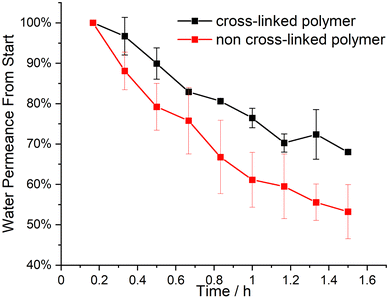 |
| | Fig. 3 Relative water permeance of the prepared BCP membranes from BCP 6 compared to their starting values, the red lines show the non-cross-linked polymer (starting at 530 L m−2 bar−1 h−1) and the black ones the cross-linked one (starting at 659 L m−2 bar−1 h−1). | |
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- K.-V. Peinemann, V. Abetz and P. F. W. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- M. Gallei, S. Rangou, V. Filiz, K. Buhr, S. Bolmer, C. Abetz and V. Abetz, Macromol. Chem. Phys., 2013, 214, 1037–1046 CrossRef CAS.
- S. Rangou, K. Buhr, V. Filiz, J. I. Clodt, B. Lademann, J. Hahn, A. Jung and V. Abetz, J. Membr. Sci., 2014, 451, 266–275 CrossRef CAS.
- S. P. Nunes, Macromolecules, 2016, 49, 2905–2916 CrossRef CAS.
- V. Abetz, Macromol. Rapid Commun., 2015, 36, 10–22 CrossRef CAS.
- K. Foroutani and S. M. Ghasemi, Macromol. Mater. Eng., 2022, 307(8), 2200084 CrossRef CAS.
- M. Q. Seah, S. F. Chua, W. L. Ang, W. J. Lau, A. Mansourizadeh and C. Thamaraiselvan, J. Environ. Chem. Eng., 2024, 12(3), 112628 CrossRef CAS.
- H.-Q. Liang, Q.-Y. Wu, L.-S. Wan, X.-J. Huang and Z.-K. Xu, J. Membr. Sci., 2014, 465, 56–67 CrossRef CAS.
- N. Kamal, S. Ahzi and V. Kochkodan, Appl. Clay Sci., 2020, 199, 105873 CrossRef CAS.
- X. Luo, Z. Wang, S. Wu, W. Fang and J. Jin, J. Membr. Sci., 2021, 621, 119002 CrossRef CAS.
- S. Y. Yang, J. Park, J. Yoon, M. Ree, S. K. Jang and J. K. Kim, Adv. Funct. Mater., 2008, 18, 1371–1377 CrossRef CAS.
- J. Sierke and A. V. Ellis, J. Membr. Sci., 2019, 581, 362–372 CrossRef CAS.
- S. Rangou, M. Appold, B. Lademann, K. Buhr and V. Filiz, ACS Macro Lett., 2022, 11, 1142–1147 CrossRef CAS PubMed.
- F. V. Frieß, Q. Hu, J. Mayer, L. Gemmer, V. Presser, B. N. Balzer and M. Gallei, Macromol. Rapid Commun., 2021, 43(3), 2100632 CrossRef.
- L. Siegwardt and M. Gallei, Chem. Eng. J., 2024, 480, 148168 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS.
- M. Mennicken, R. Nagelsdiek, H. Keul and H. Höcker, Macromol. Chem. Phys., 2004, 205, 2429–2437 CrossRef CAS.
- R. Nagelsdiek, M. Mennicken, B. Maier, H. Keul and H. Höcker, Macromolecules, 2004, 37, 8923–8932 CrossRef CAS.
- K. L. Beers, S. Boo, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1999, 32, 5772–5776 CrossRef CAS.
- S. S. Rao, S. G. Jeyapal and S. Rajiv, Composites, Part B, 2014, 60, 43–48 CrossRef CAS.
- S. Schöttner, H.-J. Schaffrath and M. Gallei, Macromolecules, 2016, 49, 7286–7295 CrossRef.
- M. Ulbricht and G. Belfort, J. Membr. Sci., 1996, 111, 193–215 CrossRef CAS.
- K. Matyjaszewski, D. A. Shipp, J.-L. Wang, T. Grimaud and T. E. Patten, Macromolecules, 1998, 31, 6836–6840 CrossRef CAS.
- R. París and J. L. D. L. Fuente, Eur. Polym. J., 2008, 44, 1403–1413 CrossRef.
- R. París and J. L. De La Fuente, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3538–3549 CrossRef.
- B. Zhang, X. Lv, A. Zhu, J. Zheng, Y. Yang and Z. An, Macromolecules, 2018, 51, 2776–2784 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
- C. Y. Lin, M. L. Coote, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 12762–12774 CrossRef CAS PubMed.
- M. Plank, F. V. Frieß, C. V. Bitsch, J. Pieschel, J. Reitenbach and M. Gallei, Macromolecules, 2023, 56, 1674–1687 CrossRef CAS.
- K. M. Persson, V. Gekas and G. Trägårdh, J. Membr. Sci., 1995, 100, 155–162 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab