
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMn2+ activated Ca-α-SiAlON – broadband deep-red luminescence and sensitization by Eu2+, Yb2+ and Ce3+†
Atul D.
Sontakke
 *a,
Arnoldus J.
van Bunningen
a,
Sadakazu
Wakui
ab and
Andries
Meijerink
*a
*a,
Arnoldus J.
van Bunningen
a,
Sadakazu
Wakui
ab and
Andries
Meijerink
*a
aCondensed Matter & Interfaces, Debye Institute for Nanomaterials Science, Utrecht University, Princetonplein 1, 3584 CC Utrecht, The Netherlands. E-mail: a.d.sontakke@uu.nl; a.meijerink@uu.nl
bNichia Corporation, 491 Oka, Kaminaka-Cho, Anan-Shi, Tokushima 774-8601, Japan
First published on 22nd February 2021
Abstract
The covalent character of (oxy)nitrides has enabled tuning of emission color for new Eu2+- and Ce3+-doped phosphors and widespread implementation in white light LEDs. Mn2+-doped (oxy)nitrides form a relatively unexplored class of luminescent materials with great potential as orange to near infrared (NIR) phosphor in white light and NIR LEDs. Here we report deep red broad band emission around 700 nm for Mn2+ in Ca-α-SiAlON. The emission shows a complex and gradual temperature quenching behavior and an unexpectedly large blue shift of the band maximum from 740 nm at 75 K to 670 nm at 475 K. This peculiar luminescence behavior is explained by disorder in the local (N,O)7 polyhedral coordination of the Mn2+ ions resulting in a variation in emission maximum and quenching temperature for differently coordinated Mn2+ ions. Interestingly, the present results deviate from previous reports on narrow band orange emission from Mn2+ in Ca-α-SiAlON. Careful analysis reveals that the orange emission likely originates from an AlN:Mn2+ impurity phase. Sensitization of the Mn2+ emission by strongly absorbing Eu2+, Yb2+ and Ce3+ ions was observed but with low efficiency as energy transfer to Mn2+via exchange interaction is ineffective for the relatively large (>5 Å) nearest neighbor separation in Ca-α-SiAlON. The new insights on Mn2+ emission show that for efficient orange/red emission in (oxy)nitrides, a tetrahedral coordination is required for Mn2+ while deep red/NIR emission can be expected for higher coordination numbers but is prone to thermal quenching. Sensitization of the Mn2+ emission is efficient only for exchange interaction and requires short (<5 Å) nearest neighbor separation.
A Introduction
Nitride and oxynitride compounds have emerged as efficient hosts for phosphor applications, and are widely commercialized.1 Although their synthesis is non-trivial and requires special processing conditions, they have the ability to achieve superior structural rigidity and their higher bond covalency offers unique optical properties that are difficult to achieve in oxides and other traditional hosts. This has primarily been exploited to achieve efficient red emitting phosphors, such as (Ca,Sr)AlSiN3:Eu2+ (CASN:Eu2+ or CSASN:Eu2+),2,3 (Sr,Ba)2Si5N8:Eu2+,4,5 SrLiAl3N4:Eu2+ (SLA:Eu2+),6 Sr[Li2Al2O2N2]:Eu2+ (SALON:Eu2+),7etc. The dopant ions surrounded by a nitrogen-rich covalent anionic coordination exerts a large nephelauxetic shift, which together with strong crystal field and rigid lattice network allow longer wavelength and relatively narrow-band emission with superior thermal stability.In view of the recent search for narrow-band emitting phosphors for wider color gamut displays and higher efficacy (lumen/watt) lighting, nitride and oxynitride based phosphors attracted special attention. Hoerder et al., have demonstrated record narrow-bandwidth for Eu2+ red emission in SALON with full width at half maximum (FWHM) of about 48 nm.7 In addition to Eu2+, also Mn2+ ions are promising activator ions to achieve narrow band emission. AlN:Mn2+ reveals crisp orange photoluminescence (PL) with a FWHM of about 43 nm.8–10 Mn2+ even offers great spectral tunability depending on crystal field strength (d5-configuration), coordination symmetry and bond covalency. In tetrahedral coordination, such as in AlON, Mn2+ exhibits green 4T1 → 6A1 emission (λpeak: 515 nm, FWHM: 32–44 nm),11 whereas orange-red emission is achieved in AlN (λpeak: 600 nm, FWHM: 43 nm)10 and ZnGeN2 (λpeak: 610 nm, FWHM: 49–54 nm) because of reduced splitting between free ion states by a stronger nephelauxetic effect.12,13 In octahedral or similar stronger crystal field coordination the 4T1 → 6A1 energy gap decreases and this shifts Mn2+ emission further into the red to near-infrared (NIR) region and also its spectral shape relatively broadens (FWHM > 60 nm).14–16 In CASN: Mn2+, two emissions bands (λpeak: 548 nm, 627 nm) were reported and attributed to Mn2+ ions occupying pentahedral Ca site and tetrahedral Al site respectively, with a combined FWHM of 100 nm.15 For a similar compound, MgAlSiN3:Mn2+ an even broader emission in deep-red to NIR region (λpeak: 754 nm, FWHM: 150 nm) was observed arising from Mn2+ ions at the pentahedral Mg site [MgN5].16
Recently in few studies, Mn2+ doped Ca-α-SiAlON was reported to exhibit orange emission with a relatively narrow bandwidth (λpeak: 600 nm, FWHM about 45 nm).17–19 However, when codoped with sensitizer ions, such as Eu2+, no sensitized Mn2+ orange emission was observed.20 Ca-α-SiAlON:Eu2+ exhibits bright yellow d–f emission. On Mn2+ codoping, the Eu2+ emission exhibited a steady decrease revealing energy transfer related quenching, but the absence of sensitized Mn2+ emission was striking.19,20 Moreover, it is anticipated that the Mn2+ ions occupy the seven coordinated Ca2+ polyhedral sites in Ca-α-SiAlON, which would lead a rather broader Mn2+ emission spectral shape.20 The reported narrow band emission would require Mn2+ ions to occupy tetrahedral sites, such as Al or Si, which are fairly small in Ca-α-SiAlON and thus are less likely to incorporate Mn2+ dopants.
Ca-α-SiAlON is a solid-solution of Ca, Si, Al, O and N and is iso-structural to α-Si3N4 (Fig. 1).21 Amongst several polymorphs of Si3N4, α and β structural phases based SiAlON compositions, when activated using appropriate dopants exhibit excellent phosphor properties.22–24 The structures in both phases consist of a three-dimensional network of interconnected tetrahedral [SiN4] units, but primarily differ in their stacking orientation along c-axis. The α phase exhibits ABCD-type stacking over ABAB-type stacking in β phase. This leads to a continuous set of channels along c-direction in β phase, but in α phase, these channels are blocked and instead form voids. Unlike the pure substitutional composition in the β phase (Si6−zAlzOzN8−z, where z = 0–4.2), the α phase offers two interstitial sites (voids) of crystallographically identical nature per unit cell. These sites can be occupied by a charge-stabilizing cation, Me (Fig. 1) and give rise to chemical compositions MexSi12−(m+n)Alm+nOnN16−n. In α-SiAlON, m + n (Si–N) bonds are replaced by m (Al–N) and n (Al–O) bonds, and the charge difference is compensated by incorporation of Me cations. Here, Me can be Li, Mg, Ca, Sr, Y or Ln ions and x = m/ν, where ν is charge on Me cation.25 The Me cations occupy interstitial sites available in lattice structure and are coordinated with seven anions (N,O) with three different Me-(N,O) distances.
 | ||
| Fig. 1 Crystal structure of α-Si3N4 and Ca-α-SiAlON along c-axis. The brown, blue and gray spheres represent Ca, Si/Al, and N/O ions, respectively. The coordination polyhedron of Ca and Si tetrahedron are presented in right side. | ||
The Me-α-SiAlON compounds show high solubility for several dopant ions, such as Eu2+, Ce3+, Pr3+, Sm2+, Tb3+, Dy3+, Yb2+, etc.26–29 Previous studies suggested Me = Ca and m ∼ 2 exhibits superior phosphor properties.26,28 Among several dopant ions, Eu2+, Ce3+ and Yb2+ exhibit intense f–d type allowed transitions and have extensively been investigated as LED phosphors.26–28 As mentioned earlier,17–19 a few studies have reported Mn2+-doped Ca-α-SiAlON, however the spectroscopic observations contradict structural considerations, particularly the narrow-band emission profile, and therefore demand further careful investigation. In present work, we studied Mn2+ doping in Ca-α-SiAlON and investigated its photoluminescence properties in detail. Accordingly, Mn2+-doped Ca-α-SiAlON shows broad deep-red luminescence, in line with Mn2+ substitution at Ca2+ polyhedral site. The previous reports of narrow-band orange Mn2+ PL is explained by trace amounts of AlN:Mn impurities. Moreover, we investigate sensitization of the Mn2+ emission in Ca-α-SiAlON. Sensitization is observed but the wide dopant site separation (>5Å) makes sensitization of Mn2+ ions inefficient with sensitizer ions such as Eu2+, Ce3+ or Yb2+.
B Materials and methods
Synthesis
All samples investigated in the present work were obtained by the solid-state reaction method. High purity reagent grade chemicals of Si3N4 (SN-E10 grade, Ube Chemicals, Japan), AlN (99.9%, Sigma-Aldrich, Germany), CaCO3 (99.9%, Merck), BaCO3 (99.9%, Merck), MnCO3 (99.9%, Sigma-Aldrich, Germany), MnF2 (99.99%, Sigma-Aldrich, Germany), EuF3 (99.99%, Highway Chemicals, NL), YbF3 (99.99%, Highway Chemicals, NL) and CeF3 (99.99%, Highway Chemicals, NL) were used. The precursor chemicals were weighed in a stoichiometric ratio and mixed thoroughly using agate mortar-pestle. Acetone was used as dispersant during mixing. In doped samples, the respective dopant ions were incorporated by substituting equivalent metal ions (Ca2+) contents in atomic % (at%), i.e. Ca1−x−ySi9Al3ON15:xMn,y(Eu/Yb/Ce), where x = 0–15 at% and y = 0, 0.5 at%. The well-mixed powder batches were transferred into covered BN crucibles and fired at high temperature under 10% H2–90% N2 gas-flow reducing atmosphere. The sintering temperature for Ca-α-SiAlON was set at 1650 °C for 5 hours dwell time, unless specified otherwise in the text. The ramp rate was 200 K hour−1. To compare, Mn2+ doped BaSi3Al3O4N5 (5 at% Mn) and AlN (1 at% Mn) samples were also obtained using same synthesis conditions (1650 °C for 5 hours dwell time). The sintered powders were then mildly crushed to fine powders using agate mortar-pestle and used for experimental analysis. The Ca-α-SiAlON samples are identified using dopant contents throughout the manuscript and other samples are detailed according to their composition, sintering temperature and doping contents, wherever necessary in the text.Experimental methods
The obtained samples were analyzed using powder X-ray diffraction (PXRD) method for phase identification and purity. A Bruker D2 PHASER X-ray diffractometer with Co target (λKα = 1.78897 Å) was used at 30 kV operating voltage and 10 mA current. The step size resolution was 0.01 Å. Another X-ray diffractometer with Cu target (λKα = 1.5401 Å) from Philips was also used at 40 kV operating voltage and 20 mA current. Photoluminescence (PL) spectroscopy was performed using an Edinburgh Instruments FLS-920 fluorescence spectrometer. The PL and PL excitation (PLE) measurements were recorded using a 450 W Xe lamp as excitation source and a Hamamatsu R928 photomultiplier tube (PMT) detector and a liquid nitrogen cooled NIR detector. The spectral resolution was maintained at 0.5 nm in UV-visible range and 1 nm in near-infrared range for both PL and PLE measurements. For PL decay measurement, a picosecond pulsed laser diode (Edinburgh Instruments EPL 375, peak wavelength: 376.5 nm, pulse width: 65 picoseconds) was used as excitation source and the signals were recorded using Hamamatsu H74220-60 fast response PMT. The decay curves were recorded using single photon counting method with 4000 channels resolution. For normal full range (10 μs) decay curve, each channel provides 2.4 ns resolution. For higher resolution (HR) decay measurements, the channel width was reduced to 0.048 ns. Mn2+ PL decay measurement was performed by using a tunable optical paramagnetic oscillator (OPO) Opotek Opolette HE 355II pulsed laser (pulse width: 10 ns; repetition rate: 10 Hz) as excitation source, and the emission was recorded by a liquid nitrogen cooled NIR detector. For cryogenic (4.2 K) measurements, the samples were cooled down using an Oxford Instruments liquid He flow cryostat. For temperature dependence study, liquid He flow cryostat was employed in the 4.2–500 K temperature range, whereas a temperature-controlled stage from Linkam Scientific (THMS600) was used for the 300–600 K temperature range. For data analysis and plotting, Origin Lab (9.1 version) software was used. All curve fittings were performed using orthogonal distance regression algorithms. The crystal structure and radial ionic distribution analysis was carried out using the VESTA (3.4.4 version) software.C Results and discussion
The powder X-ray diffraction (PXRD) patterns of undoped and Mn2+ (10 at%) doped Ca-α-SiAlON powders are presented in Fig. 2a. The diffraction patterns of all samples reveal single-phase trigonal α-Si3N4 type crystal structure with P31c space group.21 No detectable impurity phases could be observed. In the undoped sample, the pattern reflects the Ca-α-SiAlON structure corresponding to CaSi9Al3ON15 composition with cell parameters of a = 7.850 Å and c = 5.717 Å.19 On Mn2+ doping, the peak position and shape profile shows significant differences when doped using MnCO3 (oxide) precursor, whereas, the MnF2 (fluoride) precursor doping did not reveal noticeable changes compared to the undoped sample. In oxide precursor doped samples, the diffraction peaks are broader and shift towards higher diffraction angle compared to the undoped Ca-α-SiAlON. The peak shift suggests a lattice contraction, which is expected considering the substitution of larger sized Ca2+ ions by smaller Mn2+ dopant ions.20 The peak broadening typically indicates a reduced particle size, causing a broader distribution of lattice plane distances. | ||
| Fig. 2 PXRD patterns of undoped and Mn2+ (10 at%) doped Ca-α-SiAlON obtained using oxide and fluoride doping precursors (a); respective PL–PLE spectra (b); the photographic images (powders filled in quartz disks) under day-light and UV-light (254 nm Hg vapor lamp) exposure (c); and the Mn2+ PL decay curves (d). The reference XRD pattern in (a) was calculated for Ca-α-SiAlON structure (λKα = 1.78897 Å). In (b), the excitation wavelength was 255 nm for PL measurements and the emission was monitored at 695 nm to record the PLE spectra. The insets present enlarged view (10 times) of PL–PLE curves in 300–600 nm range. | ||
Contrarily, the diffraction pattern of the fluoride precursor doped sample did not reveal a peak shift compared to the undoped Ca-α-SiAlON and the peak broadening is also insignificant. This suggests that the fluoride content affects structural properties. Normally in high temperature solid-state reaction synthesis, fluoride-based contents act as flux to aid and accelerate the sintering reaction, and are often intentionally incorporated.30–32 The PXRD results indeed suggest a better crystallinity in MnF2-doped sample over the MnCO3-based sample. It is also believed that the lack of oxygen during firing stage forces fluorine to play an anionic role in the lattice structure. Each oxygen vacancy requires two fluorine anions for charge compensation. The presence of large voids in α-SiAlON structure, where only half of them are occupied with Ca sites in studied composition, indicates that the charge compensating anionic impurities, such as excess fluorine anions, can easily be accommodated at interstitial position. The presence of excess ions (fluorine anions) may thus counter the contraction effect due to Mn2+ substitution with Ca2+. This may explain the peculiar observation of diffraction pattern of MnF2 doped Ca-α-SiAlON. However, a proper reasoning requires more detailed structural studies, and is out of the scope of present investigation.
Fig. 2b presents the PL–PLE properties of undoped and Mn2+-doped Ca-α-SiAlON powders. An intense, broad deep-red PL is observed in Mn2+-doped samples under Mn2+-ligand charge transfer (255 nm) excitation. The PL peak position is situated at around 697 nm with a full width at half-maximum (FWHM) of about 200 nm (∼480 meV). The emission is assigned to the 4T1 → 6A1 transition based on the d5 Tanabe–Sugano diagram. Note that, based on the deviation of the local coordination from octahedral symmetry, the 4T1 state will be split but it is customary to describe the Mn2+ emission with the irreducible representation labels for octahedral symmetry even when the actual site symmetry is lower. The Mn2+ PL shape profile is independent of the doping precursor type (MnCO3 or MnF2) suggesting that the Mn2+ local coordination remains unaffected (ESI,† Fig. S1). It is noticed that the Mn2+ PL intensity is slightly higher in fluoride precursor doped sample. This difference becomes more pronounced for excitations in the d–d absorption bands (λ > 325 nm). This is clearly visible from the enlarged view of PLE spectra (insets, Fig. 2b), where the d–d excitation bands are more pronounced in MnF2 doped sample than for the MnCO3 doped sample. The results indicate that the fluoride precursor is more effective in Mn2+ incorporation in the Ca-α-SiAlON lattice and also improves the PL properties. In line with the PL, the PLE shape of d–d transitions in both fluoride and oxide doping precursor samples are similar (ESI,† Fig. S2). The PLE spectrum shows weak (spin- and parity forbidden) broad band excitation between 450 and 600 nm corresponding to 6A1 → 4T1,4T2 transitions and a weak sharp excitation line due to the 6A1 → 4A1,4E transition around 445 nm. Note that the wavelength of the sharp 6A1 → 4A1,4E excitation line is at longer wavelength than typically observed (∼410–425 nm) for Mn2+ in oxides because of the stronger nephelauxetic effect. A weak UV-blue PL is also observed and attributed to the host Ca-α-SiAlON as it is also present in undoped sample. The intensity of this host emission is weaker in Mn2+-doped samples.
The photographic images of undoped and Mn2+-doped Ca-α-SiAlON powders under daylight and under UV-light (254 nm) are presented in Fig. 2c. All three samples exhibit a clear white body color. The spin-and-parity forbidden d–d transitions in Mn2+ are too weak to cause visible coloration even at 10 at% doping concentration. Under UV-irradiation, the Mn2+-doped samples exhibit bright red colored glow, whereas the undoped Ca-α-SiAlON powder exhibits a faint violet-gray appearance (the blue-colored tint at peripheral border is originating from quartz disks used to hold the powders). Fig. 2d presents the Mn2+ PL decay curves in oxide and fluoride precursor doped Ca-α-SiAlON. The decay curves in both samples show similar relaxation kinetics, which further supports the earlier claim that the Mn2+ local coordination in Ca-α-SiAlON doped with either MnCO3 or MnF2 is similar.
The overall results in Fig. 2 suggest that the Mn2+ doping in Ca-α-SiAlON is more effective using fluoride (MnF2) doping precursor, and therefore for further investigations, we only used fluorides as doping precursors. To study the effect of doping concentration on PL properties, Ca-α-SiAlON doped with 1–15 at% Mn2+ contents were prepared. Phase purity of prepared samples was confirmed using PXRD measurements (ESI,† Fig. S3). Fig. 3a presents the Mn2+ PL in Ca-α-SiAlON as a function of doping concentrations. The PL intensity shows a steep increase from 1 at% to 5 at% doping, but then decreases slowly for further increase in doping concentrations. From PLE spectra, the optimum Mn2+ concentration is found to be 5 at% under charge transfer excitation (<300 nm), whereas it is 10 at% under d–d transitions excitation (ESI,† Fig. S4). The normalized PL spectra are presented in Fig. 3b. The PL exhibits small redshift with increase in Mn2+ concentration. A concentration dependent PL redshift is often obvious and similar observations have also been reported in Eu2+ (or Ce3+) doped Ca-α-SiAlON.26,33 It may arise from energy transfer among optically active ions or due to subtle differences in crystal field by progressive substitution of host cations with dopant ions, or both.
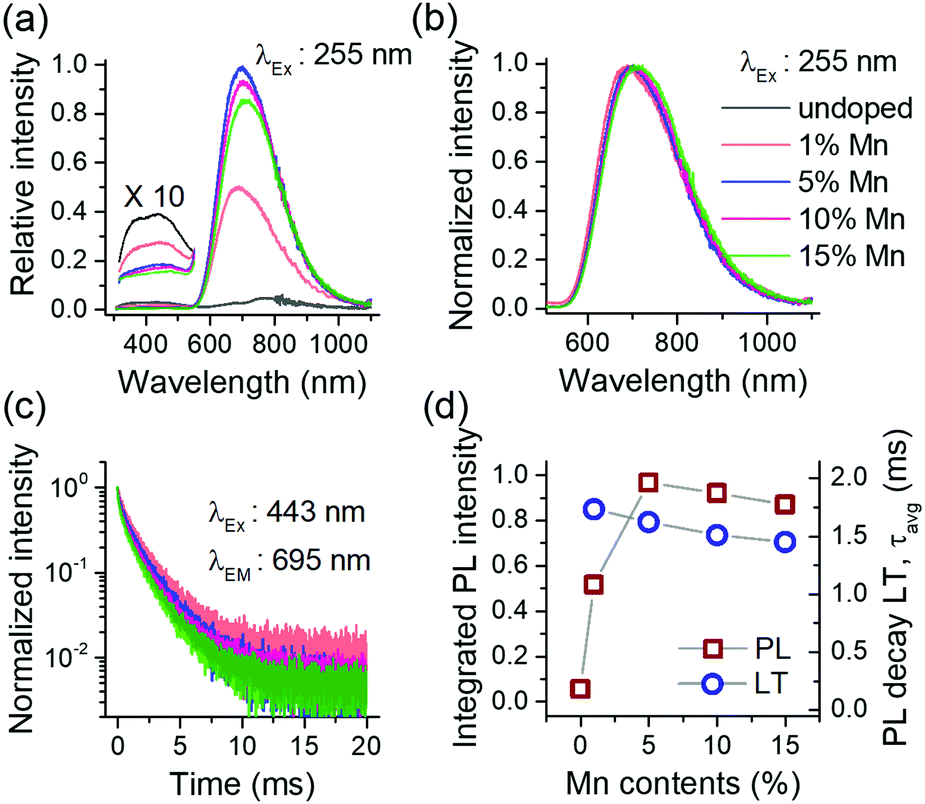 | ||
| Fig. 3 Concentration dependent PL (a); PL spectra in normalized scale (b); PL decay curves (c); and the plot of integrated PL intensity and luminescence decay times vs. concentration in Mn2+ activated Ca-α-SiAlON (d). In (a), (b) and (c), the line colors as indexed in (b) identify respective concentrations. | ||
The Mn2+ PL decay curves for different doping concentration is presented in Fig. 3c. All decay curves reveal non-exponential decay kinetics and exhibit faster relaxation with increase in concentration. The average decay lifetime (LT) was derived from the decay curve fitting using multi-exponential function (ESI,† Fig. S5). The slow decay component in least doped sample (Mn2+ concentration 1 at%) indicates that the radiative LT is about 2–2.5 ms for Mn2+ PL in Ca-α-SiAlON. This value is reasonable when compared to oxide (>5 ms to several tens of ms) and nitride (a few ms) hosts.8,16,34 Typically, selection rules become more relaxed as covalency increases as it leads to increased mixing of electronic states. This explains the trend towards shorter Mn2+ PL life times from fluorides to oxides to nitrides. Fig. 3d presents a plot of integrated PL intensity and LT with doping concentration. The PL intensity first increases till a 5 at% doping level and then decreases for a further increase in doping contents under 255 nm excitation. The LT exhibits a steady decrease with increase in concentration. It is interesting to note that both PL and LT slowly decrease with concentration, indicating that the Mn–Mn interactions are rather weak in Ca-α-SiAlON and prevent strong concentration quenching.
Fig. 4a presents the temperature dependence of Mn2+ PL in Ca-α-SiAlON under charge transfer (275 nm) excitation. It shows that the PL intensity steadily decreases above 25 K, and the shape profile slightly broadens with increase in temperature. A careful analysis of PL peak shift reveals that it exhibits an initial redshift till ∼50 K and then continuously blueshifts for further increase in temperature. The normalized PL spectra show the PL shift and broadening effect more clearly (Fig. 4b). Accordingly, the PL peak exhibits a whopping 70 nm shift (740 nm to 670 nm) for temperature increase from 75 K to 475 K. Comparatively, the broadening effects appear small, i.e. FWHM is about 185 nm (∼420 meV) at 4.2 K, 197 nm (∼480 meV) at 300 K, and is about 198 nm (∼510 meV) at 475 K. The PL broadening with temperature primarily occurs due to increased vibronic (Stokes and anti-Stokes emissions) transitions originating from thermal population of higher vibrational levels.35,36 The vibronic effect can clearly be seen from the increased intensity of higher energy side emission in PL spectra (Fig. 4a) despite the overall PL quenching with increase in temperature. The relative contribution of vibronic transitions together with crystal field variations (lattice expansion/contraction) with temperature and other temperature-induced effects, such as lower PL quenching temperatures for Mn2+ emitting at longer wavelengths, determine the PL peak shift. The lower quenching temperature of deep red/NIR emitting Mn2+ centers can explain the unusually large blue shift between 75 and 475 K and will be discussed below.
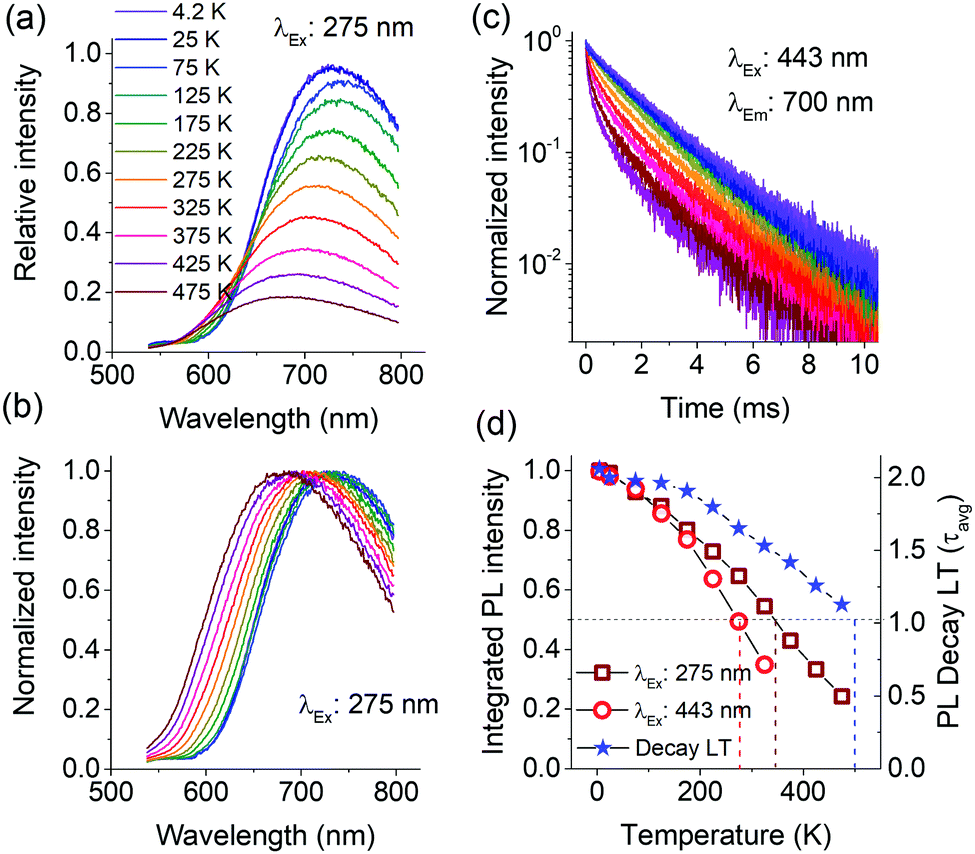 | ||
| Fig. 4 Temperature dependent PL of Mn2+ (10 at%) doped Ca-α-SiAlON under 275 nm excitation (a); PL in normalized scale (b); PL decay curves (c); and the plot of integrated PL intensity (under 275 nm and 443 nm excitations) and the PL decay time with temperature (d). In (a), (b) and (c), the line colors are as indexed in (a). In (d), the integrated PL values were derived from 535–800 nm spectral range. | ||
Interestingly, even at 4.2 K, in the PL spectra no vibronic fine structure is observed, suggesting a larger Huang–Rhys factor for Mn2+ PL in Ca-α-SiAlON (ESI,† Fig. S6).37 This is also in agreement with the large Stokes shift observed in luminescence spectra for Ca-α-SiAlON activated with Eu2+ and other ions.26 This has been attributed to the size mismatch of dopant ions with the site occupied in the α-SiAlON lattice structure, which allows extended configurational changes during excitation–relaxation dynamics in dopants’ electronic structure.38
The large Stokes shift also reflects a low energy threshold for configuration crossover between excited and ground states, thus allowing thermally activated PL quenching at relatively lower temperature.39–42Fig. 4d presents the temperature dependence of integrated PL intensities under charge transfer (275 nm) and d–d excitation (443 nm), as well as the PL decay times. The plot shows that the PL exhibits a monotonous decrease above 25 K. Under charge transfer excitation, the T50% of PL intensity occurs at ∼350 K, but still the PL does not completely quench even till 475 K. A typical thermal quenching behavior exhibits a sharp decrease in PL (or decay time) above the threshold quenching temperature.39–43 In present case, the PL decrease is continuous. This observation suggests that there is a range of dopant sites exhibiting different quenching temperatures. To substantiate this, the temperature dependence of PL intensity under d–d excitation (443 nm) and of the PL decay curves were recorded. The PL under 443 nm excitation shows similar gradual decrease with increase in temperature, but the decrease is relatively faster with T50% occurring at 280 K (Fig. 4d). Additionally, the peak shift under 443 nm excitation is larger compared to charge transfer excitation (ESI,† Fig. S7). Fig. 4c presents the temperature dependence of PL decay curves under 443 nm excitation. The decay curve at low temperature (4.2 K) does not show perfect single exponential decay kinetics, but instead is composed of multi-exponential decay components. With increase in temperature, the non-exponential nature becomes more prominent and exhibits faster relaxation kinetics, characteristic of temperature induced PL quenching. The emission LT derived using multi-exponential fits to decay curves is plotted in Fig. 4d. The LT also exhibits a decrease with temperature, but the decrease is slower and T50% occurs at 500 K. Overall, the onset of thermal quenching of the Mn2+ emission well below room temperature makes this material not suitable for commercial application in LEDs.
Ideally, thermal quenching affects the PL intensity and emission LT in the same manner for a specific quenching mechanism.35,41,42 The present observations of excitation wavelength dependent PL quenching and differences in quenching temperature of PL and emission LT suggest a complex quenching behavior. The complex behavior can be explained by the presence of disorder giving rise to Mn2+ ions with differences in local coordination affecting both the emission wavelength and quenching temperature.44 The [Ca(N,O)7] polyhedron in Ca-α-SiAlON possesses three different metal–anion distances.45 The mixed anionic coordination of α-SiAlON and the size difference of dopant ions with Ca ions, can give rise to variations in the local Mn2+ coordination and thus crystal field variations among different Mn2+ ions occupied in polyhedral sites. This also can explain the monotonous PL quenching above 25 K accompanied with PL blue shift as the longer wavelength Mn2+ PL emission has a smaller activation energy for thermal quenching and is quenched at lower temperatures. This is consistent with the configuration coordinate diagram crossover model showing lower activation energy for red-shifted emission transitions (ESI,† Fig. S8).39,40 Accordingly, the PL spectrum at low temperature consists of multiple Mn2+ emissions arising from several [Mn(N,O)7] sites with differences in (N,O) coordination. Upon increasing the temperature, the low energy (long wavelength) emission contributions in PL spectrum systematically decrease. The near exponential decay curve profile at 4.2 K suggests that the crystal field variations are small and that the PL is efficient at 4.2 K. As temperature rises, the quenching of lower activation energy emission induces a progressive nonlinearity in decay curves (Fig. 4c). The presence of multiple Mn2+ emissions arising from several different sites also gives rise to a broader Mn2+ PL profile even at 4.2 K. The temperature induced quenching of longer wavelength emissions explains the observed limited thermal broadening in overall PL profile (Fig. 4b) and the significant blue shift of the emission band maximum. To confirm the presence of multiple sites, PLE spectra were recorded at 4.2 K monitoring different emission regions (ESI,† Fig. S9). The PLE spectra revealed small changes as a function of monitoring wavelengths, specifically for the sharp excitation bands at around 440 nm. Nevertheless, the overlap in the broad Mn2+ emission bands makes it difficult to individually isolate each component and do true site selective spectroscopy.
The results and analysis presented above clearly indicate that the Mn2+ ions in Ca-α-SiAlON occupy the [Ca(N,O)7] polyhedral site with disorder in the (N,O) coordination leading to crystal field variations explaining the broad deep-red PL and complex temperature behavior. Surprisingly, in previous studies on Mn2+ activated Ca-α-SiAlON, many reports proposed narrow-band orange Mn2+ emission centered at around 600 nm.17–19 As our results showed completely different observations, we were intrigued by the literature reports and therefore carefully investigated the cause for the reported orange Mn2+ PL in Ca-α-SiAlON. We have also experienced presence of similar orange PL in several oxynitride/nitride compositions singly doped with Mn2+ ions, specifically in Al-based compositions. Careful inspection reveals that the peak position and the relatively narrow band profile of these orange emissions show close similarity to that of the Mn2+ emission of AlN:Mn. In AlN, Mn2+ ions have tetrahedral coordination, which together with high Mn–-N covalency related shift (Nephelauxetic effect), leads narrow-band orange PL at around 600 nm.6–8 Interestingly, the previous reports on Ca-α-SiAlON:Mn2+ clearly showed the presence of AlN impurity phase in the samples,17–19 which may result from either an excess AlN contents or incomplete synthesis (reaction temperature too low or reaction time too short). While optimizing the synthesis conditions for Ca-α-SiAlON, we also observed a similar but very weak emission at orange region in incompletely synthesized sample.
To provide further support for the assignment of the narrow orange emission to Mn2+ emission in impurity phase AlN:Mn2+, single phase AlN:Mn2+ and Al-based oxynitrides were synthesized. Fig. 5a shows the XRD patterns of Mn-doped AlN, BaSi3Al3O4N5 (synthesized at 1650 °C) and an incompletely synthesized Ca-α-SiAlON (synthesized at 1400 °C). Both AlN and BaSi3Al3O4N5 exhibit good phase purity. BaSi3Al3O4N5 usually accompany a small amount of oxide impurity (BaSi2Al2O8), but its influence on the PL properties is negligible.30,46 No AlN impurity was detected in the BaSi3Al3O4N5 PXRD pattern indicating that an AlN impurity phase is present in less than a few %, the detection limit of impurity phases in PXRD. Especially impurity phases of light elements (low X-ray scattering or atomic form factor) are harder to detect. Ca-α-SiAlON (synthesized at 1400 °C) revealed mixed compound with several unidentified impurity phases due to incomplete sintering reaction. The room temperature (300 K) PL properties of these samples are presented in Fig. 5b. The single phase AlN:Mn sample shows the typical bright orange PL originating from Mn2+ in tetrahedral nitride coordination. In BaSi3Al3O4N5:Mn, a prominent blue emission at 430 nm (host emission) is observed along with a weak shoulder at 600 nm. A similar weak emission at 600 nm is observed in underreacted Ca-α-SiAlON, accompanied with a broad near-infrared emission (assigned to emission from Mn2+ ions). Fig. 5c shows the cryogenic (4.2 K) PL measurements of the orange PL in all three samples. In AlN, the PL exhibits sharp peaks representing vibronic fine structure. This fine structure is well-known and serves as a fingerprint for the presence of AlN:Mn2+. An identical fine structure is observed for orange PL of other two samples. The vibronic fine-structure reflects coupling with host lattice vibrations, similar to those observed in Raman spectra.47 The observation of identical fine-structure in the orange emission observed for Ca-α-SiAlON:Mn2+ and BaSi3Al3O4N5:Mn2+ suggests that the orange PL is indeed originating from same compound, i.e. AlN:Mn, present as an impurity phase in these Al-based oxynitrides. Indeed, it can be expected that the bright orange emission from AlN:Mn can be observed in other Al-based oxynitrides because of the high efficiency and because AlN phases can go easily undetected in PXRD, especially in compositions with heavier elements such as Ba which have larger X-ray scattering cross-sections. Cryogenic measurements are a powerful tool to verify the presence of AlN:Mn2+ because of the characteristic fine structure in the low temperature emission spectrum.
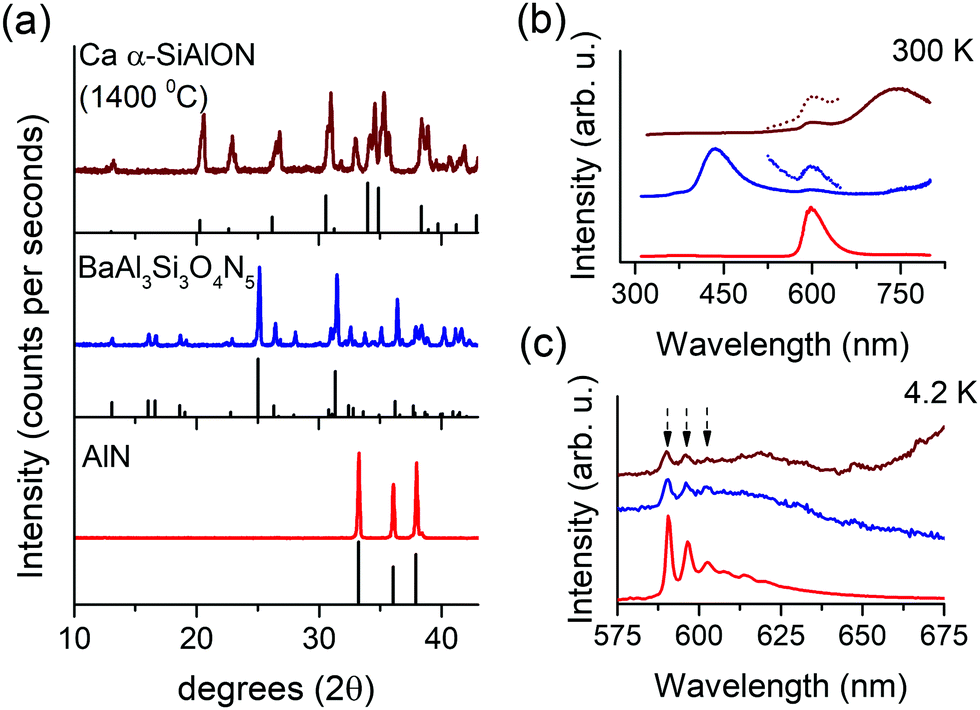 | ||
| Fig. 5 PXRD patterns of Ca-α-SiAlON (1400 °C), BaAl3Si3O4N5 and AlN (a), Mn2+ PL in respective samples at 300 K (b) and at 4.2 K (c) under 275 nm excitation. In (a), the gray colored patterns represent reference diffraction patterns (λKα = 1.5401 Å). The samples are identified using line colors as indexed in (a). In (c), the dotted arrows point position of vibronic side bands. | ||
As discussed above the PXRD pattern of BaSi3Al3O4N5:Mn did not reveal detectable presence of AlN traces, but still in PL, although weak, the orange emission could be observed. This is explained by the bright luminescence of AlN:Mn. As a result, in Mn2+-doped compounds having weak or no emission, a presence of AlN impurity, even in small amount, can lead prominent orange PL, which can easily be misinterpreted and attributed to the primary compound. Such effect has previously been reported for Eu2+ PL in CaZnOS, where the emission was arising from impurity phase CaS:Eu2+ and the pure CaZnOS:Eu2+ is non-luminescent.48 This effect can be further enhanced by solubility differences. Mn2+ is a large cation with respect to Al or Si, and in nitride tetrahedral coordination, its solubility is small. Cherepy et al. proposed that about 0.6% Mn2+ ions can be uniformly incorporated in AlN lattice (Al–N bond length about 0.18 nm).8 In Ca-α-SiAlON or other low field-strength metal ions containing compositions, the [(Al/Si)N4] tetrahedron is even smaller (Al/Si–N bond length about 0.17 nm) and thus it is more difficult for Mn2+ ions to substitute on tetrahedral sites. In absence of appropriate doping sites, such as Mg, Zn, Ca, etc., Mn2+ doping and/or emission is highly inefficient in several compounds, such as BaSi3Al3O4N5 (Ba2+ site is too large for Mn2+ ions). In lower purity BaSi3Al3O4N5 compound, the intensity of orange PL is even higher over that in Fig. 5b. We have observed prominent orange Mn2+ PL in the Ba-stabilized composition (known as Ba-s-phase) BaAlSi5O2N7, space group Imm249 and in SrAl2Si2O2N3, exhibiting AlN as impurity phase (ESI,† Fig. S10). All these samples show identical PL vibronic fine structure to that of AlN and their PL decay kinetics is also similar.
Another reason to doubt the assignment of orange PL to Mn2+ in Ca-α-SiAlON (or respective other oxynitride compounds) is its inability of getting sensitized when codoped with sensitizer ions, such as Eu2+. Ni et al. and Zhang et al. investigated the Eu2+–Mn2+ codoped Ca-α-SiAlON.19,20 Ni et al. showed the presence of Mn2+ narrow-band orange PL at 600 nm in singly doped Ca-α-SiAlON, but in Eu2+–Mn2+ codoped sample, only Eu2+ emission was observed. We previously observed similar results in Ba-s-phase, where the Mn2+ singly doped sample exhibits prominent 600 nm emission, but in codoped sample, only Eu2+ emission appears (ESI,† Fig. S10 and S11). This is due to the fact that the 600 nm emission is coming from impurity AlN phase, thus preventing any short range non-radiative sensitization interaction with donor ions (Eu2+) incorporated in primary compound. Zhang et al.20 showed quenching of Eu2+ PL with increase in Mn2+ codoping in Ca-α-SiAlON, but no sensitized Mn2+ emission was reported. As the donor emission exhibited quenching upon Mn2+ codoping, it is evident that the energy transfer is taking place. In the present investigation, we showed that Mn2+ ions exhibit broad deep-red PL in Ca-α-SiAlON, thus it is expected that the sensitized luminescence, if present should be observed at deep-red spectral region in the codoped samples.
To investigate the possibility of sensitizing the Mn2+ emission in Ca-α-SiAlON we co-doped with different sensitizers. Fig. 6a shows the PL properties of Eu2+ singly doped and Eu2+–Mn2+ codoped Ca-α-SiAlON. The Eu2+ singly doped sample shows bright yellow emission and the PLE exhibits typical Eu2+ broadband profile. In Eu2+–Mn2+ codoped sample, the Eu2+ PL intensity decreases, but a prominent Mn2+ sensitized luminescence is missing. The deep-red PL region of codoped sample indeed shows a small increase in intensity as can be seen from the normalized PL spectra in Fig. 6b. The intensity further improves at cryogenic temperature (4.2 K) PL measurements, as the Mn2+ emission is partially thermally quenched at 300 K. Still, the sensitized Mn2+ emission intensity is fairly weak. Similar small elevation of deep-red region PL in Eu2+–Mn2+ codoped Ca-α-SiAlON could be witnessed in earlier work of Zhang et al.,20 but its weak, non-prominent nature at room-temperature might have left it unnoticed. The donor (Eu2+) PL decay curves are presented in Fig. 6c, shows a slightly faster decay in codoped sample, confirming the presence of Eu2+–Mn2+ non-radiative energy transfer. The decay time decreases from 586 ns in Eu2+ singly doped Ca-α-SiAlON to 556 ns in Eu2+–Mn2+ codoped Ca-α-SiAlON. The inset shows high-resolution decay curves of the initial decay kinetics, suggesting absence of any fast energy transfer quenching, typically observed for Eu2+–Mn2+ energy transfer by exchange interactions.35 Based on the donor decay analysis, the energy transfer efficiency is about 4.7% for 10 at%. Mn2+ codoping in 0.5 at% Eu2+-doped Ca-α-SiAlON.
 | ||
| Fig. 6 PL–PLE spectra of Eu2+ singly doped and Eu2+–Mn2+ codoped Ca-α-SiAlON (a); normalized spectra at 300 K and 4.2 K (b); and Eu2+ PL decay curves monitored at 575 nm (c). Inset of (c) shows enlarged view of normal decay curves (channel width 2.4 ns) in combination with high-resolution decay curves (channel width 0.048 ns, broader translucent lines). The doping concentration for Eu2+ was 0.5 at% and for Mn2+ was 10 at%. The excitation and emission wavelengths for PL and PLE measurements were 412 nm and 575 nm, respectively. | ||
The results show that sensitization of Mn2+ emission by Eu2+ ions is inefficient in Ca-α-SiAlON. The energy transfer interaction between Eu2+ and Mn2+ is governed by short-range interactions, such as exchange (or dipole–quadrupole) interaction.50 We have recently shown that exchange interaction dominates the sensitization mechanism in Ba2MgSi2O7 by carefully analyzing the high-resolution donor decay kinetics.35 Accordingly, a fast nanosecond sensitization occurs due to exchange mediated energy transfer between nearest neighbor (<5 Å) Eu2+–Mn2+ ion pairs, and the transfer rate significant decreases for the next nearest neighbors (>5 Å) ions following exponential distance dependence. As both Eu2+ and Mn2+ ions occupy Ca2+ site in Ca-α-SiAlON, the Ca–Ca separation provides the nearest-possible neighboring distance. Fig. 7a presents the Ca-α-SiAlON structure, showing the Ca–Ca interionic separation. Accordingly, the closest Ca–Ca separation is about 5.3 Å, and the next nearest neighbor lie at 5.7 Å along c-axis to the unit cell. This suggests that the Ca-α-SiAlON is a dilute crystal structure with nearest neighbor distances too large for efficient exchange interactions. Another reason for inefficient sensitization is the poor spectral overlap between Eu2+ emission with Mn2+ absorption in Ca-α-SiAlON (Fig. 7b). Fig. 7b also shows the emission spectra of Yb2+ and Ce3+, which possess better spectral overlap with Mn2+ absorption in Ca-α-SiAlON and can be expected to improve Mn2+ sensitization. Fig. 7c shows the normalized PL–PLE spectra of 0.5 at% Yb2+ and Ce3+ singly doped Ca-α-SiAlON together with 10 at% Mn2+ codoped samples. As expected, the sensitized Mn2+ emission intensity slightly improved for Yb2+ donor ion sample compared to Eu2+ and even stronger improvement is observed in Ce3+ donor ion codoped Ca-α-SiAlON. In Ce3+–Mn2+ codoped Ca-α-SiAlON, the sensitized Mn2+ emission can distinctly be observed at 300 K. Nevertheless, the sensitized emission intensity is still weak compared to donor emission even at 10 at% Mn2+ codoping and having resonant spectral overlap. This confirms our conclusion that a large inter-ionic separation (>5Å) between donor–Mn2+ ions is primarily responsible for inefficient Mn2+ sensitization in Ca-α-SiAlON.
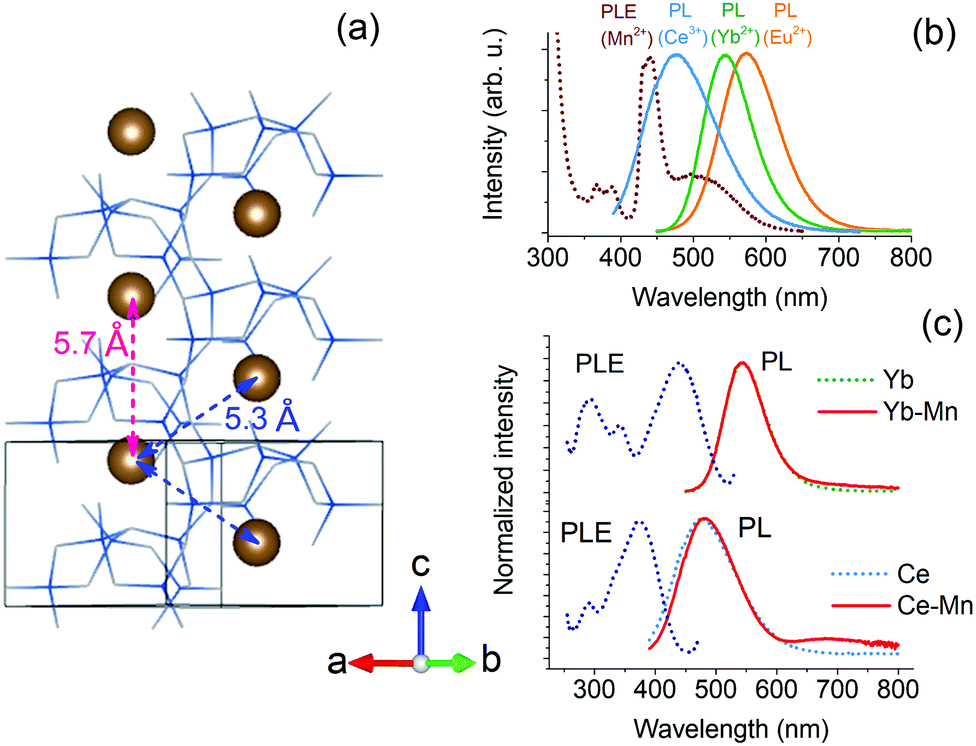 | ||
| Fig. 7 Ca-α-SiAlON crystal structure depicting Ca–Ca separation (a); spectral overlap of Mn2+ absorption (dotted line) with emissions (solid lines) of Eu2+, Yb2+ and Ce3+ donor ions (b); and normalized PL–PLE spectra of Yb2+ and Ce3+ singly doped with respective Mn2+ codoped Ca-α-SiAlON at 300 K (c). The doping concentration for Yb2+ and Ce3+ was 0.5 at%, and for Mn2+ was 10 at%. The excitation and emission wavelengths for PL and PLE measurements were 412 nm and 545 nm, respectively for Yb2+-doped samples, and 375 nm and 495 nm for Ce3+-doped samples, respectively. | ||
D Conclusions
We have presented an in-depth study of the luminescence of Mn2+ in Ca-α-SiAlON. At low temperatures deep red/NIR emission is observed between 600 and 900 nm. The emission is assigned to 4T1 → 6A1 emission for Mn2+ substituted in the [Ca(N,O)7] polyhedron. Deep red emission is explained by the strong nephelauxetic effect in nitride coordination, shifting the emission to longer wavelengths than typically observed for Mn2+ d–d emission.The emission shows unusual properties: the bandwidth is large (FWHM about 200 nm), thermal quenching occurs gradually over a wide temperature range (instead of the usual sharp drop) and the band maximum shows a remarkably large 70 nm blue shift upon raising the temperature. All these observations are explained by disorder in the local O–N coordination around Mn2+ in the polyhedron. This gives rise to variations in the crystal field. Mn2+ ions experiencing stronger crystal fields show longer wavelength emission with lower quenching temperatures. The role of crystal field inhomogeneity is further confirmed by the non-exponential emission decay curve at 4.2 K, which becomes increasingly non-exponential at higher temperatures. Interestingly, the present observation of deep red emission is different from previous reports on narrow band orange emission for Mn2+ in Ca-α-SiAlON. Based on careful analysis, including comparison of vibrational fine structure in low temperature spectra, it is concluded that the orange emission in fact originates from a small amount of AlN:Mn2+ impurity phase.
The absorption strength of the spin- and parity-forbidden d–d transitions of Mn2+ is low and successful application as a phosphor requires combination with a sensitizer. Sensitization of the Mn2+ emission was attempted by co-doping with Eu2+, Yb2+ and Ce3+, all having strong parity allowed f–d transitions. Sensitization was observed but proved to be inefficient as the nearest sensitizer–Mn2+ distance is over 5 Å, too large for efficient energy transfer via exchange interaction.
The new insights show that efficient deep red emission from a Mn2+-doped (oxy)nitride is possible for Mn2+ in polyhedral coordination with a crystal field splitting larger than for tetrahedral coordination. Thermal quenching is an issue and needs to be prevented by limiting excited state relaxation. To allow for sensitization the Mn2+ ions need to be incorporated in a host lattice that allows for short separation (<5 Å) between sensitizer and Mn2+ as exchange interaction is required for efficient energy transfer. Applying these design rules will contribute to the discovery of efficient deep red or NIR phosphors based on Mn2+ d–d emission in (oxy)nitrides.
Author contributions
ADS and AM conceptualized the original research idea. ADS and AJB carried out experimental work. After formal analysis and discussion with all, ADS wrote the original draft and all authors contributed in revision and editing of final draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Nichia Corporation (Japan) is gratefully acknowledged.References
- T. Takeda, R.-J. Xie, T. Suehiro and N. Hirosaki, Prog. Solid State Chem., 2018, 51, 41 CrossRef CAS.
- K. Uheda, N. Hirosaki and H. Yamamoto, Phys. Status Solidi, 2006, 203, 2712 CrossRef CAS.
- K. Uheda, et al. , Electrochem. Solid-State Lett., 2006, 9, H22 CrossRef CAS.
- H. A. Höppe, H. Lutz, P. Morys, W. Schnick and A. Seilmeier, J. Phys. Chem. Solids, 2000, 61, 2001 CrossRef.
- R. Mueller-Mach, G. Mueller, M. R. Krames, H. A. Höppe, F. Stadler, W. Schnick, T. Jüstel and P. Schmidt, Phys. Status Solidi, 2005, 202, 1727 CrossRef CAS.
- G. J. Hoerder, M. Seißald, D. Baumann, T. Schröder, S. Peschke, P. C. Schmid, T. Tyborsky, P. Pust, I. Stoll, M. Bergler, C. Patzig, S. Reibaus, M. Krause, L. Berthold, T. Höche, D. Johrendt and H. Huppertz, Nat. Commun., 2019, 10, 1824 CrossRef.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891 CrossRef CAS.
- N. J. Cherepy, S. A. Payne, N. M. Harvey, D. Åberg, Z. M. Seeley, K. S. Holliday, I. C. tran, F. Zhou, H. Paul Martinez, J. M. Demeyer, A. D. Drobshoff, A. M. Srivastava, S. J. Camardello, H. A. Comanzo, D. L. Schlagel and T. A. Lograsso, Opt. Mater., 2016, 54, 14 CrossRef CAS.
- X.-J. Wang, R.-J. Xie, B. Dierre, T. Takeda, T. Suehiro, N. Hirosaki, T. Sekiguchi, H. Li and Z. Sun, Dalton Trans., 2014, 43, 6120 RSC.
- J. Xu, N. Cherepy, J. Ueda and S. Tanabe, Mater. Lett., 2017, 206, 175 CrossRef CAS.
- R.-J. Xie, N. Hirosaki, X.-J. Liu, T. Takeda and H.-L. Li, Appl. Phys. Lett., 2008, 92, 201905 CrossRef.
- Q.-H. Zhang, J. Wang, C.-W. Yeh, W.-C. Ke, R.-S. Liu, J.-K. Tang, M.-B. Xie, H.-B. Liang and Q. Su, Acta Mater., 2010, 58, 6728 CrossRef CAS.
- M. Sheng, J. Wang, J. Fan, H. Lian, Y. Zhang and J. Lin, J. Mater. Chem. C, 2015, 3, 9306 RSC.
- C. J. Duan, W. M. Otten, A. C. A. Delsing and H. T. Hintzen, J. Solid State Chem., 2008, 181, 751 CrossRef CAS.
- Z. Zhang, A. C. A. Delsing, P. H. L. Notten, J. Zhao, P. Dorenbos and H. T. Hintzen, ECS J. Solid State Sci. Technol., 2013, 2, R70 CrossRef CAS.
- S. Liu, S. Zhang, N. Mao, Z. Song and Q. Liu, J. Am. Ceram. Soc., 2020, 103, 6793 CrossRef CAS.
- J. Ni, Q. Liu, J. Wan, G. Liu, Z. Zhou, F. Xu, X. Zeng and R.-J. Xie, J. Lumin., 2018, 202, 514 CrossRef CAS.
- J. Ni, Q. Liu, Z. Zhou and G. Liu, RSC Adv., 2017, 7, 42211 RSC.
- J. Ni, Z. Z. Zhou, X. K. Xu, Q. Liu, F. R. Shen and L. H. He, Mod. App. Matrl. Sci., 2020, 2, 276 Search PubMed.
- Y.-Q. Zhang, X.-J. Liu, Z.-R. Huang, J. Chen and Y. Yang, J. Lumin., 2012, 132, 2561 CrossRef CAS.
- G. Z. Cao and R. Metselaar, Chem. Mater., 1991, 3, 242 CrossRef CAS.
- T. Suehiro, N. Hirosaki, R.-J. Xie and M. Mitomo, Chem. Mater., 2005, 17, 308 CrossRef CAS.
- N. Hirosaki, R.-J. Xie, K. Kimoto, T. Sekiguchi, Y. Yamamoto, T. Suehiro and M. Mitomo, Appl. Phys. Lett., 2005, 86, 211905 CrossRef.
- S. Li, L. Wang, D. Tang, Y. Cho, X. Liu, X. Zhou, L. Lu, L. Zhang, T. Takeda, N. Hirosaki and R.-J. Xie, Chem. Mater., 2018, 30, 494 CrossRef CAS.
- J. W. T. van Rutten, H. T. Hintzen and R. Metselaar, Ceram. Int., 2001, 27, 461 CrossRef CAS.
- R.-J. Xie, N. Hirosaki, M. Mitomo, Y. Yamamoto, T. Suehiro and K. Sakura, J. Phys. Chem. B, 2004, 108, 12027 CrossRef CAS.
- Y.-Q. Li, N. Hirosaki, R.-J. Xie, J. Li, T. Takeda, Y. Yamamoto and M. Mitomo, J. Am. Ceram. Soc., 2009, 92, 2738 CrossRef CAS.
- R.-J. Xie, N. Hirosaki, M. Mitomo, K. Uheda, T. Suehiro, X. Xu, Y. Yamamoto and T. Sekiguchi, J. Phys. Chem. B, 2005, 109, 9490 CrossRef CAS.
- M. Mitomo, T. Endo, K. Ueda and M. Komatsu, EU Pat., EP1264873B1, 2011 Search PubMed.
- J.-Y. Tang, Y.-M. He, L.-Y. Hao and X. Xu, J. Mater. Res., 2013, 28, 1598 Search PubMed.
- B. J. Adamcyzyk, M. G. Brik, V. Nagirnyi, I. Romet, D. Michalik, T. Pawlik and M. Sopicka-Lizer, J. Alloys Compd., 2020, 845, 155468 CrossRef.
- S. Okamoto and H. Yamamoto, J. Electrochem. Soc., 2011, 158, J363 CrossRef CAS.
- H.-L. Li, G.-H. Zhou, R.-J. Xie, N. Hirosaki, X.-J. Wang and Z. Sun, J. Solid State Chem., 2011, 184, 1036 CrossRef CAS.
- M. Muller, S. Fischer and T. Jüstel, RSC Adv., 2015, 5, 67979 RSC.
- A. D. Sontakke, A. J. van Bunningen, F. T. Rabouw, S. Meijers and A. Meijerink, J. Phys. Chem. C, 2020, 124, 13902 CrossRef CAS.
- M. de Jong, L. Seijo, A. Meijerink and F. T. Rabouw, Phys. Chem. Chem. Phys., 2015, 17, 16959 RSC.
- T. Wylezich, A. D. Sontakke, V. Castaing, M. Suta, B. Viana, A. Meijerink and N. Kunkel, Chem. Mater., 2019, 31, 8957 CrossRef CAS.
- J. W. H. van Krevel, J. W. T. van Rutten, H. Mandal, H. T. Hintzen and R. Metselaar, J. Solid State Chem., 2002, 165, 19 CrossRef CAS.
- K. V. Ivanovskikh, J. N. Ogieglo, A. Zych, C. Ronda and A. Meijerink, ECS J. Solid State Sci. Technol., 2013, 2, R3148 CrossRef CAS.
- J. Ueda, A. Meijerink, P. Dorenbos, A. J. J. Bos and S. Tanabe, Phys. Rev. B, 2017, 95, 014303 CrossRef.
- T. Sendon, R. J. A. van Dijk-Moes and A. Meijerink, Light: Sci. Appl., 2018, 7, 8 CrossRef.
- B. Malysa, A. Meijerink and T. Jüstel, Opt. Mater., 2018, 85, 341 CrossRef CAS.
- Y. Zhang, L. Mei, H. Liu, J.-C. Chang, W.-R. Liu and Z. Huang, Mater. Chem. Phys., 2019, 228, 215 CrossRef CAS.
- A. D. Sontakke, J.-M. Mouesca, V. Castaing, A. Ferrier, M. Salaun, I. Gautier-Luneau, V. Maurel, A. Ibanez and B. Viana, Phys. Chem. Chem. Phys., 2018, 20, 23294 RSC.
- F. Izumi, M. Mitomo and J. Suzuki, J. Mater. Sci. Lett., 1982, 1, 533 CrossRef CAS.
- J.-Y. Tang, W.-J. Xie, K. Huang, L.-Y. Hao, X. Xu and R.-J. Xie, Electrochem. Solid-State Lett., 2011, 14, J45 CrossRef CAS.
- A. R. Evans and D. B. Fitchen, Phys. Rev. B: Solid State, 1970, 2, 1074 CrossRef.
- B. Budde, H. Luo, P. Dorenbos and E. van der Kolk, Opt. Mater., 2017, 69, 378 CrossRef CAS.
- C. J. Duan, W. M. Otten, A. C. A. Delsing and H. T. Hintzen, J. Alloys Compd., 2008, 461, 454 CrossRef CAS.
- A. L. N. Stevels and J. M. P. J. Verstegen, J. Lumin., 1976, 14, 207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: PL and PLE spectra, XRD patterns and PL decay curves, etc. See DOI: 10.1039/d1ma00036e |
| This journal is © The Royal Society of Chemistry 2021 |