
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally stable SiO2@TiO2 core@shell nanoparticles for application in photocatalytic self-cleaning ceramic tiles†
Elias P.
Ferreira-Neto
ab,
Sajjad
Ullah
 *bcd,
Vitor P.
Martinez
a,
Jean M. S. C
Yabarrena
a,
Mateus B.
Simões
a,
Amanda P.
Perissinotto
a,
Heberton
Wender
d,
Fabio S.
de Vicente
e,
Paul-Ludwig M.
Noeske
f,
Sidney J. L.
Ribeiro
b and
Ubirajara P.
Rodrigues-Filho
*a
*bcd,
Vitor P.
Martinez
a,
Jean M. S. C
Yabarrena
a,
Mateus B.
Simões
a,
Amanda P.
Perissinotto
a,
Heberton
Wender
d,
Fabio S.
de Vicente
e,
Paul-Ludwig M.
Noeske
f,
Sidney J. L.
Ribeiro
b and
Ubirajara P.
Rodrigues-Filho
*a
aInstitute of Chemistry of São Carlos, University of São Paulo (USP), 13560-970, São Carlos, SP, Brazil. E-mail: ubirajara@usp.br
bInstitute of Chemistry-São Paulo State University (UNESP), 14800-060, Araraquara-SP, Brazil. E-mail: sajjadullah@uop.edu.pk
cInstitute of Chemical Sciences, University of Peshawar, 25120, Peshawar, KP, Pakistan
dInstitute of Physics, Federal University of Mato Grosso do Sul (UFMS), Av. Costa e Silva S/N, 79070-900 Campo Grande, MS, Brazil
eInstitute of Geosciences and Exact Sciences, Department of Physics, São Paulo State University (UNESP), 13500-970, Rio Claro, SP, Brazil
fFraunhofer Institute for Manufacturing Technology and Advanced Materials, 28359, Bremen, Germany
First published on 1st February 2021
Abstract
Photocatalyst-coated self-cleaning ceramic tiles are in high demand for indoor and outdoor applications aimed at keeping a clean environment. Their industrial processing, however, often requires firing at temperature (1000–1200 °C) much higher than the thermal stability limits of common photocatalysts (<1000 °C) which results a significant loss in self-cleaning activity of the tiles. To address this issue, we have coated commercial ceramic tiles with thermally stable core@shell SiO2@TiO2 particles, which even after single-fire industrial treatment (1000–1140 °C), exhibit excellent self-cleaning activity, much higher than that of control tiles prepared with commercial benchmark P25 TiO2 photocatalyst. Importantly, the photocatalytic activity of SiO2@TiO2 particles, in both powder form and as coatings on ceramic tiles, enhanced with the increase in calcination temperature (to as high as 1000–1140 °C) which is in sharp contrast to the normal photocatalytic behavior of unsupported TiO2. This article explores in details the exceptionally high and industrially relevant thermal stability of silica-supported anatase nanocrystals (5 nm) (SiO2@TiO2) against phase transition and crystallite growth and brings new insight into the effect of core@shell configuration on the thermal stability and photoactivity of SiO2@TiO2 particles. A comprehensive discussion on the relationship between core@shell structure, thermal stability and photoactivity is presented. These SiO2@TiO2 particles with ideal physicochemical characteristics (small phase-pure anatase nanocrystals with higher resistance towards crystallite growth, phase transformation or surface-area loss upon calcination) are ideal photocatalytic materials for efficient photodegradation of organic pollutants for effective environmental remediation and other applications that involve high-temperature processing such as self-cleaning coatings and photocatalytic ceramics.
1. Introduction
Due to its high photocatalytic efficiency, TiO2 still holds a prominent position among heterogeneous photocatalysts and is thus one of the most widely used semiconductor photocatalysts for environmental remediation processes,1–5 the design of self-cleaning surfaces6–8 and solar energy conversion.9–12 Most of these applications closely depend on the physical properties13–19 such as crystallinity20 and crystalline form,21,22 surface properties (area, pores-volume, roughness)23 and particle size or morphology23,24 of titania powder or films.It is well-known that, among other factors mentioned above, the photocatalytic properties of TiO2 strongly depend on its crystalline phase.21,25–31 Except for the few studies that report higher photoactivity for rutile32,33 or somewhat promising activity for the least-studied brookite phase,34 most of the literature studies agree on anatase being the most active photocatalytic form of TiO2.21,25,26,28–31,35 The superior photocatalytic activity of anatase compared to rutile is due to: (i) indirect and slightly wider (by 0.1 eV) band gap of anatase26 which lowers the rate of recombination of the photogenerated e−–h+ pair,26,36 as suggested by experimentally measured lifetimes of charge carries which are one order longer in anatase than rutile29 and/or higher photoluminescence yield in rutile,37 (ii) faster enough electron mobility in anatase (than rutile) to allow effective bulk transport or diffusion of charge carries through the crystalline structure to the surface in anatase, as supported by a study of photoactivity-film thickness relationship of the anatase/rutile-based epitaxial films21,38 and the lower calculated effective mass of charge carriers in anatase,26 (iii) unique ability of anatase to promote formation of peroxo species on its surface22 and (iv) the generation and diffusion of photogenerated mobile ˙OH radical to bulk solution only in anatase, making it possible to photodegrade target molecules within the ˙OH diffusion zone, in addition to those adsorbed on the surface of anatase.28,31 Thus, it is important to develop highly phase-selective synthetic methods for anatase with small particle size and good thermal stability to ensure better photocatalytic properties of the system.24
Similarly, the crystallinity of TiO2 is important for its photocatalytic activity since suppression of electron–hole recombination closely depends on the atomic arrangement in solid materials.39 The sol–gel method is one of the most widely utilized methods for the preparation of TiO2 which involves the hydrolysis-polycondensation of the molecular precursors (a metal salt or alkoxide) to produce metal hydroxides.1,24,40,41 The sol–gel method, however, generally results in amorphous products after the hydrolysis-polycondensation step42 which often require post-synthesis treatment (such as calcination at higher temperature (>500 °C)43,44) to obtain materials with good crystallinity, less crystal structure defects, and other desired characteristics. Since crystal defects serve as recombination centers for charge carriers, annealing of the photocatalyst particles often results in higher photocatalytic activity.13,17
Unfortunately, however, calcination can also result in a possible (i) decrease in surface area and pore-volume, (ii) uncontrolled nanoparticles growth and sintering43 or increase in crystallite size and (iii) irreversible anatase-to-rutile (A → R) transformation,44–48 changes that are detrimental to the photocatalytic performance. The A → R transformation, often accompanied by an increase in crystallite size, occurs because bulk rutile is thermodynamically more stable than bulk anatase. The A → R transformation becomes of particular concern when TiO2-based materials are subjected to high-temperature calcination that causes nanoparticles sintering. The interface-nucleation49,50 is the dominant phase transformation mechanism when amorphous TiO2 samples are thermally treated above 400 °C to crystallize them. During this process, the anatase crystals grow in size due to sintering and rutile starts to nucleate at the interface of aggregated anatase and the rate of A → R transformation closely depends on initial particle size and degree of aggregation and sinterization.49,51–53
The design and development of photocatalytic self-cleaning ceramic tiles is an important example of titania-based photocatalysis where A → R transformation and other detrimental effects caused by thermal treatments become the bottle-neck in the way of practical applications of the technology. Titania-coated ceramic tiles demonstrate photocatalytic, self-cleaning and antibacterial properties as well as UV-induced hydrophilicity.54 Such properties of titania-coated self-cleaning surfaces (external structures of buildings, for instance) allow pollution control in heavily polluted environments on one hand and prevent deterioration of the aesthetical aspects on the other. Additionally, their application in coverings for floor and skim coats for walls is a promising option for sterilization and photo-inactivation of microorganisms in the indoor environments. Despite numerous reports on the development of titania-coated photoactive ceramic tiles with interesting properties, commercial processing or viability and performance of such materials is still hindered by the low thermal stability of titania nanomaterials (<500 °C),1,55 as compared to the high firing temperatures (900–1200 °C) employed in the ceramic industry.54,56 As expected, such harsh treatments often lead to A → R transformation and loss of surface area and thus greatly reduce the photocatalytic performance of titania nanoparticles in the self-cleaning surface/tiles.57 Thus, research efforts are being made to develop thermally stable photocatalysts capable of enduring the harsh treatments during the single-fire thermal processing of ceramic tiles,54 without undergoing phase transformation or loss in photoactivity.
The task of obtaining materials with such high thermal stability (up to 800–1000 °C) and desired physicochemical properties is challenging, but recent studies1,53,58 have demonstrated the possibility of preparing nanosized and yet thermally stable photocatalytic materials through a careful choice of support material (such as SiO2 in this study) or crystallographic control that allow better dispersion of the nanophotocatalysts and restrain the movement of photocatalysts’ nanocrystallites under high-temperature conditions. Silica is the support material of choice due to its rich and well-known surface chemistry,59 easy synthesis by the widely-investigated Stöber method,60 low cost, high thermo-, mechanical-stability and high adsorption capacity.24 Moreover, the surface silanol (Si–OH) groups of SiO2 are active and provide sites for effective functionalization59,61 and the formation of interfacial Ti–O–Si bond in TiO2–SiO2 mixed oxide materials which has been found to affect the optical62–64 and thermal properties1,65,66 of the silica-supported TiO2 materials. Additionally, SiO2 is optically transparent in the wavelength region (UV-visible) of interest for photocatalysis.
Keeping this strategy in mind, the present study reports the preparation of thermally stable SiO2@TiO2 photocatalysts materials. Importantly, we show how the enhanced thermal stability of silica-supported anatase nanocrystals affects their photocatalytic performance when the SiO2@TiO2 samples are subjected to high-temperature thermal treatment. Based on a detailed characterization of the samples using an array of complementary characterization techniques, a comprehensive discussion on the relationship between core@shell structure, thermal stability and photoactivity is presented. Additionally, we demonstrate the successful application of the prepared thermally stable SiO2@TiO2 photocatalyst on commercial ceramic tiles using single-fire treatment under both benchmark (up to 1000 °C) and industrial (up to 1140 °C) conditions. These SiO2@TiO2-based photocatalytic ceramic tiles show superior photocatalytic performance than that of control tiles coated with commercial P25 TiO2 photocatalyst. We advocate that the unique physicochemical characteristics of SiO2@TiO2 make them ideal photocatalysts for efficient environmental remediation processes and the design of self-cleaning surfaces such as ceramic tiles.
2. Experimental
2.1 Reagents
Titanium(IV) isopropoxide (TiP, 97%) and tetraethylorthosilicate (TEOS, 98%) were purchased from Sigma-Aldrich (USA). Commercial TiO2 photocatalyst (Aeroxide TiO2 P25) was obtained from Evonik, Germany. HPLC grade anhydrous ethanol and isopropanol were supplied by Panreac (Spain). Ammonium hydroxide (28%) and crystal violet (CV, 1% solution) dye were obtained from QHEMIS (SP, Brazil). All chemicals were used without further purification.2.2 Synthesis of SiO2 and SiO2@TiO2 particles
The Stöber SiO2 particles60 and SiO2@TiO2 core@shell particles were prepared by a sol–gel method according to our previous report2 using the corresponding alkoxides as molecular precursors. Briefly, 0.5 g of Stöber SiO2 particles were dispersed in 100 mL of ethanol–isopropanol mix solvent (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)), followed by addition of 750 μL of titanium(IV) isopropoxide, allowing the mixture to stir during 20 h in an air-tight Teflon container before being hydrolyzed by addition of a 9 mL water–solvent mixture (1:2 (v/v)). The resulting SiO2@amorphous-TiO2 particles were subjected to a hydrothermal treatment (110 °C, 24 h) to selectively crystallize the amorphous TiO2 into phase-pure anatase with mesoporous shell character. For comparative study, unsupported TiO2 particles were also prepared by the same method in the absence of SiO2 particles in the reaction mixture. Further details of the synthesis can be found in ref. 2. To study the effect of core@shell configuration on their thermal stability (phase transformation, crystallite size growth) and other morphological features, the as-prepared SiO2@TiO2 particles were subjected to thermal treatment at different temperatures (600, 800 and 1000 °C) for 1h under ambient air at a heating rate of 15 °C min−1, as discussed in details later.
:1 (v/v)), followed by addition of 750 μL of titanium(IV) isopropoxide, allowing the mixture to stir during 20 h in an air-tight Teflon container before being hydrolyzed by addition of a 9 mL water–solvent mixture (1:2 (v/v)). The resulting SiO2@amorphous-TiO2 particles were subjected to a hydrothermal treatment (110 °C, 24 h) to selectively crystallize the amorphous TiO2 into phase-pure anatase with mesoporous shell character. For comparative study, unsupported TiO2 particles were also prepared by the same method in the absence of SiO2 particles in the reaction mixture. Further details of the synthesis can be found in ref. 2. To study the effect of core@shell configuration on their thermal stability (phase transformation, crystallite size growth) and other morphological features, the as-prepared SiO2@TiO2 particles were subjected to thermal treatment at different temperatures (600, 800 and 1000 °C) for 1h under ambient air at a heating rate of 15 °C min−1, as discussed in details later.
2.3 Preparation of SiO2@TiO2-coated ceramic tiles
Unfired commercial white-colored ceramic tiles were kindly provided by Artec Pisos e Revistimentos Company located in Cordeirópolis, SP, Brazil. Typically, A 20 × 20 mm tile piece was spray-coated with aqueous suspension (3.4% (wt/vol)) of SiO2@TiO2 using a SagymaPro SW-770 airbrush (Sagyma, China). Spray-coating deposition was carried out using compressed air at a controlled flow rate of 7 L min−1, depositing a single layer of photocatalyst particles on ceramic tiles (estimated mass deposited = 0.25 mg cm−2). For comparison of photocatalytic performance, ceramic tiles spray-coated with benchmark commercial TiO2 photocatalyst (Aeroxide TiO2 P25 from Evonik, Germany) were prepared under identical conditions. The photocatalysts-coated ceramic tiles were annealed under two different firing conditions: (i) annealing in laboratory furnace involving isothermal heating at 1000 °C for 1h (referred to as laboratory-fired tiles) and (ii) industrial sintering process at Artec ceramics company (Cordeirópolis, SP, Brazil) involving single firing annealing at a temperature around 1140 °C for 20 min (referred to as industrially-fired tiles).2.4 Materials characterization
For SEM and TEM analysis, the sample powder was suspended in water and the resulting dilute suspension was deposited onto the surface of a pre-cleaned monocrystalline silicon substrate or SiO/Formvar coated TEM 300 mesh copper grid and dried under ambient air. Ceramic tiles samples were directly supported on aluminum stubs. All samples were sputter-coated with a thin layer of carbon to ensure conductivity. FEG-SEM analysis was carried out using a JEOL JSM-7500F microscope (JEOL, Japan) in secondary electron detection mode, operated at 2 kV and a working distance of 6–13 mm. TEM and HRTEM images were collected using an FEI Tecnai G2 F20 microscope (FEI, Netherlands) operated at 200 kV. In situ X-ray diffraction (XRD) measurements were carried out at the XPD beamline of the Brazilian Synchrotron Light Laboratory (LNLS, Brazil)67 under research proposal XPD-19059. Beamline experimental station setup included a Huber diffractometer (Huber, Germany), 1D Mythen detectors (DECTRIS, Switzerland) and sample holder inserted in a furnace enabling data collection under in situ heating conditions. In situ XRD measurements were performed with 7.99 keV (0.155 nm) monochromatic beam during high-temperature thermal treatment and the diffractograms were acquired under constant slow heating (5 °C min−1) up to 1000 °C with an interval of around 25 °C between each measurement. Higher quality diffraction patterns were also obtained after isothermal treatments at 600, 800 and 1000 °C for 1 h. For comparison, ex situ XRD measurements were also collected in an AXS D8 ADVANCE (Bruker, Germany) benchmark X-ray diffractometer operated at 40 kV and 40 mA and employing Ni-filtered Cu Kα radiation. Crystallite sizes were estimated using Scherrer formula68 based on diffraction peaks broadening after correction for instrumental broadening (determined by measuring under the same conditions a bulk Al2O3 standard). Rutile formation as a function of temperature was estimated using Spurr-Myers empirical relation69 based on the intensity ratio of (100) and (101) diffraction peaks of anatase and rutile, respectively. Nitrogen adsorption isotherms were measured in an ASAP 2010 Micromeritics (Micromeritics, USA) apparatus at liquid nitrogen temperature (77 K). To remove adsorbed water, powder samples were degassed in vacuum at 120 °C for 12 h. The specific surface area (ABET) values were determined from nitrogen adsorption isotherms using the Brunauer–Emmett–Teller (BET) method, while pore size distribution (PSD) analysis was carried out using DFT Plus Micromeritics software based on the classical Kelvin equation and the Harkins and Jura isotherm model for cylindrical pores.70,71 Raman spectroscopy microanalysis of the photocatalyst-coated ceramic tiles was carried out using a LabRAM HR 800 model Raman spectrophotometer (Horiba Scientific, Japan) coupled to an optical microscope and equipped with a 632.81 nm He–Ne laser. Spectra of selected micro-regions (×100 objective lens magnification) were collected in the range of 100–1000 cm−1 with an acquisition time of 20 s and 20 scans. The X-ray photoemission spectra (XPS) spectra were acquired using a Kratos AXIS Ultra instrument equipped with a monochromatic X-ray source (Al Kα, hν = 1486.6 eV) under the following acquisition parameters: base pressure of 4 × 10−8 Pa, sample neutralization with low energy electrons (kinetic energy < 5 eV), hybrid mode (electrostatic and magnetic lenses used), electrons take-off angle of 0°, pass energy of 10 or 20 eV in high-resolution spectra. The analysis area was elliptically shaped with main axes of 300 μm × 700 μm. Charge correction of the binding energy (BE) values was made with respect to the main C 1s emission peak (C–C, C–H) of adventitious carbon with BE of 285.0 eV. Using CasaXPS software, the fitting of XPS spectra was performed with a mixture of Gaussian and Lorentzian functions considering 30% of Lorentzian contribution. The photoluminescence (PL) emission spectra of the powder samples (λ(exc) = 325 nm) were acquired using a Horiba Jobin Yvon spectrofluorometer (model fluorolog-3 FL3-122).2.5 Photocatalytic activity evaluation
Before coating the SiO2@TiO2 particles onto the ceramic tiles, we studied the photodegradation of crystal violet (CV) dye in aqueous suspensions of the SiO2@TiO2 particles24 to evaluate the effect of physicochemical characteristics, especially thermal stability, on photoactivity. For this purpose, the photocatalyst powder (15 mg) dispersed in 35 mL deionized water by sonication for 30 min was mixed with 35 mL of CV dye solution (20 ppm). This dye/photocatalysts mixture, containing 0.2 g L−1 photocatalysts and 10 ppm CV, was magnetically stirred in dark for 0.5 h and then irradiated with UV-visible light from a Xe-arc lamp (Lightningcure LC8 (Hamamatsu, Japan), λemis = 300–600 nm, maximum UV light output at 365 nm = 18.5 mW cm−2) placed 7 cm away from the photoreactor. Sample aliquots before and after each 10 min of UV illumination were collected, centrifuged and their electronic absorption spectra collected. To compare the photoactivity of the different samples, the photodegradation kinetic profiles were fitted as first-order exponential decay curves and the corresponding observed rate constants (kobs) were determined using the equation: where C0 and C represents initial CV concentration and concentration after certain interval of time (t), respectively.
where C0 and C represents initial CV concentration and concentration after certain interval of time (t), respectively.
The photocatalytic self-cleaning performance of the SiO2@TiO2-coated ceramic tiles was evaluated by following the photodegradation of CV dye over-layer deposited on tile's surface.72 The dye over-layer was deposited by drop-casting 40 μL of a 0.08% CV dye solution in isopropanol on the surface of ceramic tile and allowing it to dry under ambient air for 24 h. The concentration of CV in the over-layer, deposited on the surface of photoactive tiles, as function of UV irradiation exposure time, was monitored by taking its electronic reflectance spectra using a USB 4000 spectrometer (Ocean Optics, USA.) equipped with a Vis/NIR reflectance optical fiber and an LS1 tungsten halogen lamp. A 16S Solar Light Simulator Xe arc lamp (Solar Light Co., USA) was used as light source, maintaining a sample-to-lamp distance of 7 cm and light spot diameter of 1 cm. The relative humidity during photodegradation assays was kept in the range of 60–70%. The collected reflectance spectra were converted to F(R) function using the Kubelka–Munk equation73,74 and the dye photodegradation kinetic profiles were obtained based on decrease in area of the F(R) vs. wavelength spectra (450–700 nm). For comparison, the photocatalytic activity of P25-coated ceramic tiles was also measured under identical conditions.
3 Results and discussion
3.1 Synthesis and particles structure of SiO2@TiO2
In this work, a two-step strategy2 was followed to prepare uniform SiO2@TiO2 particles with small anatase nanocrystals essentially supported on the surface of SiO2 particles, without the formation of unsupported free TiO2 particles. First, we allowed the precursor (titanium(IV) isopropoxide) to pre-adsorb on the surface of SiO2 particles suspended in ethanol–isopropanol mix solvent (3:1 (v/v)) containing a minute quantity of water (<0.05%) and then slowly hydrolyzed the titanium(IV) isopropoxide adding an alcohol–water mixture. The morphology of the resulting SiO2@TiO2 particles was studied by FEG-SEM (Fig. 1).
 | ||
| Fig. 1 Representative (a and b) FEG-SEM and (c and d) TEM images of the SiO2@TiO2 particles (average size = 260 ± 22 nm) before calcination (c) and after calcination at 1000 °C (d); HRTEM image of the shell part of the SiO2@TiO2 particles (e) before and (f) after calcination at 1000 °C; the presence of ∼5 nm nanocrystallites with interplanar distances of 0.35 nm is evident in (e). | ||
Knowing that the Stöber SiO2 particles exhibit a smooth surface,2,60 the rougher surface of SiO2@TiO2 particles confirms the formation of a TiO2 layer around SiO2 particles (Fig. 1a and b). This core@shell configuration is more evident in the TEM image of the as-prepared sample (Fig. 1c). The SiO2@TiO2 particles have an average size of 260 ± 22 nm and the TiO2 is exclusively deposited on the surface of SiO2, with no evidence of the formation of coreless or unsupported TiO2 particles. The presence of ethanol ensures partial conversion of highly reactive titanium(IV) isopropoxide to its less reactive oligomeric form,2 thus allowing control of the nucleation and deposition rates of TiO2 on the surface of SiO2 particles. Moreover, the trace amount of water present during the pre-adsorption step is expected to induce limited hydrolysis of titanium(IV) isopropoxide and condensation, or at-least H-bonding interaction, of the hydrolysed products with the surface hydroxyl group of SiO2 leads to their adsorption on the surface of SiO2, thus favouring heterogeneous nucleation process75 and avoiding the formation of unsupported TiO2 particles (via homogenous nucleation). The HRTEM image (Fig. 1e) of the as-prepared SiO2@TiO2 particles shows the presence of small nano-crystallites (5.2 ± 0.3 nm) with interplanar distances of 0.35 nm which correspond to (101) crystal planes of anatase TiO2.76 After calcination at 1000 °C, the porous TiO2 shell becomes somewhat denser and a slight increase in particle/crystal size (∼14 nm) is observed (Fig. 1d and f).
3.2 Surface chemical characterization of SiO2@TiO2 using XPS
XPS analysis was performed to investigate the surface properties of the SiO2@TiO2 core@shell nanoparticles. The Si 2p core region of the sample, adequately fitted with two components, exhibits the characteristic emission peaks of SiO2 at a binding energy (BE) of 104.1 eV77–80 with a second component appearing at 102.8 eV (Fig. 2a). This component at slightly lower BE has previously been assigned to the Si atom in Si–O–Ti bond.2,77,80 | ||
| Fig. 2 XPS spectra of SiO2@TiO2 showing (a) Si 2p and (b) O 1s core regions. The chemical bonds involving O and Si atoms that give rise to different component peaks in the XPS spectra have been indicated on the graphs. | ||
To reinforce our assignment of the Si 2p photoemission peak at 102.8 eV to Si–O–Ti cross-linking bonds at the interface of the two oxide materials, the O 1s core region was also analyzed. The O 1s core region of the as-prepared SiO2@TiO2 samples could not be satisfactorily fitted by considering the contributions of oxygen atoms from Si–O–Si and Ti–O–Ti only and an adequate fitting was obtained considering three components at BE values of 530.2, 531.6 and 533.1 eV (Fig. 2b). The XPS peak at 530.2 eV and 533.1 eV could be assigned to the O atoms in TiO2 (Ti–O–Ti) and SiO2 (Si–O–Si), respectively.2,77–81 The additional intermediate feature at 531.6 eV could be assigned to the O atom in the interfacial Si–O–Ti bond,2,77,81 thus confirming the previous assignment of Si 2p region.82 Moreover, a theoretical study using DFT calculations also suggests that the formation of interfacial Si–O–Ti bond in SiO2-TiO2 materials plays important role in determining the enhanced stability of SiO2/TiO2 interface.65
3.3 Crystalline phase and thermal stability of SiO2@TiO2
In situ XRD measurements were performed to study the structural changes and thermal stability of SiO2@TiO2 (Fig. 3b) and unsupported TiO2 (Fig. 3a) particles during high-temperature thermal treatment up to 1000 °C. These measurements allowed us to continuously monitor the A → R transformation and crystallite growth as function of temperature (Fig. 3a, b and 4). Higher quality diffraction patterns were also obtained after isothermal treatments at 600, 800 and 1000 °C for 1h (Fig. 3c and d) and relevant structural information of such annealed samples are summarized in Table 1.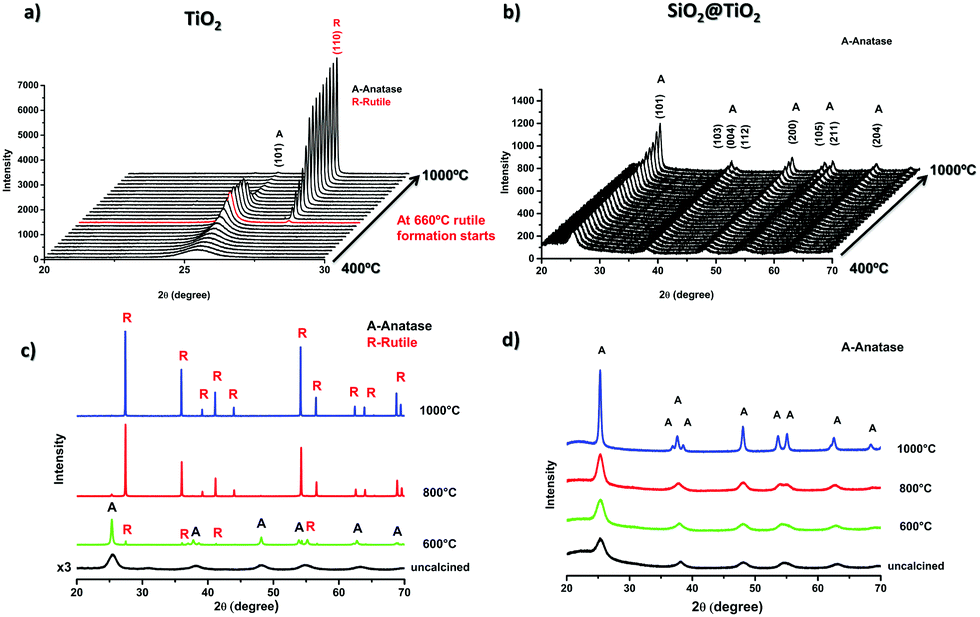 | ||
| Fig. 3 In situ powder X-ray diffractograms of (a) unsupported TiO2 and (b) SiO2@TiO2 samples collected under continuous and slow heating (5° min−1) up to 1000 °C; powder X-ray diffractograms of (c) unsupported TiO2 and (d) SiO2@TiO2 after isothermal treatments at 600 °C, 800 °C and 1000 °C for 1 h. | ||
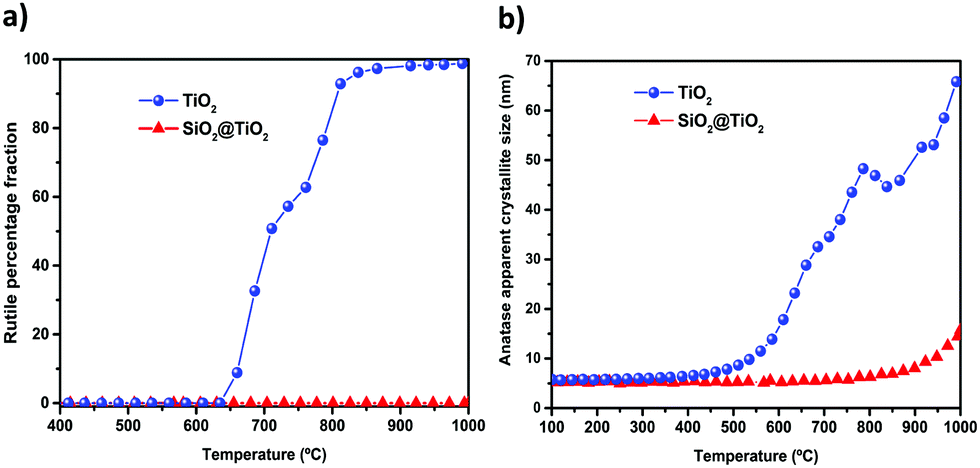 | ||
| Fig. 4 Evolution of (a) rutile formation and (b) anatase crystallite growth for the SiO2@TiO2 and unsupported TiO2 as function of calcination temperature. Data derived from in situ XRD data (Fig. 3a and b) collected under continuous and slow heating (5° min−1) of the samples. | ||
| Sample code | Annealing temperature | TiO2 phases | Crystalline size (nm) (major phase) | A BET (m2 g−1) |
|---|---|---|---|---|
| SiO2@TiO2 | As-prepared | Anatase | 5 ± 1 | 119 ± 1 |
| SiO2@TiO2 | 600 °C | Anatase | 5 ± 1 | 85 ± 1 |
| SiO2@TiO2 | 800 °C | Anatase | 7 ± 1 | 68 ± 1 |
| SiO2@TiO2 | 1000 °C | Anatase | 17 ± 4 | 24 ± 4 |
| TiO2 | As-prepared | Anatase (major) | 5 ± 1 | 217 ± 1 |
| Brookite (minor) | ||||
| TiO2 | 600 °C | Anatase (83%) | 24 ± 6 | 40 ± 2 |
| Rutile (17%) | ||||
| TiO2 | 800 °C | Rutile (98%) | >150 | <1 |
| Anatase (2%) | ||||
| TiO2 | 1000 °C | Rutile | >150 | <1 |
As shown in Fig. 3, the XRD patterns of the as-prepared SiO2@TiO2 and unsupported TiO2 both closely match that of anatase phase, thus confirming the selective crystallization of amorphous titania into phase-pure anatase nanocrystals of around 5 nm in both these samples upon hydrothermal treatment (110 °C, 24 h). However, the structural and thermal properties of these samples start to show significant differences when further subjected to calcination at higher temperatures. For instance, the unsupported TiO2 undergoes A → R transformation, possibly following an interface-nucleation mechanism,46,49,83 as evidenced by the appearance of sharp diffraction peaks assigned to the rutile phase of TiO284 (Fig. 3a and c). Such A → R transformation in TiO2 starts around 660 °C and rutile becomes the major phase at 800 °C and the only phase at 1000 °C (Fig. 4a). The A → R transformation in TiO2 upon annealing (at T > 600 °C) is accompanied by a drastic increase in crystallite size and large crystallites (rutile > 150 nm, anatase = 50 nm) are obtained at around 800 °C (Table 1 and Fig. 4b).
For the SiO2@TiO2, the anatase nanocrystals of the same initial size (∼5 nm) show remarkable thermal stability, retaining their phase-purity even after calcination at 1000 °C (Fig. 3b, d and 4a). In this case, it is only at temperatures higher than 800 °C that a slight increase in intensity and/or decrease in width of anatase diffraction peaks are observed (Fig. 3b and d), indicating a relatively small increase in crystallite size (from 5 nm to 17 nm after annealing at 1000 °C for 1 h) of silica-supported anatase (Fig. 4b). Similar results were obtained when the ex situ diffractograms of SiO2@TiO2 annealed at 600 °C, 800 °C and 1000 °C for 1h were obtained using a benchmark Bruker D8-Advance X-ray diffractometer (Fig. S1, ESI†).
Literature survey indicates that our SiO2@TiO2 particles show higher thermal stability than most of the reported thermally stable SiO2–TiO2 photocatalysts66,85–88 which, upon thermal treatments in air at temperatures higher than 900 °C suffer from at least partial A → R transformation.66,85–88 Such thermal behavior has also been observed in core–shell structured SiO2–TiO2 aerogels previously reported by our group.1 Moreover, such outstanding stability could be related an interfacial diffusion of silicon and titanium cations during hydrothermal treatment leading to Si–O–Ti bond formation not only at the interface but also in deeper atomic layers of both materials. The effect of this interdiffusion and formation of Si–O–Ti bonds was studied using DFT calculations,65 which showed an interface stabilizing effect of this bond, leading to higher mechanical stability. We may as well infer that such interfacial bonding results in higher thermal stability by inhibiting nanocrystals diffusion on the interface which is involved in grain growth as well as A → R transformation, as discussed later.
3.4. Porosity, surface area and pore size distribution
The BET specific surface area (ABET), pore structure and pore size distribution (PSD) of the as-prepared and calcined SiO2@TiO2 photocatalysts was studied through nitrogen physisorption analysis. The N2 adsorption–desorption isotherms of the SiO2@TiO2 particles (Fig. 5a) are of type IV,71 confirming the mesoporous nature of TiO2 shell. The SiO2@TiO2 particles display a bimodal PSD consisting of narrowly distributed small (average diameter = 5 nm) mesopores pertaining to anatase shell and widely distributed larger pores (>20 nm) that correspond to inter-particle spaces in agglomerated core@shell photocatalyst (Fig. 5b).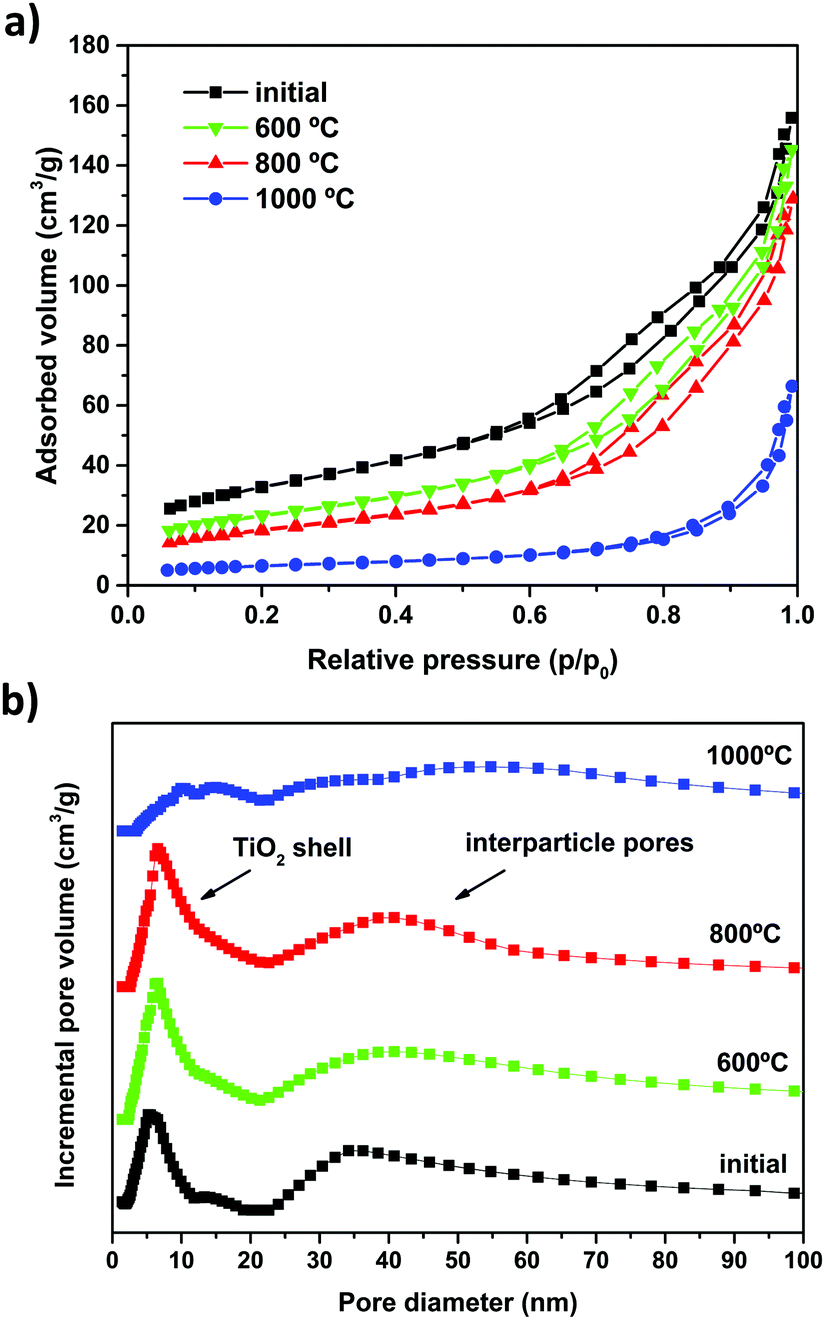 | ||
| Fig. 5 (a) N2 adsorption–desorption isotherms and (b) pore size distribution of the as-prepared SiO2@TiO2 particles and after calcination at different temperatures. | ||
A comparison of the ABET values (Table 1) and PSD analysis (Fig. 5b) before and after calcination sheds some light on the effect of high-temperature annealing on the textural properties of SiO2@TiO2 and unsupported TiO2 particles. The unsupported TiO2 particles completely lose their porosity and surface area (by as much as ∼99%, see Table 1) due to drastic sintering and crystallite growth, as previously demonstrated by XRD analysis (Fig. 3 and 4). On the other hand, the higher stability of SiO2@TiO2 is reflected in their resistance to decrease in surface area upon calcination. The SiO2@TiO2 particles retain their mesoporous structure (Fig. 5) and 60% of their ABET upon calcination at temperatures as high as 800 °C (Table 1). Upon further thermal treatment at 1000 °C, however, the TiO2 shell in SiO2@TiO2 samples loses its mesoporosity and suffers around 80% decrease in ABET (∼), changes that are related to the growth of the crystallites of silica-supported anatase (see Fig. 4) as well as the collapse of porous system at such a high temperature.
3.5. Why SiO2@TiO2 core@shell particles show enhanced thermal stability?
From the discussion presented above, we notice that the enhanced thermal stability of anatase in SiO2@TiO2 samples is manifested in its ability to resist change in its crystalline phase, crystallite size and specific surface area upon high-temperature calcination (see Table 1). It is thus important to understand how the SiO2 support affects the thermal properties of supported anatase nanocrystals.The growth of anatase nanocrystals upon thermal treatment is one of the main factors that induce conversion into rutile45,89 and the rate of anatase-to-rutile transformation closely depends on initial particle size and degree of nanoparticles aggregation.46,49,53 Considering this, the higher thermal stability of our SiO2@TiO2 compared to unsupported TiO2 can be assigned to a combined effect of different factors including (i) the formation of an interfacial Ti–O–Si bond, (ii) the formation of small anatase nanocrystals (∼5 nm), (iii) better dispersion of anatase nanocrystals and (iv) silica effect, each one of which is discussed in details below.
The presence of amorphous silica has been found to improve the thermal stability of anatase, possibly due to a surface curvature effect90,91 and/or the formation of Ti–O–Si bond.65,92 These two factors may hinder the diffusion of small anatase nanocrystals92 which is the first step in the interface nucleation mechanism of A → R transformation.93 For instance, Li et al.91 reported that the crystallite size of TiO2 was smaller for small size SiO2 core particles (24 nm) due to surface curvature effect and this surface curvature effect was less important for SiO2 core particles with diameters of 95 nm or larger. According to their model, for SiO2 particles smaller than 60 nm, surface silanol groups remain isolated due to greater surface curvature and act as isolated nucleation sites for the formation of small TiO2 crystallites (8.2 nm at Ti/Si = 0.28). For SiO2 particles larger than 60 nm, extensive hydrogen bonding between surface silanol groups leads to the formation of agglomerates and TiO2 crystals of larger sizes (22 nm at Ti/Si = 0.4).91 However, this model neither explains the formation of small (5 nm) anatase crystals nor the thermal stability of our SiO2@TiO2 particles since the SiO2 sub-micron spheres used in our work have larger diameters (>200 nm) than the threshold size of 60 nm suggested by the authors.91
Considering that XPS analysis confirmed the formation of interfacial Ti–O–Si bond in SiO2@TiO2 samples (Fig. 2), in agreement with previous theoretical and experimental studies,2,65,77,81 we suggest that the formation of this interfacial bond and the presence of amorphous SiO2 help in preventing aggregation and thus phase transformation by lowering the surface diffusion of anatase crystallites.92 Moreover, theoretical studies of the TiO2/SiO2 interface using density functional theory suggested the formation of Ti–O–Si bond and interdiffusion of Si and Ti ions between the anatase and SiO2 structures as the two interface stabilizing interactions with an interface stabilization of 1 eV and 1.5 eV, respectively.65
It is well known that the thermodynamic stability of the three phases of TiO2 (anatase, rutile, brookite) closely depends on their particles size and degree of aggregation.49,51,52 For instance, bulk anatase is less stable than bulk rutile but anatase becomes more stable in the nanoscale size regime (< 14 nm).46,52,94 This is because the contribution of surface enthalpies to total Gibbs free energy becomes significant at such small particle dimensions and the surface enthalpy of anatase ((0.4 ± 0.1) J m−2) is lower than that of rutile ((2.2 ± 0.2) J m−2).95 The lower surface energy of nanoanatase thus plays an important role in its phase stability, knowing that nanomaterials possess higher surface/volume ratios.94,96 The critical crystallite size of anatase for A → R transformation has been reported to be even higher (70–100 nm) for SiO2/TiO2 composites,92 compared to 14 nm52,94 for unsupported TiO2 cited above. Since the crystallite size of the well-dispersed anatase in our SiO2@TiO2 samples is smaller than 14 nm, even after thermal treatment at 800 °C, no phase transformation takes place.
Moreover, since A → R transformation occurs when rutile starts to nucleate at the interface of aggregated anatase crystals,46,49 the process is facilitated in case of samples containing aggregated nanoparticles. For the same reason, A → R transformation can be inhibited by reducing particles aggregation53,83,91,97 and/or keeping the particle size below 14 nm,49,51,52 for example, by supporting small TiO2 nanoparticles on silica, as discussed in this article. The hydrothermal formation of stable small (5 nm) anatase nanocrystallites, together with their improved dispersion on SiO2 surface, may thus be considered one of the reasons behind the higher thermal stability of our SiO2@TiO2 samples.49
We thus attribute the outstanding thermal stability of anatase in SiO2@TiO2 to its small crystallite size (∼5 nm) with better dispersion as well as to the suppression of coarsening and surface diffusion of anatase nanocrystals due to the presence of amorphous silica and the formation of the interfacial Ti–O–Si bond between the anatase TiO2 and silica particles, as evidenced by XPS analysis (Fig. 2). It implies that the SiO2@TiO2 particles, with their unique structural/phase stability, could be calcined at higher temperatures (1000 °C) to improve anatase crystallinity and/or reduce structural defects without any crystal growth or phase transformation. This results in a photocatalytic system exhibiting high efficiency (vide infra) and we exploit these ideal physicochemical characteristics of SiO2@TiO2 to prepare highly active self-cleaning ceramic tiles, as discussed later.
3.6 Thermal stability-photocatalytic relationship in SiO2@TiO2 particles
We noticed that the formation of core@shell structure in SiO2@TiO2 plays important role in controlling their physical properties (crystallinity, ABET, PSD) during post-synthesis annealing. To evaluate the effect of calcination or thermal stability on the photocatalytic properties, the photoactivity of SiO2@TiO2 (Fig. 6a) and unsupported TiO2 (Fig. 6b) samples as function of calcination temperatures was studied through photodegradation of CV dye (λmax = 582 nm) as a model pollutant. Direct photolysis of the CV dye by the UV light in the absence of any photocatalysts is small (∼6%), as indicated in Fig. 6a and b). An analysis of the kinetic profiles (Fig. 6a and b) and photodegradation rate constants (Fig. 6c) shows that the as-prepared uncalcined SiO2@TiO2 samples show better photoactivity than the as-prepared unsupported TiO2 samples. The higher photoactivity of SiO2@TiO2 is a combined result of the formation of porous shell consisting of well-dispersed small anatase (∼5 nm) nanocrystals, improved adsorption of the dye molecules and increased light path and hence efficient light harvesting ability of SiO2@TiO2, as demonstrated in our previous study.24 | ||
| Fig. 6 Photocatalytic activity of SiO2@TiO2 and unsupported TiO2 before and after calcination at different temperatures (600–1000 °C). The first-order exponential decay fitted kinetic profiles of CV dye removal in the presence of (a) SiO2@TiO2 and (b) unsupported TiO2 and (c) a comparison the first-order decay kinetic constants for these photocatalysts. Error bars correspond to the standard deviation of triplicate test. Direct photolysis of the CV dye by UV light in the absence of any photocatalysts has also been included in (a) and (b). | ||
Moreover, as expected from their contrasting thermal stability, the materials display very distinct photocatalytic behaviour upon annealing. A non-linear variation of photocatalytic activity with the calcination temperature is observed for the unsupported TiO2 sample (Fig. 6b). As compared to the hydrothermally prepared uncalcined TiO2, the TiO2 sample treated at 600 °C shows higher photocatalytic performance due to its increased crystallinity. However, calcination at a temperature higher than 600 °C leads to drastic decreases in photoactivity, which is related to the extensive crystallite growth, drastic loss of surface area and full conversion of the anatase (most photoactive anatase) to rutile (less photoactive) induced by thermal treatments at 800 °C and 1000 °C.
On the contrary, the photocatalytic activity of SiO2@TiO2 is significantly enhanced upon annealing to as high as 1000 °C (Fig. 6a) and is much higher than that of unsupported TiO2 samples. Again, such excellent photocatalytic behaviour is related to the higher thermal stability of anatase in SiO2@TiO2 samples which allows an increase in crystallinity of anatase without any phase transformation or drastic crystallite growth. The fact that SiO2@TiO2 annealed at different temperatures (600–1000 °C) display almost the same photoactivity and similar photodegradation rate constants (Fig. 6c) can be understood by considering the interplay between increased crystallinity (positive impact over photocatalytic activity) and decreased surface area (negative impact over photocatalytic activity) with the increase in the calcination temperature.
To further verify the effect of temperature and possible increase in crystallinity on the charge separation efficiency, photoluminescence spectra (PL) of the as-prepared and calcined SiO2@TiO2 samples were compared (Fig. S2, ESI†). The PL intensity resulting from the radiative recombination of charge carriers is higher for the as-prepared SiO2@TiO2 samples and lower for the calcined samples. Moreover, the PL intensity decreases with increase in calcination temperature, indicating lower electrons–holes recombination, possibly due to increased crystallinity of the calcined samples. These remarkable results reinforce the great potential of the prepared SiO2@TiO2 core@shell particles for technological applications that demand high-temperature processing such as self-cleaning coatings and photocatalytic ceramic tiles.
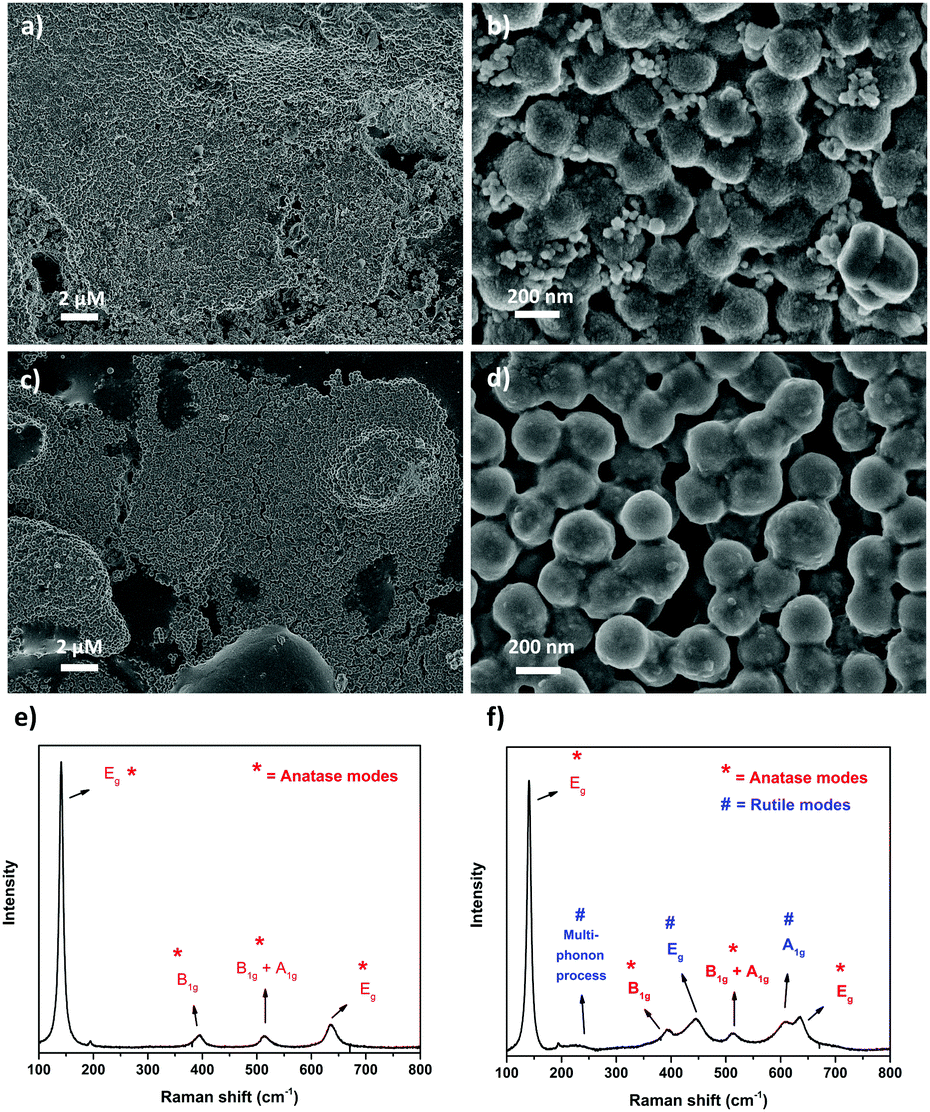 | ||
| Fig. 7 SEM images of SiO2@TiO2-coated ceramic tiles after annealing using (a and b) laboratory furnace (1000 °C, 1 h, 15 °C min−1) and (c and d) industrial single firing process (1140 °C, 20 min); Raman spectra of the respective (e) lab-fired and (f) industrially-fired SiO2@TiO2-coated ceramic tiles. The Raman modes and the corresponding crystalline phases of TiO2 have been marked (anatase = *, rutile = #) on the Raman spectra. | ||
3.7 Photocatalytic, self-cleaning ceramic tiles coated with SiO2@TiO2 particles
To test the implication of high thermal stability and excellent photocatalytic properties of SiO2@TiO2 particles in practical applications of commercial interest, the SiO2@TiO2 were spray-coated onto the surface of unfired commercial ceramic tiles. The low-magnification SEM images of SiO2@TiO2-coated tiles obtained after annealing in laboratory furnace (Fig. 7a) or industrial furnace (Fig. 7c) both show a uniform micrometric scale coating of the tiles’ surface. The higher magnification SEM image (Fig. 7b) of the coated tiles annealed in laboratory furnace clearly shows the presence of SiO2@TiO2 particles (similar to those observed for powder samples (Fig. 1b)) albeit with some neck formation between the particles. Moreover, this sample exhibits some small nanoaggregates that possibly results from the sintering of supported anatase nanocrystals (Fig. 7b). This makes sense as the industrially fired coated tile, which passes through a more abrupt and higher temperature (1140 °C) and is expected to undergo more sintering, shows no sign of any small nanoaggregate and exhibits a more extensive inter-particle neck-formation (Fig. 7d). This hypothesis is further supported by a comparison of the Raman spectra of SiO2@TiO2-coated tiles obtained after annealing in laboratory furnace (Fig. 7e) and industrial furnace (Fig. 7f). While the Raman spectrum of coated ceramic tile fired in laboratory furnace shows the characteristics Raman band of only anatase phase (marked as *) at 148 cm-1 (Eg), 396 cm-1 (B1g), 513 cm-1 (A1g/B1g) and 635 cm-1 (Eg)98,99 (Fig. 7e), additional Raman bands (marked as #) corresponding to rutile phase appear in the industrially fired ceramic tile (Fig. 7f). This suggests a higher degree of sintering in the industrially fired ceramic tiles which leads to at least partial conversion of anatase to rutile despite the high thermally stability of silica-supported anatase in SiO2@TiO2 samples. This partial conversion of anatase to rutile, however, is beneficial for the photocatalytic performance of the photoactive tiles as discussed below.To understand the effect of annealing and hence thermal stability on the photoactivity of coated photocatalytic ceramic tiles, the photodegradation of CV deposited as an over-layer on the surface of annealed tiles was studied under UV illumination (Fig. 8). In case of lab-fired tiles, the P25-coated tiles could photodegrade around only 30% of the CV dye after 30 min of UV illumination, whereas the SiO2@TiO2-coated tiles degraded over 90% of the dye in the same time (Fig. 8a). This difference in photoactivity and/or the higher self-cleaning of the lab-fired SiO2@TiO2-coated tiles is evident from the digital images shown in Fig. 8d. Similarly, the industrially-fired tiles showed excellent self-cleaning activity, with the SiO2@TiO2-coated tiles performing better than P25-coated tiles (Fig. S3, ESI†).
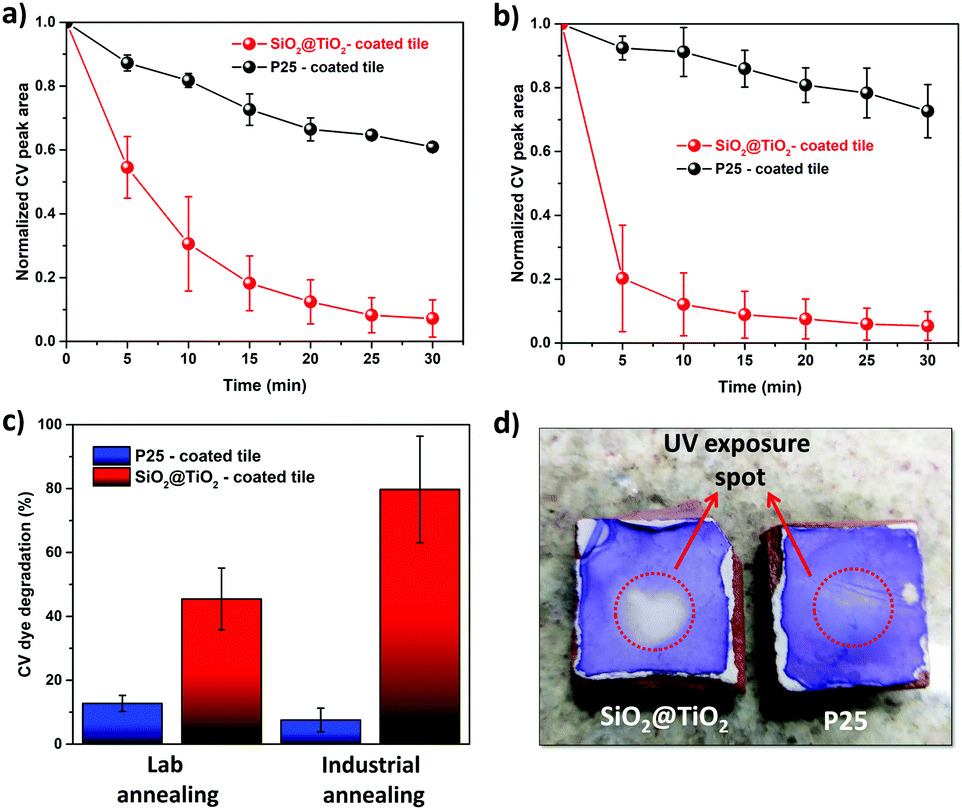 | ||
| Fig. 8 Comparison of the photocatalytic activity of annealed SiO2@TiO2-coated and P25-coated ceramic tiles. (a and b) Kinetic profiles of CV dye removal in the presence of (a) laboratory-fired and (b) industrially-fired ceramic tiles; (c) Comparison of the amount of CV dye degraded after 5 min of UV irradiation in the presence of annealed SiO2@TiO2-coated and P25-coated tiles; (d) digital photos of the laboratory-fired CV-covered photoactive tiles after 30 min exposure to UV light showing the superior self-cleaning performance of SiO2@TiO2-coated tiles. | ||
Comparing Fig. 8a and b, a prominent increase and slight decrease in photoactivity is observed for SiO2@TiO2-coated and P25-coated tiles, respectively, after industrial firing at higher temperature (∼1140 °C) even though such treatment results in more sintering and anatase to rutile conversion (Fig. 7). In fact, the SiO2@TiO2-coated industrially fired sample degraded around 80% of the dye over-layer within just 5 min of UV-illumination, as compared to less than 10% in case of P25-coated tiles (Fig. 8c). While the loss of photoactivity in P25-coated tiles upon industrial firing may be related to extensive sintering and complete transformation of the anatase fraction (∼70%)100 of P25 TiO2 to rutile (Fig. S4, ESI†), the higher activity of annealed SiO2@TiO2-coated tiles may be attributed to the formation of anatase/rutile heterojunction that allows better charge carries separation.100–103 These results testify the greater potential of SiO2@TiO2 particles with ideal physicochemical characteristics for use in the design of self-cleaning coatings and photocatalytic ceramics as well as environmental remediation. In fact, these SiO2@TiO2 coated tiles show far better photocatalytic behaviour than other TiO2-based coatings reported in literature (Table S1, ESI†).
4 Conclusions
Small anatase nanocrystals (∼5 nm), deposited on the surface of SiO2 particles in a core@shell (SiO2@TiO2) configuration using the sol–gel method, exhibit excellent thermal stability (1000 °C) which is reflected in their ability to resists change in phase, loss in surface area or increase in crystallite size during high-temperature calcination. Such higher thermal stability of silica-supported anatase is attributed to its small crystallite size (∼5 nm) with better dispersion and the formation of the interfacial Ti–O–Si bond between the anatase TiO2 and silica support particles which suppresses coarsening and surface diffusion of anatase nanocrystals during calcination. The consequence of this high thermal stability is that, unlike unsupported TiO2, the photocatalytic activity of SiO2@TiO2 particles enhances upon calcination at 1000 °C. The implication of this high thermal stability is that the SiO2@TiO2-coated photocatalytic ceramic tiles vastly outperform the photocatalytic activity of control tiles coated with commercial benchmark P25 TiO2 photocatalyst. Better dispersion, high thermal stability towards crystallite growth and phase transformation and small anatase crystallite size of the SiO2@TiO2 particle make these photocatalysts ideal candidates for effective environmental remediation and other applications that involve high temperature processing such self-cleaning coatings and photocatalytic ceramics.Conflicts of interest
There are no conflicts to declare. All authors have contributed to bring the manuscript to its current form and have given approval of the final version. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by São Paulo Research Foundation (FAPESP) [research grants no. 2013/24948-3, 2015/22828-6, 2018/01934-0 and 2018/19785-1]. Elias P. Ferreira-Neto thanks FAPESP for PhD and pos-doctoral fellowships [grants # 2013/24948-3 and # 2018/01934-0]. S. Ullah acknowledges financial support from the Higher Education Commission, Pakistan [Project No. 9286] as well as the Federal University of Mato Grosso do Sul (UFMS) and São Paulo State University (UNESP) for the support and opportunity to work as visiting researcher. The authors acknowledge the Brazilian funding agencies (CNPq and CAPES) for financial assistance. Mateus B. Simões and Amanda P. Perissinotto thank the Coordination for the Improvement of Higher Education Personnel (CAPES, Brazil) for PhD fellowships. H. Wender acknowledges the CNPq [grants no. 310066/2017-4 and 427835/2016-0] and the financial support provided by the CAPES-PrInt funding program [grant no. 88881.311921/2018-01 and # 88887.311920/2018–00]. We also thank the Brazilian National Synchrotron Light Laboratory (LNLS, Campinas Brazil) for providing the facility for in situ XRD analysis performed under re-search proposal # XPD-19059. We are grateful to Artec Pisos e Revistimentos Company (Corde-irópolis, SP, Brazil) for the donation of unfired ceramic tiles and assistance with industrial firing of the coated tiles.Notes and references
- E. P. Ferreira-Neto, M. A. Worsley and U. P. Rodrigues-Filho, J. Environ. Chem. Eng., 2019, 7, 103425 CrossRef CAS.
- E. P. Ferreira-Neto, S. Ullah, M. B. Simões, A. P. Perissinotto, F. S. de Vicente, P. L. L. M. Noeske, S. J. L. L. Ribeiro and U. P. Rodrigues-Filho, Colloids Surf., A, 2019, 570, 293–305 CrossRef CAS.
- J.-M. Herrmann, Catal. Today, 1999, 53, 115–129 CrossRef CAS.
- H. Zangeneh, A. A. L. Zinatizadeh, M. Habibi, M. Akia and M. Hasnain Isa, J. Ind. Eng. Chem., 2015, 26, 1–36 CrossRef CAS.
- P. A. K. Reddy, P. V. L. Reddy, E. Kwon, K.-H. Kim, T. Akter and S. Kalagara, Environ. Int., 2016, 91, 94–103 CrossRef CAS.
- S. Banerjee, D. D. Dionysiou and S. C. Pillai, Appl. Catal., B, 2015, 176–177, 396–428 CrossRef CAS.
- M. P. Seabra, R. R. Pires and J. A. Labrincha, Chem. Eng. J., 2011, 171, 692–702 CrossRef CAS.
- M. Gurbuz, B. Atay and A. Dogan, Int. J. Appl. Ceram. Technol., 2015, 12, 426–436 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Y. K. Kho, A. Iwase, W. Y. Teoh, L. Mädler, A. Kudo and R. Amal, J. Phys. Chem. C, 2010, 114, 2821–2829 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165 RSC.
- H. Kominami, S. Murakami, J. Kato, Y. Kera and B. Ohtani, J. Phys. Chem. B, 2002, 106, 10501–10507 CrossRef CAS.
- H. Cheng, J. Wang, Y. Zhao and X. Han, RSC Adv., 2014, 4, 47031–47038 RSC.
- A. P. Rivera, K. Tanaka and T. Hisanaga, Appl. Catal., B. Environ., 1993, 3, 37–44 CrossRef CAS.
- O.-O. Prieto-Mahaney, N. Murakami, R. Abe and B. Ohtani, Chem. Lett., 2009, 38, 238–239 CrossRef CAS.
- K. Tanaka, M. F. V. Capule and T. Hisanaga, Chem. Phys. Lett., 1991, 187, 73–76 CrossRef CAS.
- F. Pellegrino, L. Pellutiè, F. Sordello, C. Minero, E. Ortel, V.-D. Hodoroaba and V. Maurino, Appl. Catal., B. Environ, 2017, 216, 80–87 CrossRef CAS.
- X. Wang, L. Sø, R. Su, S. Wendt, P. Hald, A. Mamakhel, C. Yang, Y. Huang, B. B. Iversen and F. Besenbacher, J. Catal., 2014, 310, 100–108 CrossRef CAS.
- K. Eufinger, D. Poelman, H. Poelman, R. De Gryse and G. B. Marin, J. Phys. D: Appl. Phys., 2007, 40, 5232–5238 CrossRef CAS.
- T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, E. Sutter and M. Batzill, Sci. Rep., 2015, 4, 1–8 Search PubMed.
- J. Augustynski, Electrochim. Acta, 1993, 38, 43–46 CrossRef CAS.
- A. Vaccari, M. Campanati and G. Fornasari, Catal. Today, 2003, 77, 299–314 CrossRef.
- S. Ullah, E. P. Ferreira-Neto, A. A. Pasa, C. C. J. Alcântara, J. J. S. Acuña, S. A. Bilmes, M. L. Martínez Ricci, R. Landers, T. Z. Fermino and U. P. Rodrigues-Filho, Appl. Catal., B. Environ, 2015, 179, 333–343 CrossRef CAS.
- Z. Ding, G. Q. Lu and P. F. Greenfield, J. Phys. Chem. B, 2000, 104, 4815–4820 CrossRef CAS.
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC.
- Q. Sun and Y. Xu, J. Phys. Chem. C, 2010, 114, 18911–18918 CrossRef CAS.
- G. Odling and N. Robertson, ChemSusChem, 2015, 8, 1838–1840 CrossRef CAS.
- M. Xu, Y. Gao, E. M. Moreno, M. Kunst, M. Muhler, Y. Wang, H. Idriss and C. Wöll, Phys. Rev. Lett., 2011, 106, 138302 CrossRef.
- A. Y. Ahmed, T. A. Kandiel, T. Oekermann and D. Bahnemann, J. Phys. Chem. Lett., 2011, 2, 2461–2465 CrossRef CAS.
- W. Kim, T. Tachikawa, G. Moon, T. Majima and W. Choi, Angew. Chem., Int. Ed., 2014, 53, 14036–14041 CrossRef CAS.
- L. Li, J. Yan, T. Wang, Z. J. Zhao, J. Zhang, J. Gong and N. Guan, Nat. Commun., 2015, 6, 5881 CrossRef.
- S. J. Kim, E. G. Lee, S. D. Park, C. J. Jeon, Y. H. Cho, C. K. Rhee and W. W. Kim, J. Sol-Gel Sci. Technol., 2001, 22, 63–74 CrossRef CAS.
- A. Di Paola, M. Bellardita and L. Palmisano, Catalysts, 2013, 3, 36–73 CrossRef CAS.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C. Y. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS.
- S. Banerjee, J. Gopal, P. Muraleedharan, A. Tyagi and B. Raj, Curr. Sci., 2006, 90, 1378–1383 CAS.
- H. S. Jung and H. Kim, Electron. Mater. Lett., 2009, 5, 73–76 CrossRef CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- H. Tada and M. Tanaka, Langmuir, 1997, 13, 360–364 CrossRef CAS.
- C. J. Brinker and G. W. Scherrer, Sol-Gel Science, Academic Press, San Diego, 1990 Search PubMed.
- B. E. Yoldas, J. Mater. Sci., 1986, 21, 1087–1092 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- J. F. Porter, Y. G. Li and C. K. Chan, J. Mater. Sci., 1999, 34, 1523–1531 CrossRef CAS.
- J. Yu, J. Yu, L. Zhang and W. Ho, J. Photochem. Photobiol., A, 2002, 148, 263–271 CrossRef CAS.
- D. a. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- X. Ding and X. Liu, J. Mater. Res., 2011, 13, 2556–2559 CrossRef.
- M. Stir, T. Traykova, R. Nicula, E. Burkel, C. Baehtz, M. Knapp and C. Lathe, Nucl. Instrum. Methods Phys. Res., Sect. B. Beam Interact. with Mater. Atoms, 2003, 199, 59–63 CrossRef CAS.
- R. Nicula, M. Stir, C. Schick and E. Burkel, Thermochim. Acta, 2003, 403, 129–136 CrossRef CAS.
- K. Sabyrov, N. Burrows and R. Penn, Chem. Mater., 2012, 25, 1408–1415 CrossRef.
- K. Sabyrov, V. Adamson and R. L. Penn, CrystEngComm, 2014, 16, 1488–1495 RSC.
- H. Zhang, B. Chen and J. F. Banfield, Phys. Chem. Chem. Phys., 2009, 11, 2553–2558 RSC.
- H. Zhang and J. F. Banfield, J. Phys. Chem. B, 2000, 104, 3481–3487 CrossRef CAS.
- J. Ye, W. Liu, J. Cai, S. Chen, X. Zhao, H. Zhou and L. Qi, J. Am. Chem. Soc., 2011, 133, 933–940 CrossRef CAS.
- A. L. da Silva, M. Dondi, M. Raimondo and D. Hotza, J. Eur. Ceram. Soc., 2018, 38, 1002–1017 CrossRef CAS.
- Y.-H. Zhang and A. Reller, Mater. Sci. Eng., C, 2002, 19, 323–326 CrossRef.
- J. Feltrin, M. N. Sartor, A. De Noni Jr., A. M. Bernardin, D. Hotza and J. A. Labrincha, Cerâmica, 2013, 59, 620–632 CrossRef CAS.
- V. B. Tezza, M. Scarpato, L. F. S. Oliveira and A. M. Bernardin, Powder Technol., 2015, 276, 60–65 CrossRef CAS.
- D. Pan, Z. Han, Y. Miao, D. Zhang and G. Li, Appl. Catal., B. Environ, 2018, 229, 130–138 CrossRef CAS.
- R. K. Iler, The Chemistry Of Silica, Wiley-Interscience, New York, 1978 Search PubMed.
- A. Fink, W. Stöber and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- Y. Chen, H. Chen and J. Shi, Acc. Chem. Res., 2014, 47, 125–137 CrossRef CAS.
- M. Gärtner, V. Dremov, P. Müller and H. Kisch, ChemPhysChem, 2005, 6, 714–718 CrossRef.
- G. Lassaletta, A. Fernandez, J. P. Espinos and A. R. Gonzalez-Elipe, J. Phys. Chem., 1995, 99, 1484–1490 CrossRef CAS.
- T. Matsuda, H. Itoh, S. Tagawa, T. Ohno and H. Suzuki, Mater. Chem. Phys., 2009, 113, 119–123 CrossRef.
- A. Staykov, E. P. Ferreira-Neto, J. M. Y. S. Cruz, S. Ullah and U. P. Rodrigues-Filho, Int. J. Quantum Chem., 2018, 118, e25495 CrossRef.
- C. Kang, L. Jing, T. Guo, H. Cui, J. Zhou and H. Fu, J. Phys. Chem. C, 2009, 113, 1006–1013 CrossRef CAS.
- F. F. Ferreira, E. Granado, W. Carvalho, S. W. Kycia, D. Bruno and R. Droppa, J. Synchrotron Radiat., 2006, 13, 46–53 CrossRef CAS.
- A. R. West, Solid State Chemistry and its Applications, Wiley, Chichester [West Sussex] New York, 1984 Search PubMed.
- R. A. Spurr and H. Myers, Anal. Chem., 1957, 29, 760–762 CrossRef CAS.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207–219 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- S. Ullah, J. J. S. Acuña, A. A. Pasa, S. A. Bilmes, M. E. Vela, G. Benitez and U. P. Rodrigues-Filho, Appl. Surf. Sci., 2013, 277, 111–120 CrossRef CAS.
- R. López and R. Gómez, J. Sol–Gel Sci. Technol., 2012, 61, 1–7 CrossRef.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS.
- I. Djerdj and A. M. Tonejc, J. Alloys Compd., 2006, 413, 159–174 CrossRef CAS.
- S. Ullah, E. P. Ferreira-Neto, A. A. Pasa, C. C. J. Alcântara, J. J. S. Acuña, S. A. Bilmes, M. L. Martínez Ricci, R. Landers, T. Z. Fermino and U. P. Rodrigues-Filho, Appl. Catal. B Environ., 2015, 179, 333–343 CrossRef CAS.
- T. Gross, M. Ramm, H. Sonntag, W. Unger, H. M. Weijers and E. H. Adem, Surf. Interface Anal., 1992, 18, 59–64 CrossRef CAS.
- D. S. Jensen, S. S. Kanyal, N. Madaan, M. A. Vail, A. E. Dadson, M. H. Engelhard and M. R. Linford, Surf. Sci. Spectra, 2013, 20, 36–42 CrossRef.
- Y. Jiang, Z. Jin, C. Chen, W. Duan, B. Liu, X. Chen, F. Yang and J. Guo, RSC Adv., 2017, 7, 12856–12870 RSC.
- G. Lassaletta, A. Fernández, J. P. Espinós, A. R. González-Elipe, A. Fernandez, J. P. Espinos and A. R. Gonzalez-Elipe, J. Phys. Chem., 1995, 99, 1484–1490 CrossRef CAS.
- M. E. Simonsen, Z. Li and E. G. Søgaard, Appl. Surf. Sci., 2009, 255, 8054–8062 CrossRef CAS.
- H. Zhang and J. F. Banfield, J. Mater. Res., 2000, 15, 437–448 CrossRef CAS.
- D. T. Cromer and K. Herrington, J. Am. Chem. Soc., 1955, 77, 4708–4709 CrossRef CAS.
- R. Morozov, I. Krivtsov, V. Avdin, Z. Amghouz, S. A. Khainakov and J. R. García, J. Non-Cryst. Solids, 2016, 435, 8–16 CrossRef CAS.
- B. Braconnier, C. A. Páez, S. Lambert, C. Alié, C. Henrist, D. Poelman, J.-P. Pirard, R. Cloots and B. Heinrichs, Microporous Mesoporous Mater., 2009, 122, 247–254 CrossRef CAS.
- L. Zhao, S. Wang, Y. Wang and Z. Li, Surf. Interface Anal., 2017, 49, 173–176 CrossRef CAS.
- K. Okada, N. Yamamoto, Y. Kameshima, A. Yasumori and K. J. D. MacKenzie, J. Am. Ceram. Soc., 2004, 84, 1591–1596 CrossRef.
- N. Sudhakar, R. K. Singh, S. K. Mishra and S. Kannan, RSC Adv., 2014, 4, 49752–49761 RSC.
- A. Staykov, Y. Ooishi and T. Ishihara, J. Phys. Chem. C, 2014, 118, 8907–8916 CrossRef CAS.
- A. Li, Y. Jin, D. Muggli, D. T. Pierce, H. Aranwela, G. K. Marasinghe, T. Knutson, G. Brockman and J. X. Zhao, Nanoscale, 2013, 5, 5854–5862 RSC.
- M. Hirano, K. Ota and H. Iwata, Chem. Mater., 2004, 16, 3725–3732 CrossRef CAS.
- H. Zhang and J. F. Banfield, Chem. Mater., 2005, 17, 3421–3425 CrossRef CAS.
- H. Zhang and J. F. Banfield, J. Mater. Chem., 1998, 8, 2073–2076 RSC.
- M. R. Ranade, A. Navrotsky, H. Z. Zhang, J. F. Banfield, S. H. Elder, A. Zaban, P. H. Borse, S. K. Kulkarni, G. S. Doran and H. J. Whitfield, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(suppl 2), 6476–6481 CrossRef CAS.
- H. Zhang and J. F. Banfield, Chem. Rev., 2014, 114, 9613–9644 CrossRef CAS.
- A. Hanprasopwattana, T. Rieker, A. Sault and A. Datye, Catal. Lett., 1997, 45, 165–175 CrossRef CAS.
- G. R. Hearne, J. Zhao, A. M. Dawe, V. Pischedda, M. Maaza, M. K. Nieuwoudt, P. Kibasomba, O. Nemraoui, J. D. Comins and M. J. Witcomb, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 134102 CrossRef.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef.
- B. Ohtani, O. O. Prieto-Mahaney, D. Li and R. Abe, J. Photochem. Photobiol., A, 2010, 216, 179–182 CrossRef CAS.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS.
- C. Ai, P. Xie, X. Zhang, X. Zheng, J. Li, A. Kafizas and S. Lin, ACS Sustainable Chem. Eng., 2019, 7, 5274–5282 CrossRef CAS.
- P. Deák, B. Aradi and T. Frauenheim, J. Phys. Chem. C, 2011, 115, 3443–3446 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, Raman spectra and digital photos of the samples. See DOI: 10.1039/d0ma00785d |
| This journal is © The Royal Society of Chemistry 2021 |