Total synthesis of diazonamide A†
Robert R.
Knowles
,
Joseph
Carpenter
,
Simon B.
Blakey
,
Akio
Kayano
,
Ian K.
Mangion
,
Christopher J.
Sinz
and
David W. C.
MacMillan
*
Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, USA. E-mail: dmacmill@princeton.edu; Fax: +1 609 2585922; Tel: +1 609 2582254
First published on 24th December 2010
Abstract
A total synthesis of the marine natural product diazonamide A (1) has been accomplished. This work features a highly stereoselective synthesis of the C(10) quaternary center and the central furanoindoline core enabled by an iminium-catalyzed alkylation–cyclization cascade. Additionally, a magnesium-mediated intramolecular macroaldolization and a palladium-catalyzed tandem borylation/annulation were developed to enable the closure of the two 12-membered macrocycles of diazonamide A. This synthesis involves 20 steps in its longest linear sequence and proceeds in 1.8% overall yield.
Introduction
The diazonamides are a structurally unique class of secondary metabolites first isolated by Fenical and coworkers from the colonial marine ascidian Diazona angulata.1 Within this family, diazonamide A (1) was found to be a potent antimitotic agent, exhibiting low nanomolar GI50 values towards a diverse panel of human cancer cell lines.1a, 2aCells treated with 1 fail to develop a mitotic spindle, a phenotype commonly associated with tubulin-binding agents such as taxol and vinblastine. However, the cellular receptor of 1 was identified by Harran and Wang to be ornithine δ-amino-transferase (OAT), a mitochondrial matrix protein previously unknown to play any role in spindle regulation.2b Given the documented viability of OAT-null organisms, it was subsequently proposed that OAT-dependence may be a differential characteristic of mitosis in cancerous and healthy cells. If correct, 1 would represent a cancer-selective therapeutic agent2c and a valuable molecular probe of the oncology associated with OAT-dependent mitosis.As a consequence of this intriguing bioactivity and its remarkable molecular structure, diazonamide A has been the target of intense and sustained synthetic interest for nearly two decades.3 The structure of 1 was misassigned in the original isolation report,1 but was later corrected by Harran and coworkers in 2001 following a seminal synthesis of the nominal structure.4 The correct structure of 1 is characterized by two 12-membered macrocycles that are conjoined through a triaryl-substituted quaternary carbon stereocenter embedded in an unusual furanoindoline core. Together with a large degree of unsaturation, these structural elements comprise an exceptionally compact and rigid framework whose inner atoms possess almost no rotational degrees of freedom. While many groups have reported partial syntheses of 1 over the past decade, Nicolaou5a–d and Haran5e have reported the only three completed total syntheses of 1 to date, while Magnus6a and Sammakia6b have recently outlined formal syntheses.
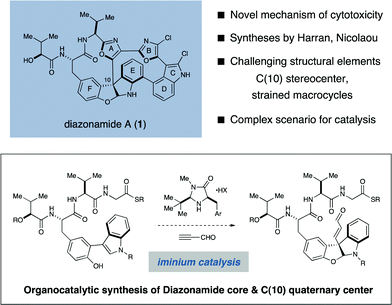
Design plan
Retrosynthetically, the principal challenge associated with the synthesis of 1 is the stereoselective installation of the C(10) quaternary carbon stereocenter. Collective consideration of earlier synthetic efforts offer support for this view, as this aspect of the target structure has not been successfully addressed in any of the three completed total syntheses.7 With the specific aim of developing catalytic asymmetric solutions to the C(10) problem, we initiated a program directed towards the total synthesis of diazonamide A. In this communication we describe the realization of these efforts, which features a highly stereoselective synthesis of the C(10) quaternary center with concomitant installation of the central furanoindoline enabled by an iminium-catalyzed diastereoselective conjugate addition/phenol cyclization cascade (depicted above).8 In addition, we present novel solutions to the syntheses of the two large ring systems of 1, including a magnesium-mediated intramolecular macroaldolization in the construction of the left-hand twelve-membered ring, as well as a palladium-catalyzed tandem borylation/annulation to furnish the intact right-hand macrocycle. The details of our synthetic sequence are described herein.Results and discussion
Our synthesis began with the Suzuki coupling of boronic ester 2 and iodophenol 3 under the action of catalytic Pd(dppf)Cl2 to yield biaryl 4 in 82% yield (Scheme 1). The methyl ester of 4 was then hydrolyzed with aqueous lithium hydroxide and the resulting carboxylic acid was coupled to an N-TFA salt of valine·glycine dipeptide thioester, providing access to 5, the substrate required for our proposed key organocatalytic transformation.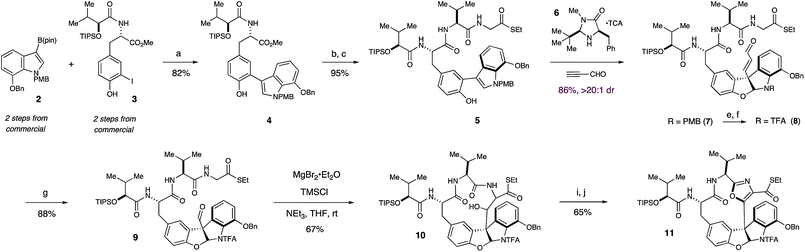 | ||
| Scheme 1 Organocatalytic synthesis of the furanoindoline core and Lewis acid mediated synthesis of left-hand macrocycle of diazonamide A. Reagents and conditions: (a) 5 mol% Pd(dppf)Cl2, K3PO4, dioxane, 40 °C (b) LiOH, THF, MeOH, H2O, rt (c) EDC, HOBt, (S)-S-ethyl 2-(2-amino-3-methylbutanamido)ethanethioate·TFA, NaHCO3, DMF, rt (d) Table 1, entry 5 (e) DDQ, CH2Cl2, pH 7 buffer, 0 °C (f) TFAA, DMAP, CH2Cl2, 0 °C (g) O3, CH2Cl2, MeOH, −78 °C (h) MgBr2·Et2O, TMSCl, NEt3, THF, rt (i) Dess–Martin, CH2Cl2, rt (j) DAST, C6H6, rt. | ||
In accord with our design plan, treatment of 5 with 30 mol% of imidazolidinone catalyst 6·TFA and excess propynal in dichloromethane and methanol at −70 °C resulted in full conversion to 7, though as a modest 4.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers at the critical C(10) position (Table 1, entry 1).
:1 mixture of diastereomers at the critical C(10) position (Table 1, entry 1).
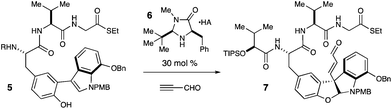
| Entry | Catalyst·HA | T/°C | solvent: MeOH | % convb | drb |
|---|---|---|---|---|---|
| a Optimizations performed on 0.05 mmol scale for 24 h. b Determined by 1H-NMR analysis of the crude reaction mixture. c Isolated yield for reaction performed on 15 mmol scale. | |||||
| 1 | 6·TFA | −70 | CH2Cl2(20:1) |
>95 | 4.2:1 |
| 2 | 6·TCA | −70 | CH2Cl2(20:1) |
>95 | 7.7:1 |
| 3 | 6·TCA | −70 | CHCl3(20:1) |
>95 | 8.2:1 |
| 4 | 6·TCA | −70 |
PhMe (20:1) |
>95 | 8.3:1 |
| 5 | 6·TCA | −50 |
CHCl3:PhMe (10:10:1) |
86c | >20:1 |
| 6 | rac·6TCA | −50 |
CHCl3:PhMe (10:10:1) |
80 | 1:1 |
During subsequent optimization studies, it was found that the use of 6·TCA and the replacement of dichloromethane with chloroform or toluene resulted in improved selectivity with no associated loss of reactivity (Table 1, entries 2–4). Interestingly, the use of a ternary solvent mixture comprised of toluene, chloroform and methanol ultimately proved superior, to deliver 7 in 86% isolated yield and >20:1 dr after 24 h at −50 °C (Table 1, entry 5). Notably, use of racemic imidazolidinone catalyst 6·TCA results in a 1:1 mixture of diastereomers at C(10), demonstrating that the observed stereoselectivity is entirely amine catalyst-controlled (Table 1, entry 6). In addition to being well suited to address this specific synthetic problem, we believe this is the most complex and challenging setting in which organocatalysis has been employed to date.
With the diazonamide core in hand, the indoline protecting group was exchanged to ensure the stability of the nascent aminal towards a subsequent ozonoylsis, which cleanly furnished aldehyde 9 in 84% yield. Aldehyde 9 was specifically targeted as the substrate for a proposed intramolecular aldol reaction to furnish a 13-membered, ring-opened form of the left-hand macrocycle. This unconventional macroaldolization approach was targeted after prior experimental efforts suggested that closure of the intact 12-membered ring via a traditional macrolactamization would not proceed efficiently. More specifically, we reasoned that creating a larger, more conformationally flexible macrocycle that could then undergo ring contraction with concomitant formation of the central oxazole would allow ring strain to be offset by the net gain in aromaticity. While such aldol reactions are rare,9 a careful evaluation of Lewis acids, amine bases and solvents led to the discovery that treatment of thioester 9 with MgBr2·Et2O, TMSCl and triethylamine in THF at room temperature readily effected soft enolization, allowing an efficient, intramolecular macroaldolization to deliver 10 as a single diastereomer in 67% yield (Scheme 1). Notably, the addition of TMSCl was necessary to circumvent the possibility of retro-aldol pathways, a characteristic concern of magnesium aldol processes.10
While alcohol 10 could be readily oxidized with the Dess–Martin periodinane to its corresponding ketone, subsequent cyclodehydration to furnish the central oxazole ring proved unsuccessful using many standard protocols. However, we were pleased to discover that direct treatment of the β-ketothioester with an excess of DAST in benzene led to a highly efficient cyclodehydration, furnishing intact macrocycle 11 in 61% yield over two steps.11 At this stage, the structure and relative stereochemistry of 11 were confirmed through X-ray crystallographic analysis of its corresponding carboxylic acid (supporting information†).
With the synthesis and stereochemistry of the left-hand macrocycle secured, we next turned our attention to the second and more challenging 12-membered ring of diazonamide A. With intermediates similar to 11, Harran and Nicolaou both employed an elegant photochemical Witkop cyclization to furnish the D-E biaryl bond and close the right hand macrocycle.5 With a goal of developing a distinctive solution to this challenging construction, we elected to explore an approach wherein the same biaryl bond could be formed utilizing a palladium-catalyzed cross-coupling protocol. In assessing this approach, we were wary of attempting to carry a sensitive aryl stannane or boronic ester through the synthetic steps prior to the proposed annulation. Rather, we elected to install the requisite coupling functionality immediately prior to ring closure via reaction conditions that would then facilitate subsequent annulation in a single, tandem operation, in analogy to the well-known Stille–Kelly protocol.12
Towards these ends, the thioester functionality of 11 was activated with AgTFA and displaced by amine 12 to yield ketoamide 13 in 88% yield.13Furanoindoline 13 was in turn subjected to the Wipf conditions14 for oxazole cyclodehydration, furnishing bisoxazole 14 in 86% isolated yield (Scheme 2). Deprotection of the benzyloxy and trifluoroacetamide groups of 14 with BBr3, followed by treatment of the crude product with excess PhNTf2 provided bromo-bistriflate 15 in 80% yield over two steps. After extensive optimization, we were able to realize a successful tandem borylation/annulation, as treatment of 15 with (Bpin)2, KF, and Pd(PPh3)4 in a 5% aqueous solution of dioxane under microwave heating at 120 °C yielded the desired biaryl bond of macrocycle 16 in 50% yield.15 Mass spectrometric analysis of the progressing reaction suggests that this reaction proceeds through rapid and selective borylation of the aryl bromide, followed by a slower coupling between the nascent boronate and the aryl triflate.16
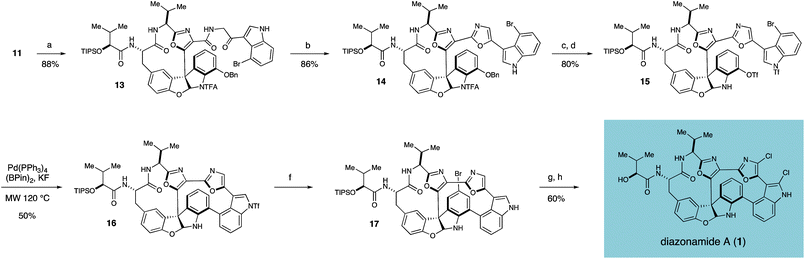 | ||
| Scheme 2 Construction of the second macrocyclic ring, dichloro installation and completion of the total synthesis of diazonamide A (1). Reagents and conditions: (a) AgTFA, NaHCO3, 2-amino-1-(4-bromo-1H-indol-3-yl)ethanone·TFA, DMF, 40 °C (b) PPh3, C2Cl6, NEt3, CH2Cl2, 0 °C (c) BBr3, CH2Cl2, −78 °C (d) PhNTf2, NEt3, CH2Cl2, rt (e) (Bpin)2, KF, 70 mol% Pd(PPh3)4, dioxane:H2O (20:1), MW 120 °C (f) NBS, THF; then LiOH, THF, MeOH, H2O (g) NCS, THF, 40 °C; then H2, Pd(OH)2, THF, 40 °C (h) TASF, DMF, rt. | ||
With the carbon framework of diazonamide A fully installed, selective introduction of the aryl chlorides was accomplished by first brominating the activated indolineE-ring, followed by in situhydrolysis of the indolyl triflate to yield 17 in 83% isolated yield. Subsequent chlorination of the desired oxazole and indole functionalities of 17 was achieved upon exposure to NCS, and the aryl bromide was selectively excised by hydrogenolysis over Pearlman's catalyst in a two-step, one-pot procedure. Finally, treatment with TASF removed the remaining silyl ether to furnish synthetic diazonamide A (1), whose spectral characteristics were identical to the natural product in all respects. The completed synthesis as it stands was accomplished in 20 steps in the longest linear sequence and in 1.8% overall yield.
Conclusions
In summary, we have completed a 20 step total synthesis of diazonamide A from commercial materials, wherein a series of synthetic solutions to many of the challenging structural features of 1 were realized. In particular, the use of asymmetric iminium catalysis provided an efficient and highly stereoselective synthesis of the C(10) center and the complete furanoindoline core. A complete account of our studies towards the synthesis of diazonamide A will be reported in due course.Acknowledgements
Financial support was provided by the NIHGMS (R01 GM078201) and kind gifts from Merck Research Laboratories, Amgen and Abbott Laboratories. R.R.K and J.C. are grateful to Eli Lilly for a graduate fellowship. J.C is grateful to Bristol-Myers Squibb and to Princeton University for graduate fellowships. A.K. is grateful for an Eisai visiting scholarship. I.K.M. is grateful for a NSF predoctoral fellowship. C.J.S. is grateful to the NIH for postdoctoral fellowship support (NIH-F32-CA-091635-01). We acknowledge Dr. Alan Lough (University of Toronto) for crystal structure analysis.Notes and references
- (a) N. Lindquist, W. Fenical, G. D. Van Duyne and J. Clardy, J. Am. Chem. Soc., 1991, 113, 2303–2304 CrossRef CAS; (b) R. Fernandez, M. J. Martin, R. Rodriguez-Acebes, F. Reyes, A. Francesch and C. Cuevas, Tetrahedron Lett., 2008, 49, 2282–2285.
- (a) Z. Cruz-Monserrate, H. C. Vervoort, R. Bai, D. J. Newman, S. B. Howell, G. Los, J. T. Mullaney, M. D. Williams, G. R. Pettit, W. Fenical and E. Hamel, Mol. Pharmacol., 2003, 63, 1273–1280 CrossRef CAS; (b) G. Wang, L. Shang, A. W. G. Burgett, P. G. Harran and X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2068–2073 CrossRef CAS; (c) N. S. Williams, A. W. G. Burgett, A. S. Atkins, X. Wang, P. G. Harran and S. L. McKnight, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2074–2079 CrossRef CAS.
- M. Lachia and C. Moody, Nat. Prod. Rep., 2008, 25, 227–253 RSC.
- (a) J. Li, S. Jeong, L. Esser and P. G. Harran, Angew. Chem., Int. Ed., 2001, 40, 4765–4769 CrossRef CAS; (b) J. Li, A. W. G. Burgett, L. Esser, C. Amezcua and P. G. Harran, Angew. Chem., Int. Ed., 2001, 40, 4770–4773 CrossRef CAS.
- (a) K. C. Nicolaou, M. Bella, D. Y.-K. Chen, X. Huang, T. Ling and S. A. Snyder, Angew. Chem., Int. Ed., 2002, 41, 3495–3499 CrossRef CAS; (b) K. C. Nicolaou, D. Y.-K. Chen, X. Huang, T. Ling, M. Bella and S. A. Snyder, J. Am. Chem. Soc., 2004, 126, 12888–12896 CrossRef CAS; (c) K. C. Nicolaou, P. B. Rao, J. Hao, M. V. Reddy, G. Rassias, X. Huang, D.-K. Chen and S. A. Snyder, Angew. Chem., Int. Ed., 2003, 42, 1753–1758 CrossRef CAS; (d) K. C. Nicolaou, J. Hao, M. V. Reddy, P. B. Rao, G. Rassias, S. A. Snyder, X. Huang, D. Y.-K. Chen, W. E. Brenzovich, N. Giuseppone, P. Giannakakou and A. O'Brate, J. Am. Chem. Soc., 2004, 126, 12897–12906 CrossRef CAS; (e) A. W. G. Burgett, Q. Li, Q. Wei and P. G. Harran, Angew. Chem., Int. Ed., 2003, 42, 4961–4966 CrossRef CAS.
- (a) C.-M. Cheung, F. W. Goldberg, P. Magnus, C. J. Russell, R. Turnbull and V. Lynch, J. Am. Chem. Soc., 2007, 129, 12320–12327 CrossRef CAS; (b) C.-K. Mai, M. F. Sammons and T. Sammakia, Angew. Chem. Int. Ed., 2010, 49, 2397–2400 CAS.
- The Sammakia formal synthesis reports a highly diastereoselective synthesis of the C(10) center, while the Magnus synthesis is moderately stereoselective. See ref. 6.
- (a) J. F. Austin, S. G. Kim, C. J. Sinz, W. J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482–5487 CrossRef CAS; (b) S. B. Jones, B. L. Simmons and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 13606–13607 CrossRef CAS.
- For notable examples, see: (a) C. M. Hayward, D. Yohannes and S. J. Danishefsky, J. Am. Chem. Soc., 1993, 115, 9345–9346 CrossRef; (b) D. Meng, P. Bertinato, A. Balog, D. Su, T. Kamenecka, E. J. Sorensen and S. J. Danishefsky, J. Am. Chem. Soc., 1997, 119, 10073–10092 CrossRef CAS.
- D. A. Evans, J. S. Tedrow, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2002, 124, 392–393 CrossRef CAS.
- To the best of our knowledge, this is first instance of DAST being used as a reagent for this transformation.
- T. R. Kelly, Q. Li and V. Bhushan, Tetrahedron Lett., 1990, 31, 161–164 CrossRef CAS.
- S. V. Ley and P. R. Woodward, Tetrahedron Lett., 1987, 28, 3019–3020 CrossRef CAS.
- P. Wipf and C. P. Miller, J. Org. Chem., 1993, 58, 3604–3606 CrossRef CAS.
- A.-C. Carbonelle and J. Zhu, Org. Lett., 2000, 2, 3477–3480 CrossRef CAS.
- Similar strategies have been reported to be unsuccessful. (a) M. C. Bagley, L. S. Hind and C. J. Moody, Tetrahedron Lett., 2000, 41, 6897–6900 CrossRef CAS; (b) J. E. M. Booker, A. Boto, G. H. Churchill, C. P. Green, M. Ling, G. Meek, J. Prabhakaran, D. Sincair, A. J. Blake and G. Pattenden, Org. Biomol. Chem., 2006, 4, 4193–4205 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, structural proofs, and spectral data for all new compounds are provided. See DOI: 10.1039/c0sc00577k |
| This journal is © The Royal Society of Chemistry 2011 |