Amine directed Pd(II)-catalyzed C–H bond functionalization under ambient conditions†
Benjamin
Haffemayer
,
Moises
Gulias
and
Matthew J.
Gaunt
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: mjg32@cam.ac.uk
First published on 15th October 2010
Abstract
Pd(II)-catalyzed C–H bond functionalizations that proceed under ambient conditions are a pivotal part of the future of chemical synthesis. Herein, an amine directed Pd(II)-catalyzed C–H bond functionalization strategy is described that generates a diversity of molecular frameworks via C–H carbonylation, C–H arylation and C–H amination. The new reactions work for a variety of substrates and are tolerant of delicate stereogenic centres and functionality. Furthermore, the new reactions can be sequenced to generate complex architectures from simple building blocks via iterative Pd(II)-catalyzed C–H bond functionalization.
The development of metal-catalyzed C–H functionalization processes that generate molecules with the architectures commonly found in natural products or medicines remains a significant synthetic challenge.1 A broad range of C–H transformations can be catalyzed by a variety of transition metals under more forcing conditions, but reports of similar processes that proceed at ambient temperature are less common and often restricted to electron rich arenes and heteroarenes.2 One of the goals in our laboratory is the development of a synthesis toolbox comprising mild metal catalyzed C–H bond functionalization reactions that will facilitate complex molecule synthesis.3 Herein, we report the development of a Pd(II)-catalyzed C–H bond functionalization strategy for the β-arylethylamine motif, a structure that frequently occurs in natural products and medicinal agents (Fig. 1).4 Specifically, the β-arylethylamines undergo Pd(II)-catalyzed C–H carbonylation, C–H arylation, C–H amination, and iterative C–H functionalization to products with diverse structures, offering numerous opportunities for further synthetic application.
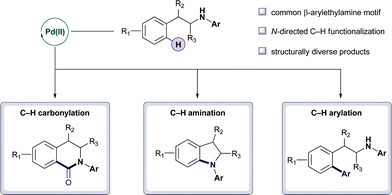 | ||
| Fig. 1 Diversity generation using Pd(II)-catalyzed C–H bond functionalization. | ||
The amine group has a rich history of facilitating cyclometallation reactions with Pd(II)-salts.4 It is therefore surprising that few Pd(II)-catalyzed C–H bond functionalization reactions directed by amines or their derivatives have been reported.3b,5–7,8a,b,9 In relation to the functionalization of the β-arylethylamine motif, Vicente et al. reported a carbopalladation of the phenylalanine methyl ester (Fig. 2A);10 notably, the free-amine directed palladation proceeded at room temperature, but to the best of our knowledge no catalytic reactions have been reported. Indeed, our own extensive efforts failed to facilitate catalytic transformations on this scaffold. We speculated that introduction of an aryl group onto the amine system would tune the nucleophilicity of the nitrogen atom such that it would reduce the likehood of bis-amino-Pd(II) complex formation that would have prevented carbopalladation.3b Furthermore, the aryl group would increase the acidity of the aryl N–H bond in the cyclopalladation complex and may assist the deprotonation step that must accompany the C–H bond functionalization. Accordingly, we prepared amine1a, bearing the 4-methoxyphenyl group (PMP), and found that carbopalladation proceeded at room temperature providing a dimeric complex 1a•Pd. When the palladacycle was subjected to a CO atmosphere in the presence of benzoquinone,2d,11 it underwent carbonylation to dihydro-2-quinolone 2a (Fig. 2B), a synthetically versatile intermediate.6,12,13
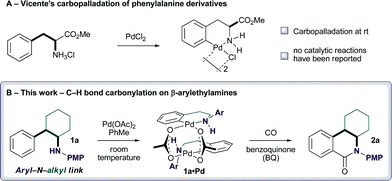 | ||
| Fig. 2 Amine directed Pd(II)-catalyzed C–H functionalization. | ||
Having secured the desired reactivity, we next assessed the catalytic aspect of the carbonylative process.2d,6,11 After a brief screen of conditions (Table 1), we found an optimum reaction required treatment of 1a with 10 mol% Pd(OAc)2, 2 equivalents of t-BuO2Ac, 2 equivalents of benzoquinone, under 1 atmosphere of CO and O2, and with AcOH as solventat room temperature (entry 7). The use of AcOH as solvent appears to suppress the CO mediated reduction of the Pd(II) to palladium black. It is also notable that only N-aryl compounds resulted in product formation (entries 8–11), and no product was observed in the absence of Pd(OAc)2.
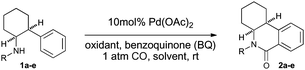
| Entry | R | Oxidant | Solvent | Additive | Conv (yield 2 %) |
|---|---|---|---|---|---|
| 1 | PMP (1a) | — | PhMe | — | trace |
| 2 | PMP (1a) | — | THF | — | trace |
| 3 | PMP (1a) | — | CH2Cl2 | — | trace |
| 4 | PMP (1a) | — | AcOH | — | 20 |
| 5 | PMP (1a) | O2 | AcOH | — | 30 |
| 6 | PMP (1a) | O2 | AcOH | NaOAc | 81 |
| 7 | PMP (1a) | O2, t-BuO2Ac | AcOH | NaOAc | 100 (85) |
| 8 | Ac (1b) | O2, t-BuO2Ac | AcOH | NaOAc | — |
| 9 | i-Pr (1c) | O2, t-BuO2Ac | AcOH | NaOAc | — |
| 10 | Bn (1d) | O2, t-BuO2Ac | AcOH | NaOAc | — |
| 11 | H (1e) | O2, t-BuO2Ac | AcOH | NaOAc | — |
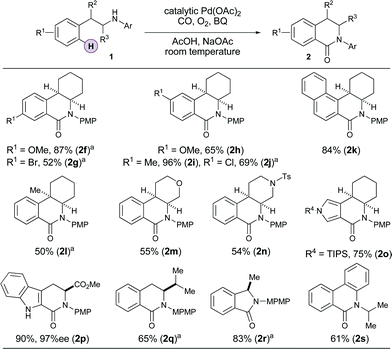
With optimal conditions in hand, the scope of the catalytic reaction was assessed (Table 2). The carbonylation reaction proceeds smoothly onto electron rich arenes (2f, 2h, 2i) and haloarenes also produce the desired products in good yields (2g, 2j), demonstrating the orthogonality of this process with the functionality required for conventional Pd(0)-catalyzed cross coupling reactions.
|
Substrates bearing sterically hindered quaternary centers adjacent to the amine group (2l) also form the carbonylated product in reasonable yield, although we found it necessary to increase the temperature of the reaction to 50 °C. Tetrahydropyran (2m) or piperidine (2n) ring systems also work well, and piperidine2n in particular represents an attractive scaffold for medicinal chemistry purposes. The nature of the arene can also be varied with naphthalene (2k), pyrrole (2o) and indole (2p) derived heteroarenes giving good yields of the carbonylated product. Indeed, tryptophan derived 2p was obtained without erosion of enantiomeric purity highlighting its potential in complex molecule synthesis applications. Interestingly, straight chain amines required us to use a more sterically hindered aryl group to suppress addition of the amine to benzoquinone, but with this modified group the C–H carbonylation proceeds in good yields (2q–r). In all of these cases (2f–r) the N-aryl group is ‘exo’ to the cyclizing system, however if the N-aryl group is accommodated in an ‘endo’ sense to the ring formation then an alkyl group can be included as the other nitrogen substituent. This conserves the crucial alkyl–N-aryl linkage that facilitates the ambient temperature C–H carbonylation process in good yield (2s). We note that the C–H carbonylation tactic does not work with arenes displaying strongly electron withdrawing groups (see SI for details).
Employing the same catalytic carbopalladation tactic, we were able to further extend the Pd(II)-catalyzed C–H bond functionalizations on the β-arylethylamine scaffold to an arylation process.14 We showed that Pd(II)-catalyzed C–H arylation of 1 proceeds at room temperature with aryl boronic acids in good yields and provides facile access to complex β-arylethylamines 3 (Scheme 1).
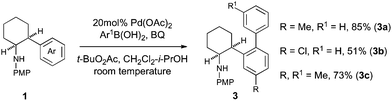 | ||
| Scheme 1 Pd(II)-catalyzed C–H bond arylation at room temperature. | ||
Linking the arylation and carbonylation reactions delivered an iterative C–H bond functionalization tactic that would be suitable for complex molecule synthesis.15Pd(II)-catalyzed C–H arylation of 1i proceeds as expected to form 3a,16 and Pd(II)-catalyzed C–H bond carbonylation furnishes the highly substituted dihydro-2-quinolone framework 3d in 63% yield over two steps (Scheme 2).
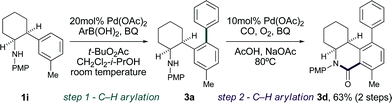 | ||
| Scheme 2 Iterative Pd(II)-catalyzed C–H bond functionalization. | ||
Finally, we have performed preliminary experiments towards a C–H amination process to form on the same β-arylethylamine framework.5a,b In contrast to our carboazole work,3bPd(II)-catalyzed C–H bond amination was not observed under mild conditions with a range of oxidants that would enable a Pd(II)/(IV) mechanism. However, after extensive screening, we found that a catalytic C–H amination on 1a was possible through a Pd(II)/(0) manifold in the presence of Na2CO3/PivOH and Cu(OAc)2 at elevated temperatures, to give 4a in 86% yield. The use of pivalic acid is crucial and no turnover was observed when it was removed or replaced with acetic acid.17 It is possible that this buffered system is required for the deprotonation of the N–H bond as part of the C–N bond forming step. Indolines 4b and 4c were also obtained in good yields demonstrating the generality of this new C–H amination process, and further expanding the utility of C–H bond functionalization on β-arylethylamines (Scheme 3).18
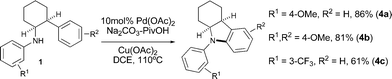 | ||
| Scheme 3 Iterative Pd(II)-catalyzed C–H bond functionalization. | ||
Conclusions
In summary, we have developed a versatile C–H bond functionalization platform for β-arylethylamines that generates a diversity of architectures. Remarkably, Pd(II)-catalyzed C–H bond carbonylation and C–H arylation proceed at room temperature and tolerate complex functionality and stereocentres. We further demonstrated a Pd(II)-catalyzed C–H bond amination to indolines from the same scaffold, albeit at elevated temperatures. We are currently further investigating the capacity of this new C–H bond functionalization platform and will report these results in due course.Acknowledgements
We gratefully acknowledge GSK and EPSRC for a studentship (to B.H.), the Marie Curie Foundation for IEF (M.G.), the Royal Society for University Research Fellowship and Philip & Patricia Brown for Next Generation Fellowship (to M.J.G.), Johnson Matthey for the loan of Pd(OAc)2 and the EPSRC Mass Spectrometry service (University of Swansea). We are grateful to Dr Steve Stanway (GSK) for useful discussion.Notes and references
- For reviews of transition-metal-catalyzed C–H activation of arenes, see: (a) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS; (b) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (c) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS; (d) L.-C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35; (e) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (f) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (g) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (h) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS; (i) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS; (j) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (k) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC.
- For selected examples of C–H activation at room temperature, see: (a) B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468 CrossRef CAS; (b) T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 4978–4979 CrossRef CAS; (c) T. Nishikata, A. R. Abela and B. H. Lipshutz, Angew. Chem., Int. Ed., 2010, 49, 781 CAS; (d) C. E. Houlden, M. Hutchby, C. D. Bailey, J. G. Ford, S. N. G. Tyler, M. R. Gagne, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830–1833 CrossRef CAS; (e) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972–4973 CrossRef CAS; (f) H.-B. Zhang, L. Liu, Y.-J. Chen, D. Wang and C.-J. Li, Adv. Synth. Catal., 2006, 348, 229 CrossRef CAS; (g) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS; (h) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS; (i) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef.
- (a) E. M. Beck, N. P. Grimster, R. Hatley and M. J. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528–2529 CrossRef CAS; (b) J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef CAS.
- For a review, see: J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2572 Search PubMed.
- For Pd(II)-catalyzed C–H bond functionalizations on N-triflyl β-arylethylamines, see (a) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452–6455 CrossRef CAS; (b) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520–7521 CrossRef CAS; (c) C. Vickers, T. S. Mei and J.-Q. Yu, Org. Lett., 2010, 12, 2511 CrossRef CAS.
- For amine directed C–H carbonylation, see K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342–14343 Search PubMed.
- For other examples of amine directed Pd-catalyzed C–H activation reactions, see (a) A. Lazareva and O. Daugulis, Org. Lett., 2006, 8, 5211–5213 CrossRef CAS . See also ref. 4, 5, 7a, 7c, and 10a.
- For examples of Pd-catalyzed amination of aromatic C–H bonds see: (a) W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560 CrossRef CAS; (b) K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931 CrossRef CAS; (c) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS; (d) M. Yamamoto and S. Matsubara, Chem. Lett., 2007, 36, 172 CrossRef CAS; (e) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058–14059 CrossRef CAS; (f) T.-S. Mei, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806–10807 CrossRef CAS.
- For recent examples of allylic C–H bond amination, see (a) K. J. Fraunhoffer and M. C. White, J. Am. Chem. Soc., 2007, 129, 7274 CrossRef CAS; (b) S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316 CrossRef CAS; (c) G. S. Liu, G. Y. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733 CrossRef CAS; (d) M. R. Luzung, C. A. Lewis and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 7025–7029 CrossRef CAS.
- (a) J. Vicente, I. Saura-Llamas, J.-A. Garcia-Lopez and B. Calmuschi-Cula, Organometallics, 2007, 26, 2768–2776 CrossRef CAS; (b) J. Vicente, I. Saura-Llamas and J.-A. Garcia-Lopez, Organometallics, 2009, 28, 448–464 CrossRef CAS.
- For examples of Pd-catalyzed C–H carbonylation, see (a) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686–693 CrossRef CAS; (b) R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082–14083 CrossRef CAS . See also ref. 2d.
- Isoquinolones can be accessed via applications of the Bischler–Napieralski reactions. See L. Kurti and B. Czako, in Strategic applications of named reactions in organic synthesis, Elsevier, 2005, pp. 62–63 Search PubMed.
- See supporting information for stoichiometric reactions from palladacycle 1a•Pd.
- For Pd(II)-catalyzed ortho-directed C–H arylation with aryl boronic acids, see: (a) D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676–17677 CrossRef CAS; (b) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS.
- For iterative metal-catalyzed C–H bond functionalizations in complex molecule synthesis, see: (a) E. M. Beck, R. Hatley and M. J. Gaunt, Angew. Chem., Int. Ed., 2008, 47, 3004 CrossRef CAS.
- The PMP group can be removed from isoquinolone 2a by oxidative cleavage using cerium ammonium nitrate in 75% yield. See SI for details. See also M. Yamauchi, M. Masao, T. Miuara and M. Murakami, J. Am. Chem. Soc., 2010, 132, 54–55 Search PubMed.
- For a discussion on the role of PivOH in Pd-catalyzed C–H bond functionalization, see: (a) M. LaFrance and K. J. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS; (b) M. LaFrance, D. Lapointe and K. Fagnou, Tetrahedron, 2008, 64, 6015 CrossRef CAS.
- We observe between 10 and 20% of indole in addition to the yield of the indoline stated. See SI for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectra for all new compounds. See DOI: 10.1039/c0sc00367k |
| This journal is © The Royal Society of Chemistry 2011 |