
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of sulfation on a tough hybrid hydrogel network
Sander
Driesen†
 ab,
Valentino
Atella†
ab,
Kristi
Kiick
cd,
Louis M.
Pitet
*b and
Geert-Jan
Graulus
*a
ab,
Valentino
Atella†
ab,
Kristi
Kiick
cd,
Louis M.
Pitet
*b and
Geert-Jan
Graulus
*a
aBiomolecule Design Group, Institute for Materials Research (imo-imomec), Hasselt University, Agoralaan Building D, 3590 Diepenbeek, Belgium. E-mail: geertjan.graulus@uhasselt.be
bAdvanced Functional Polymers Group, Institute for Materials Research (imo-imomec), Hasselt University, Agoralaan Building D, 3590 Diepenbeek, Belgium. E-mail: louis.pitet@uhasselt.be
cDepartment of Biomedical Engineering, 590 Avenue 1743, University of Delaware, Newark, DE 19713, USA
dDepartment of Materials Science and Engineering, University of Delaware, 201 DuPont Hall, DE 19716, USA
First published on 17th March 2025
Abstract
Hybrid hydrogels can mimic the exceptional stiffness of tough native tissues (e.g., articular cartilage). However, many of these tough hybrid hydrogels currently lack bioactive moieties. Therefore, our work focuses on introducing sulfated alginate into a tough poly(acrylamide-co-acrylic acid)/alginate hybrid hydrogel network. This modification introduces the potential for effective tissue interactions and allows further diversification through chemical transformations. These hydrogels are synthesized via the radical-mediated polymerization and covalent crosslinking of acrylamide and acrylic acid. The covalent network is fortified with a second ionically crosslinked sulfated alginate network. FTIR, 13C-NMR, and elemental analysis confirmed a degree of sulfation of 42.5%. Mechanical testing showed that hydrogels with a sulfated alginate content of 2 wt% exhibit comparable compressive stiffness (up to 230 kPa) to native articular cartilage. Cyclical mechanical testing revealed the network's resilience and remarkable toughness. These results suggest the hydrogels’ potential as cartilage mimics and support their additional investigation in vitro.
Introduction
Hydrogels are three-dimensional crosslinked polymeric networks swollen with relatively large amounts of water (50–95 wt%), making them ideal for various biomedical applications.1,2 Moreover, hydrogels often exhibit favorable properties such as biocompatibility, resemblance to living tissue, and ease of use.1,2 These features render them capable of mimicking the extracellular matrix (ECM). Thus, they play a crucial role in many tissue engineering strategies, a multidisciplinary field focused on regenerating damaged tissues.3,4 Moreover, the specific physicochemical characteristics associated with hydrogels can be fine-tuned to the intended application using chemical strategies.3Articular cartilage is a frequent target in tissue engineering due to its limited regenerative ability, which stems from the lack of vascularization and finite cellular content.3,5–7 Moreover, current treatment options, such as cartilage surgery, are impeded by complicated procedures, low quality of the regenerated cartilage, and postsurgical infections. Therefore, there is a need for tissue engineering applications that allow cartilage tissue regeneration combined with non-invasive delivery methods.5,6
The main role of cartilage is to provide a low-friction surface inside joints to allow unobstructed motion and prevent bone–bone impact. Moreover, the composition and structure of cartilage tissue is depth-dependent. In general, cartilage consists of water (70–85 wt%), various collagen types (10–18 wt%), proteoglycans (5–9 wt%), and chondrocytes (3–6 wt%).5,6 The load-bearing properties of cartilage that arise from these compositional components include high stiffness (≥1 MPa), high tensile strength (15–35 MPa), and compressive strength (14–59 MPa).5,6 An important factor in designing a cartilage scaffold is that it can effectively dampen the mechanical energy around the damaged area. To achieve this, the hydrogel must be adequately tough to mimic the high stiffness of native cartilage.5,6
Many studies have demonstrated impressive mimics of native cartilage in terms of mechanics. However, their comprehensive performance varies widely due to issues with cell viability, adhesion and proliferation, shape fidelity, controllable porosity, toxic gelation agents, and gelation time.5 Furthermore, many of these networks are built upon impractically complex compositions. The most successful categories of mechanically robust hydrogels include nanocomposite hydrogels, sliding ring hydrogels, and double network (DN) hydrogels. Among these, DN hydrogels have been developed with exceptionally high mechanical strength and toughness.5,8,9 The superior mechanical properties emerge from the interpenetration of two individual polymer networks with contrasting properties. The first highly cross-linked brittle network provides energy dissipation via the breakage of so-called sacrificial bonds. In contrast, the second weakly cross-linked network will absorb external stress, offering shape fidelity.8,9 In the early stages of DN hydrogel research, covalently crosslinked networks were routinely employed. When stretched, the presumed rupture of covalent bonds resulted in irreversible damage and a precipitous, permanent reduction in mechanical strength.8,9 Therefore, dynamic and reversible physical bonding strategies have been adopted to circumvent permanent damage. Such dynamic crosslinking strategies include ionic interactions, van der Waals forces, hydrogen bonds, hydrophobic associations, etc. In contrast to the permanent breaking of covalent bonds, dynamic crosslinking enables continuous energy dissipation. The reversibility and recoverability of the non-covalent bonds, combined with extremely high mechanical strength and toughness, make the physically/chemically cross-linked hybrid hydrogels one of the most used types in cartilage tissue engineering.8,9
Generally, the polymers used for tissue engineering can be divided into synthetic or natural materials. The most widely used synthetic polymers are polyesters, vinyl polymers, and polyethylene glycol (PEG).10 Of these synthetic materials, a few specific polymers are attractive for cartilage scaffolds, namely PEG, polycaprolactone (PCL), poly(vinyl alcohol) (PVA), and poly(acrylamide). Poly(acrylamide) forms stable, biocompatible, and bioinert hydrogels and has been applied as a filler for damaged cartilage tissue.10,11 Furthermore, incorporating co-monomers, such as acrylic acid, has been reported to create acrylamide copolymers with different properties.10 Polymers based on acrylic acid and acrylamide are superabsorbent and have been used for various biomedical and tissue engineering applications.12 The work described here builds on previous concepts by introducing additional functionality and using acrylate/acrylamide networks in hybrid hydrogels. Covalently crosslinked poly(acrylamide) networks have already been combined with ionically crosslinked networks to yield physically/chemically cross-linked hybrid hydrogels. Seminal work employing poly(acrylamide)/alginate hybrid hydrogel was first reported by Suo et al. in 2012.13 Their findings show that combining poly(acrylamide) and alginate networks exhibit a maximum fracture energy of 8700 J m−2 and extensibility beyond 20 times the initial length. These findings demonstrate that these hydrogels are extremely tough and could be mechanically suitable as a cartilage scaffold.
Alginates are unbranched linear copolymers composed of 1,4-linked mannuronic acid (M) and guluronic acid (G) and are typically isolated from brown algae. Alginate gelation occurs when polyvalent cations like Ca2+ interact selectively with G blocks to form ionic crosslinks.14,15 In addition, alginate has considerable advantages, such as being biobased and biocompatible, having suitable porosity, and exhibiting facile gelation.16 Alginates have also been used as synthetic extracellular matrices for cell encapsulation and proliferation.16,17
However, other factors besides selecting optimal materials to match the mechanical properties of cartilage are also crucial for engineering a cartilage tissue scaffold. Cell differentiation, cell adhesion, and integration into native cartilage are all crucial factors contributing to tissue regeneration.5,18 These bioactive properties have been achieved using cell adhesion ligands and epitopes for cell–surface interactions.5,18 Proteins, peptides, or growth factors attached to a hydrogel scaffold can also improve tissue regeneration.
A major component of the native cartilage ECM is chondroitin sulfate (CS), which is a sulfated glycosaminoglycan (GAG) composed of alternating N-acetylgalactosamine and glucuronic acid chains, with varying sulfation along the polysaccharide chain.19 CS is responsible for many of the important biomechanical properties of cartilage, such as stiffness and elasticity.19–21 Furthermore, they also play a vital role in the development, maintenance, and pathophysiology of tissues and may serve as receptors, co-receptors, and reservoirs of proteins and growth factors through electrostatic interactions.19,22–24 In addition, CS has been used for medical purposes for more than 40 years and is sold as an over-the-counter dietary supplement in North America and as a prescription drug in Europe.20
We reasoned that introducing sulfate moieties on an alginate network may mimic CS. Sulfated alginate can serve as a reservoir and a slow-release system for growth factors aimed toward tissue regeneration.22,24 This has been shown by Gionet-Gonzales et al., as sulfate alginate hydrogels could bind recombinant and cell-secreted growth factors.25 Moreover, Mhanna et al. showed that introducing sulfated alginate into a hydrogel network promotes proliferation while maintaining chondrogenic expression.22
Multiple strategies have been described for the sulfation of alginate.26–28 One common method is the chlorosulfonic acid-mediated sulfation of alginate in formamide, routinely employed due to high yields and low batch-to-batch variation.26 It has already been shown that adding these sulfate moieties results in enhanced proliferation and long-term viability of chondrocytes, further enhancing the cartilage tissue engineering capabilities of alginate-containing hydrogels.22,24
The design and synthesis of sulfated hybrid hydrogels for cartilage tissue engineering has yet to be reported. There is a clear need for new advanced cartilage tissue treatments, as the current treatments for osteoarthritis remain restricted. We studied whether sulfate groups could be introduced along the alginate backbone while maintaining the relatively high toughness of the hybrid hydrogels. To this end, this work describes the design, preparation, and mechanical performance of a poly(acrylamide-co-acrylic acid)/sulfated alginate hybrid hydrogel network. Sulfation with chlorosulfonic acid is explored to yield sulfated alginate. This network is then combined with poly(acrylamide-co-acrylic acid) to yield a tough sulfated hybrid hydrogel network, potentially suitable as a matrix for regenerating articular cartilage.
Experimental
Materials
Sodium alginate, N,N′-methylene bisacrylamide (MBAA, 99+%), and ammonium persulfate (APS, 98+%) were purchased from Sigma-Aldrich. Chlorosulfonic acid and formamide were purchased from Thermo Fisher Scientific. Acrylamide (AAm, 99%), tetramethylethylenediamine (TEMED, 99%), calcium chloride (CaCl2, 96%), and calcium sulfate (CaSO4, 98+%) were purchased from Acros Organics. Acrylic acid (AA, 98%) was purchased from J&K Scientific. Deuterated water (D2O) was purchased from Cambridge Isotope Laboratories. All chemicals were used as received unless stated otherwise.One-step hydrogel synthesis
Various (sulfated) poly(AAm-co-AA)/alginate hybrid hydrogel network formulations were synthesized (Table 1). The process starts with forming a homogeneous solution of the network components. First, sodium alginate (1, 2, or 3 wt%) was dissolved in the desired volume of demineralized water at room temperature. Then, AAm and AA, with various AAm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AA ratios (80:20 or 90:10), were added to the solution while stirring. Then, in the first crosslinking step, both the poly(AAm-co-AA) and alginate networks were formed. The poly(AAm-co-AA) network was formed via radical copolymerization/crosslinking. To this end, the crosslinker, MBAA (0.05 mol% relative to AAm & AA), the radical initiator APS (0.80 mol% relative to AAm & AA), and the accelerator TEMED (76.37 mol% relative to APS) were added. Simultaneously, the formation of the alginate network was induced by the addition of CaSO4 (13 wt% relative to alginate). The solution was then transferred to a PMMA mold and left to gel overnight.
:acrylic acid ratio, 80:20 and 90:10, respectively
:AA ratios (80:20 or 90:10), were added to the solution while stirring. Then, in the first crosslinking step, both the poly(AAm-co-AA) and alginate networks were formed. The poly(AAm-co-AA) network was formed via radical copolymerization/crosslinking. To this end, the crosslinker, MBAA (0.05 mol% relative to AAm & AA), the radical initiator APS (0.80 mol% relative to AAm & AA), and the accelerator TEMED (76.37 mol% relative to APS) were added. Simultaneously, the formation of the alginate network was induced by the addition of CaSO4 (13 wt% relative to alginate). The solution was then transferred to a PMMA mold and left to gel overnight.
:acrylic acid ratio, 80:20 and 90:10, respectively
| Gel | Water (wt%) | Alginate (wt%) | Acrylamide (wt%) | Acrylic acid (wt%) |
|---|---|---|---|---|
| 1A | 75 | 1 | 19.2 | 4.8 |
| 2A | 75 | 2 | 18.4 | 4.6 |
| 3A | 75 | 3 | 17.6 | 4.4 |
| 1B | 75 | 1 | 21.6 | 2.4 |
| 2B | 75 | 2 | 20.7 | 2.3 |
| 3B | 75 | 3 | 19.8 | 2.2 |
Two-step hydrogel synthesis
Various poly(AAM-co-AA)/alginate hybrid hydrogel network formulations (Table 1) were synthesized. The process starts with forming a homogeneous solution of the network components. First, sodium alginate (1, 2, or 3 wt%) was dissolved in the desired volume of demineralized water at room temperature. Then, AAm and AA monomers, with various AAm:AA ratios (80:20 or 90:10), were added to the solution while stirring. The formation of the poly(AAM-co-AA) network proceeds following the methods described in the one-step method, but without the simultaneous addition of CaSO4. The solution was then stirred for ∼1 min before transferring to a PMMA mold. The solution was incubated overnight under ambient conditions to crosslink into hydrogels. In the second crosslinking step, the alginate network was ionically crosslinked. The hydrogels were carefully removed from the PMMA mold and immersed into a 0.3 M aqueous solution of CaCl2 for 2 hours.
Mechanical analysis
Uniaxial tensile tests and uniaxial unconfined compression tests were performed using an autograph AGS-X and accompanying software TrapeziumX, purchased from Shimadzu Corporation, Japan. All hydrogels for compression testing were made in cylindrical PMMA molds (diameter: 8 mm; height: 3 mm), while all hydrogels for tensile testing were made in dog bone PMMA molds (gauge length: 10 mm; width: 3.5 mm; height: 2.15 mm). Compression tests were conducted with a load cell of 5 kN and a strain speed of 1 mm min−1 up to 90% strain. Cyclic compression tests were performed in triplicate for 20 cycles with a downward strain speed of 5 mm min−1 up to 90% strain, followed by an upward strain speed of 10 mm min−1 down to 0% strain. Tensile testing was conducted with a load cell of 500 N and a strain speed of 50 mm min−1 until gel rupture. Compression and tensile testing data were processed with Igor Pro 8 software from WaveMetrics. The tensile and compressive moduli were calculated by determining the slope of the linear region (10–20% strain) of the stress–strain curve.Equilibrium water content (EWC)
Hybrid hydrogel samples were freeze-dried for 24 h and weighed. Subsequently, the dried gels were swollen in deionized water (23 °C or 37 °C) and weighed at time points (30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 24 h, 28 h, and 30 h) until equilibrium was reached.
With WS, the weight of the swollen hydrogels, and WD, the weight of the freeze-dried hydrogel.
Mass swelling ratio (q)
Upon reaching a constant equilibrium mass after swelling, the gels were freeze-dried overnight. The mass swelling ratio (q) is defined using the following equation:with mt the mass at time t and m0 the dried mass.

Solid-gel content
Hybrid hydrogel samples were freeze-dried (24 h) and then weighed. Subsequently, the dried gels were swollen in deionized water (23 °C or 37 °C) for 30 h. Samples were then freeze-dried again before being weighed once more.
| Gel content (%) = 100 − solid content (%) |
With WI, the initial weight after the first freeze-drying, and WF, the final weight after the second time of freeze-drying.
Sodium alginate sulfation with chlorosulfonic acid
Sodium alginate was dissolved in a 20 vol% solution of chlorosulfonic acid in formamide. The solution was then heated to 60 °C and left to react for 4 h under continuous stirring. Afterward, the sulfated alginate was precipitated in cold acetone and filtrated before dissolving once again in demineralized water. The solution was then neutralized with NaOH (2 M) and dialyzed (MWCO 6–8 kDa) with demineralized water (48 h), and finally, lyophilized.13C-NMR spectroscopy
Samples were prepared by dissolving 40–80 mg of the product in 1.2 ml deuterated water (D2O). Tetramethylsilane (TMS) was used as an internal standard. 13C-NMR spectra were recorded on a JEOL instrument operating at 400 MHz under standard quantitative conditions. Analysis of measurements was performed using MNova software from Mestrelab Research.FTIR spectroscopy
The spectra were recorded using a Spectrum 3 FTIR spectrometer equipped with a diamond ATR prism purchased from PerkinElmer, U.S.A. The spectra were collected in the spectral region from 4000 cm−1 to 600 cm−1 at a resolution of 4 cm−1 for 16 scans. FTIR data was processed with Igor Pro 8 software from WaveMetrics.Size exclusion chromatography
Size exclusion chromatography (SEC) was performed to assess the effects of the alginate purification process. Samples were prepared by dissolving 5 mg of the product in 5 ml of a 0.1 M NaNO3 aqueous solution. Aqueous SEC measurements were made on a Shimadzu Prominence LC purchased from Shimadzu Corporation, Japan. Measurements were performed using water (0.1 M NaNO3) as the mobile phase on a Tosoh. G4000PWXL column (7.8 × 300 mm) and a flow rate of 0.4 mL min−1. Eluograms were converted to molar mass (MW) distributions using calibration data from poly(ethylene glycol) standards. SEC data was processed with Igor Pro 8 software from WaveMetrics.Elemental analysis
The carbon, hydrogen, nitrogen, and sulfur content of the alginate was measured by the elemental analysis method via a FLASH 2000 CHNS/O analyzer purchased from Thermo Fisher Scientific, U.S.A. The degree of sulfation (DS), the number of sulfate groups per monomer, was calculated using the following formula:27,29where [S] was the sulfur content (%) of sulfated alginate obtained from the element analysis.

Statistical analysis
Data is reported as mean ± standard deviation (SD) or median [lower limit; upper limit]. For each experiment, three samples were analyzed unless indicated (§). Statistical differences (p-value ≤ 0.05) between the two groups were determined using the two-sided Student's t-test. All statistical analyses were done with Igor Pro 8 software from WaveMetrics.Results and discussion
Optimization of the one- and two-step hybrid hydrogel networks
Six different poly(AAM-co-AA)/alginate hydrogel formulations (Table 1) were synthesized using a one- or two-step solution-gel method. The mechanical properties of these formulations were evaluated in tensile and (cyclic) compression tests.Tensile strength comparison
The tensile tests were performed in triplicate at a strain rate of 50 mm min−1 until gel rupture for all hydrogels. Moreover, all six hydrogel formulations were synthesized via the one- and two-step solution gel method. The respective tensile moduli (kPa) and max. strain values (%) are summarized in Table 2, while Fig. 1 shows the respective stress–strain curves and moduli plots.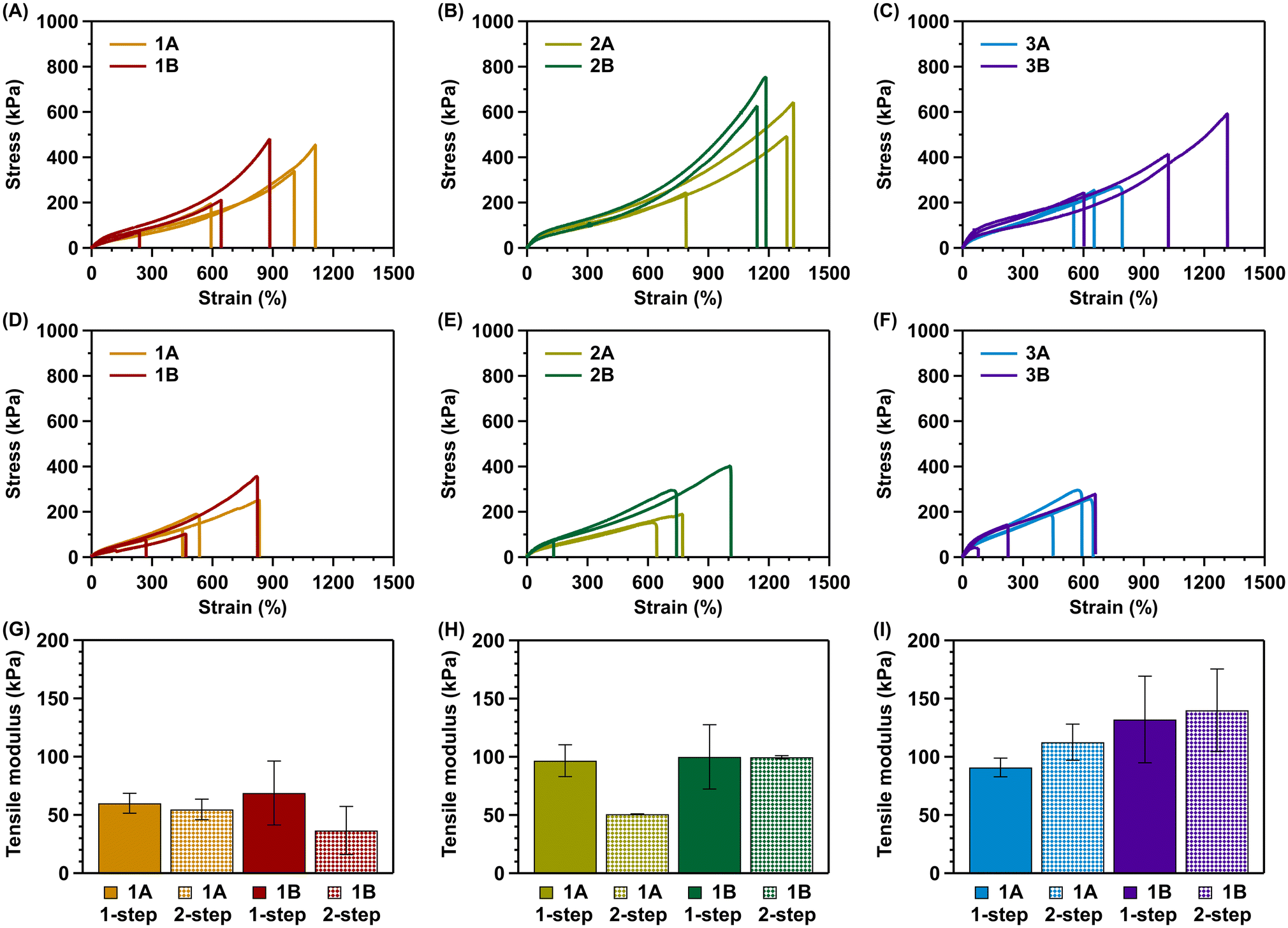 | ||
| Fig. 1 Tensile strength and tensile modulus comparison of the one-step vs. the two-step method. (A–C) Tensile stress–strain curves of the A and B variants of formulations 1, 2 and 3, synthesized via the one-step method, respectively. (D–F) Tensile stress–strain curves of the A and B variants of formulations 1, 2 and 3, synthesized via the two-step method, respectively. (G–I) The tensile modulus of the A and B variants of formulations 1, 2, and 3, synthesized via the one-step vs. the two-step method, respectively. | ||
| 1-step | 2-step | |||
|---|---|---|---|---|
| Modulus (kPa) | Max strain (%) | Modulus (kPa) | Max strain (%) | |
| 1A | 60 ± 9 | ∼900 | 55 ± 9 | ∼600 |
| 2A | 97 ± 14* | ∼1130 | 51 ± 0*,§ | ∼700 |
| 3A | 91 ± 8 | ∼670 | 113 ± 16 | ∼550 |
| 1B | 69 ± 27 | ∼590 | 37 ± 21§ | ∼640 |
| 2B | 100 ± 28§ | ∼1160 | 100 ± 1§ | ∼860 |
| 3B | 132 ± 37 | ∼980 | 140 ± 35§ | ∼440 |
The hydrogel formulations can be distinguished based on three key factors. Their alginate content (1, 2, or 3 wt%), the ratio between monomers (AAm:AA – 80:20 or 90:10), and the synthesis method (one-step or two-step method).
For all hydrogel formulations, the alginate content was set at 1, 2, or 3 wt%. In general, it is expected that an increasing alginate content results in stronger but more brittle hydrogels. A similar trend was observed during tensile tests, as the average tensile modulus increases with increasing alginate content (Table 2 and Fig. 1G–I). This trend can be observed for both monomer ratios, as well as for both synthetic/preparation protocols. Moreover, the extensibility is the highest for the 2 wt% alginate hydrogels and decreases for 3 wt% alginate hydrogels (Table 2 and Fig. 1A–F). As expected, the hydrogels become more brittle above the optimal alginate content of 2 wt% for both monomer ratios and synthesis methods.
For all hydrogel formulations, the AAm:AA content was set at an 80:20 or 90:10 ratio. This moderate variation was made to identify an optimal formulation, generating tough and extensible hydrogels. While there are slight differences, the results are absent of an unambiguous trend in modulus and extensibility for the A and B formulations (Table 2 and Fig. 1A–I). Both variations yield workable, extensible hydrogels.
Lastly, all hydrogel formulations were synthesized according to both the one-step and two-step gelation methods described in the Experimental section. As can be observed (Table 2 and Fig. 1G–I), the average tensile modulus is similar for all formulations except for formulation 2A, which shows a statistically significant (p < 0.05) difference between the methods. In general, the synthesis method does not influence the average tensile modulus of the hybrid hydrogels. However, a clear difference can be observed when looking at the extensibility of the hydrogel formulations, with the one-step method yielding more extensible hydrogels in most cases, except for formulation 1B (Table 2 and Fig. 1A–F).
These results clearly show that the one-step method results in more extensible hybrid hydrogel networks while maintaining remarkable toughness. Furthermore, the properties are also consistent with results reported in the literature for poly(acrylamide)/alginate DN hydrogel systems.13,30–33 Tensile moduli reported in the literature vary from lower ranges (50–70 kPa) to higher ranges (150–500 kPa).13,30–33 The results reported here are values within the upper end of the lower ranges reported in the literature. Therefore, it can be concluded that the elastic properties displayed by the various formulations are exemplary of the properties to be expected from physically/chemically crosslinked hybrid hydrogels.
Compressive strength comparison
The compressive tests were performed in triplicate at a strain rate of 1 mm min−1 until 90% strain or rupture was reached for all hydrogels. Moreover, all six hydrogel formulations were synthesized via the one- and two-step solution gel method. The respective moduli (kPa) and max. stress values (MPa) are summarized in Table 3, while Fig. 2 shows the respective stress–strain curves and moduli plots. | ||
| Fig. 2 Compressive strength and compressive modulus comparison of the one-step vs. the two-step method. (A–C) Compressive stress–strain curves of the A and B variants of formulations 1, 2 and 3, synthesized via the one-step method, respectively. (D–F) Compressive stress–strain curves of the A and B variants of formulations 1, 2 and 3, synthesized via the two-step method, respectively. (G–I) Compressive (comp.) modulus of the A and B variants of formulations 1, 2, and 3, synthesized via the one-step vs. the two-step method, respectively. | ||
| 1-step | 2-step | |||
|---|---|---|---|---|
| Modulus (kPa) | Max stress (MPa) | Modulus (kPa) | Max stress (MPa) | |
| 1A | 223 ± 23* | 7.9 [6.2–9.6] | 93 ± 43* | 1.6 [0.4–2.7] |
| 2A | 244 ± 58 | 4.6 [2.5–7.2] | 150 ± 72 | 2.5 [1.5–2.5] |
| 3A | 376 ± 48* | 8.8 [8.6–12.4] | 149 ± 27* | 4.7 [3.3–5.1] |
| 1B | 234 ± 45* | 18.3 [10.1–20.6] | 123 ± 5* | 1.8 [1.5–3.3] |
| 2B | 277 ± 87 | 10.3 [4.6–10.8] | 185 ± 93 | 3.4 [2.5–3.8] |
| 3B | 304 ± 25* | 10.2 [6.0–12.3] | 163 ± 19* | 2.2 [2.1–2.7] |
As previously mentioned, an increasing sodium alginate content results in stronger, more brittle hydrogels. This is also evident from compressive tests, as the average compressive modulus increases based on an increasing sodium alginate content (Table 3 and Fig. 2G–I). Both monomer ratios follow the trend, as do both synthesis methods. Furthermore, no clear trend can be observed for the maximum stress based on differences in sodium alginate content.
Considering the differences in AAm:AA content, slight differences were also observed during compression. However, there is no clear trend to be observed when looking at the moduli of the A and B formulations (Table 3 and Fig. 2G–I).
Lastly, the one-step or two-step solution gel methods were evaluated in terms of their mechanical performance under compression. As can be observed (Table 3 and Fig. 2G–I), the average compressive modulus varies consistently for most formulations, except for formulations 2A and 2B. For all other formulations, the difference between the methods is statistically significant (p < 0.05). Here, the synthesis method affects the average compressive modulus of the hybrid hydrogels. Furthermore, a clear difference is observed in the maximum stress of the hydrogel formulations (Table 3 and Fig. 2A–F). The one-step method gives rise to higher stress in all cases.
The one- and two-step methods are notably similar in one aspect. Differences in maximum stress between formulations are not logically matched by differences in compressive modulus. This could be explained by compressive stress consistent with plastic deformation from ∼50–75% strain onwards. The deviation from the linear relation between stress and strain in the elastic region explains this discrepancy between the maximum stress reached and the compressive modulus. However, the results suggest that the one-step method provides tougher hybrid hydrogels. This can be attributed to the lower water content (75 wt%) of hydrogels prepared via this method since there is no additional water uptake during ionic crosslinking, in contrast to the two-step method. Secondly, by using the one-step method, the cross-linking of the alginate network will be more effective, as the distribution of cations is more uniform throughout the gel when compared to the diffusion-based two-step method. Furthermore, the properties are also consistent with the results reported in the literature for various poly(acrylamide)/alginate DN hydrogel systems.34–37 The maximum stress reported in the literature varies over a wide range from relatively low values (0.1–0.2 MPa) to moderate values (1.5–2.5 MPa) and even high values (11.5–12.5 MPa).34–37 The results reported here fall within these ranges, although they are not on par with the highest reported values. Nevertheless, all six formulations, when synthesized via the one-step method, reach a compressive toughness that lies within or is close to the compressive modulus of native cartilage (0.23–0.85 MPa).5,7 Among the six formulations, both 2 and 3 display the most promising mechanical properties for cartilage tissue engineering.
Degree of swelling
Based on the mechanical analysis, swelling tests were performed in triplicate using hybrid gels synthesized via the one-step method. Measurements were performed to determine the equilibrium water content (EWC), mass swelling ratio (MSR), and gel content (GC). All gels were freeze-dried, swollen to equilibrium, and freeze-dried again. The respective graphs are shown in Fig. 3.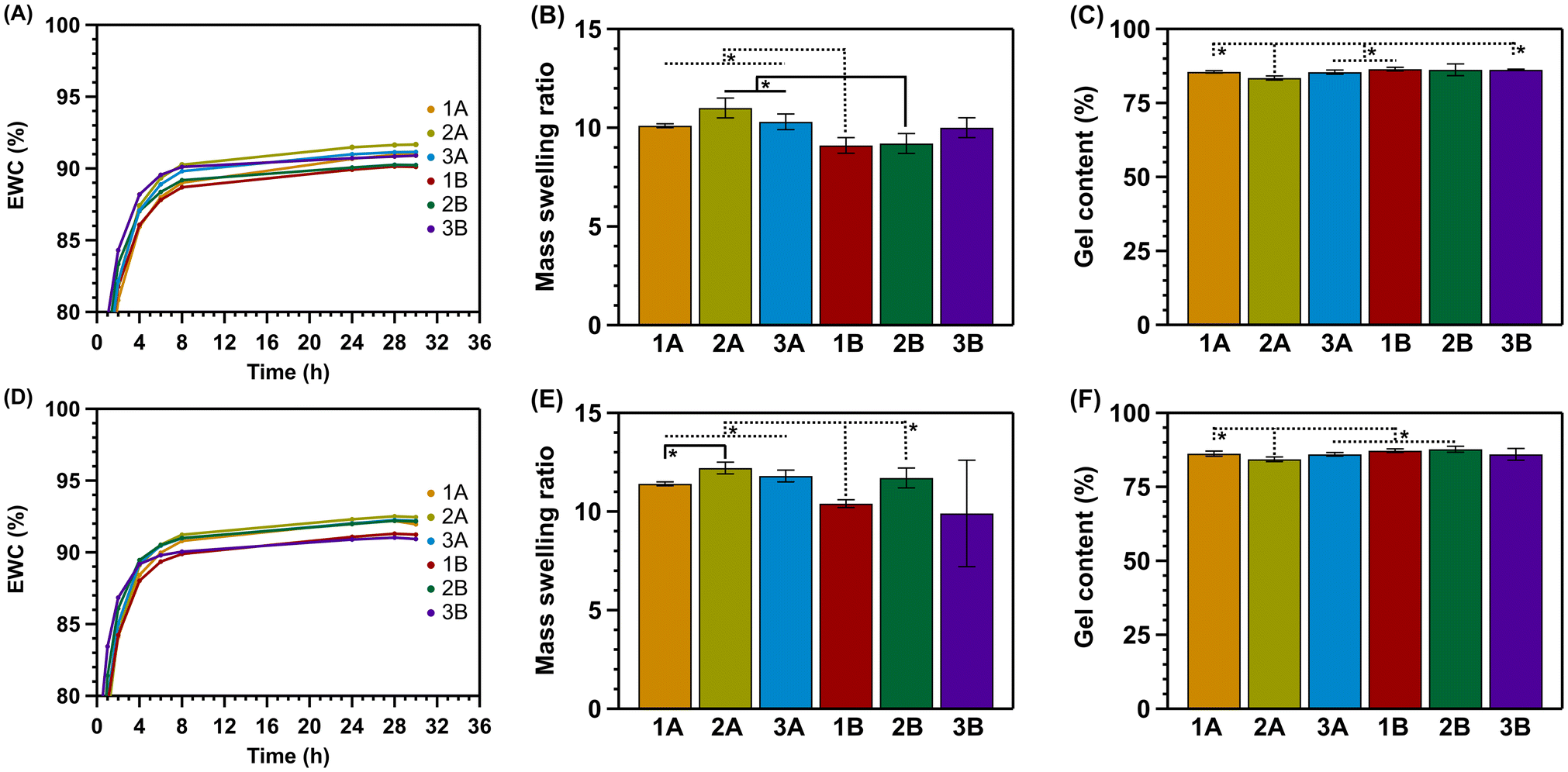 | ||
| Fig. 3 Swelling tests of the one-step solution gel-method hydrogels. (A–C) Equilibrium water content, mass swelling ratio, and gel content of all six hydrogel formulations synthesized via the one-step method, swollen at RT = 23 °C, respectively. (D–F) Equilibrium water content, mass swelling ratio, and gel content of all six hydrogel formulations synthesized via the one-step method, swollen at 37 °C, respectively. Significant differences at indicated * (p < 0.05). | ||
Based on the literature, most hydrogels reach their EWC after 24 hours of swelling.38 This is consistent with our findings (Fig. 3A and D). After 28 hours of swelling, no further increase in weight was observed. All hydrogels reached an EWC between 90 and 93%, with gel 2A showing the highest EWC at 92.5%. Moreover, the EWC was found to be slightly higher for all gels at 37 °C. As the temperature increases, the polymer chains in the network become more flexible and mobile. This increased mobility leads to greater expansion and hydration of the gels, resulting in a higher EWC.
The swelling ability was further assessed by determining the mass swelling ratio. This gives an indication of how much water the gels absorb relative to their dry weight. For all hydrogels, the MSR was between 9 and 12 times their original weight (Fig. 3B and E). In accordance with the EWC, all hydrogels show a slightly higher MSR at 37 °C.
Lastly, the gel content was determined. This value indicates the polymerized/crosslinked percentage of the formed network. For all hydrogels, the GC was between 84 and 87% (Fig. 3C and F). Here, there are only slight differences between gels swollen at RT or 37 °C.
From these swelling experiments, it is clear that there are minimal differences in hydrogel swelling based on the composition. Moreover, all hydrogels retain between 84 and 87% of the polymerized/crosslinked network.
Picking the most suitable hydrogel network
When the results of both the tensile and compression tests are considered, formulations 2A and 2B display the most promising mechanical properties. These formulations render hydrogels that offer extensibility, high tensile strength, and high compressive strength, showing rounded and versatile behavior when stress is applied. The versatility to be stretched and compressed is particularly useful for cartilage tissue engineering, as cartilage tissue has to allow for unhindered movement in multiple dimensions, not limited to either compression or extension.39,40 Furthermore, the results of the tensile and compression tests show a general increase in the toughness of the hydrogels as the sodium alginate content increases for both synthesis methods. This is expected as the high toughness and strength are derived from the brittle yet rigid sacrificial ionic alginate network. Unzipping the ionic crosslinks supplies an energy dissipation mechanism; the number of load-bearing polymer chains increases as the alginate network is unzipped. Nevertheless, the hydrogels do not completely break compression because the stretchable poly(AAM-co-AA) stabilizes the deformation once the ionic cross-links are broken.41,42 Therefore, an increase in the relative amount of this rigid network will increase the overall stiffness and toughness of the DN hydrogel. Furthermore, the monomer ratio of AAm:AA did not affect the mechanical properties. Therefore, formulation 2A was chosen, as its higher acrylic acid content offers future biofunctionalization opportunities.
Sulfation of sodium alginate
Sodium alginate sulfation was performed to introduce sulfate moieties into our hybrid hydrogel system, promoting the material to mimic the natural function of sulfated glycosaminoglycans (i.e., chondroitin sulfate). Therefore, sodium alginate was reacted with chlorosulfonic acid, resulting in sulfated alginate (Fig. 4A).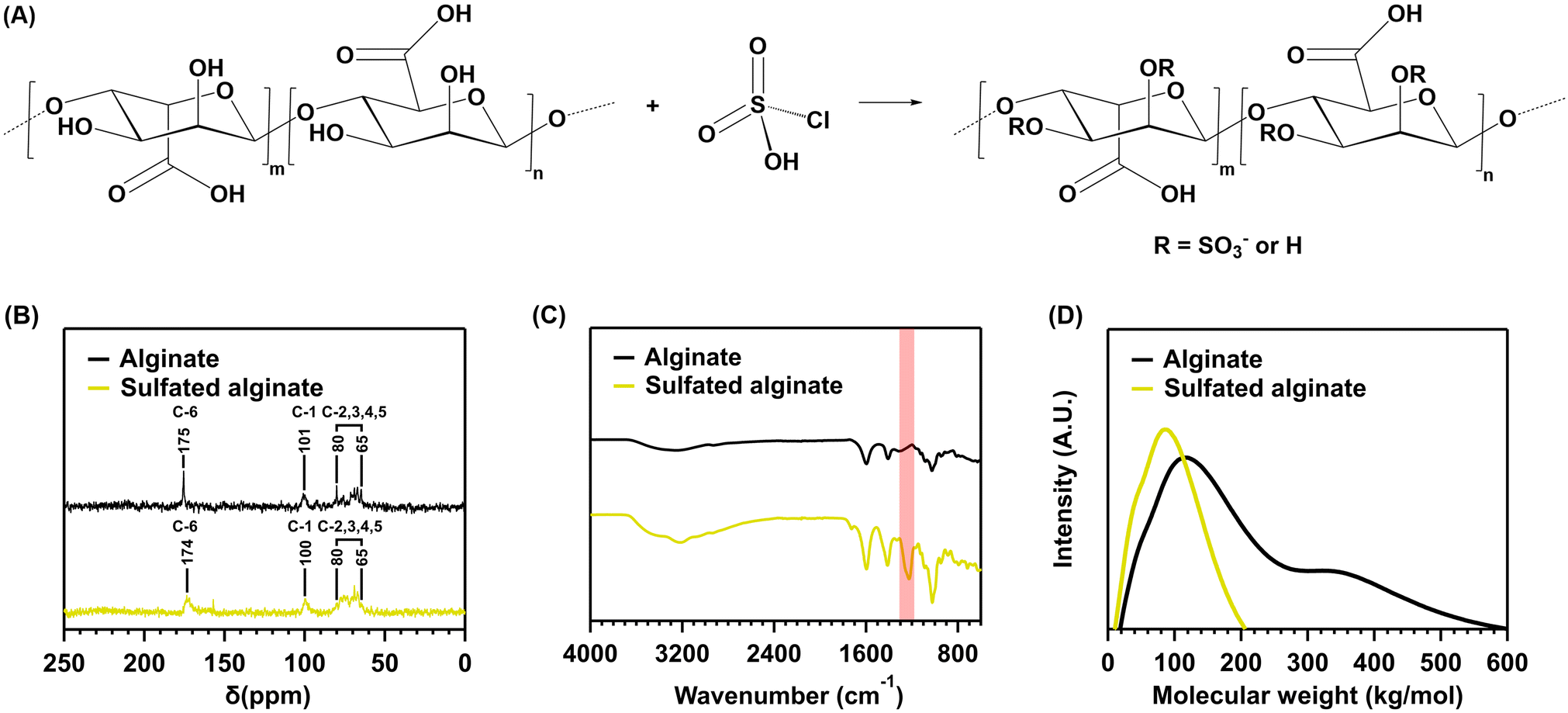 | ||
| Fig. 4 (A) Sulfation reaction between sodium alginate and chlorosulfonic acid, resulting in sulfated alginate. Possible sulfate groups are present at the R-positions attached to C-2,3. (B) The 13C-NMR-spectra of sodium alginate and the reaction product of sulfation using chlorosulfonic acid. (C) The FTIR-spectra of sodium alginate and the reaction product of sulfation using chlorosulfonic acid. (D) The SEC chromatogram of sodium alginate and the reaction production of sulfation using chlorosulfonic acid. | ||
The 13C-NMR and FTIR spectra, SEC chromatogram and elemental analysis suggest a successful reaction between sodium alginate and chlorosulfonic acid. The 13C-NMR spectrum (Fig. 4B) displays peaks corresponding to the carbonyl carbon (C-6) at δ = 175 and 174 ppm for the starting sodium alginate (I) and the reaction product (II), respectively. The anomeric carbon (C-1) appears at δ = 101 and 100 ppm, respectively. The remaining carbon atoms (C-2,3,4,5) provide signals in the range δ = 80–65 ppm for both spectra. However, the intensity of the peaks in the δ = 80–65 ppm range is skewed towards the lower field position of 65 ppm for the reaction product (II). This indicates a downfield shift of C-2,3, consistent with a bond to relatively electronegative sulfate groups.26,27 However, due to incomplete sulfation, signals for C-2,3,4,5 remain, diminishing the strength of the shift. Furthermore, the FTIR spectrum (Fig. 4C) of the reaction product displays the characteristic peaks of alginate at 3570–3100 cm−1 (O–H), 1635 cm−1 and 1419 cm−1 (COO), 1050–1250 cm−1 (C–O–C), 820 cm−1 and 946 cm−1 (C–H). In addition to these peaks, a characteristic sulfate peak is present at 1225 cm−1 (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), further suggesting the addition of sulfate moieties. The element analysis also supports this conclusion, as the reaction product contains 9.56% sulfur, indicating a degree of sulfation (DS) of 0.85. This infers that, of the two hydroxyl groups each repeating unit contains, on average, 0.85 are replaced by sulfate groups. Consequently, a total conversion of 42.5% was achieved. Moreover, a decrease in MW can be noticed after the reaction of sodium alginate with chlorosulfonic acid (Fig. 4D).
O), further suggesting the addition of sulfate moieties. The element analysis also supports this conclusion, as the reaction product contains 9.56% sulfur, indicating a degree of sulfation (DS) of 0.85. This infers that, of the two hydroxyl groups each repeating unit contains, on average, 0.85 are replaced by sulfate groups. Consequently, a total conversion of 42.5% was achieved. Moreover, a decrease in MW can be noticed after the reaction of sodium alginate with chlorosulfonic acid (Fig. 4D).
Despite the successful functionalization reaction, the DS is lower than expected. Ronghua et al. reported a DS of 1.41 for the reaction with 20 vol% chlorosulfonic acid.26 Lower DS are reported in the literature for the reaction with chlorosulfonic acid. However, the vol% used in these reports is also much lower. Baei et al. report a DS of 0.45 and 0.67 for the reaction with 2 and 3 vol% chlorosulfonic acid.24 Daemi et al. report a DS of 0.9 for the reaction with 3.5 vol% chlorosulfonic acid.43 The DS reported here is thus more in line with the reactions using 2–3.5 vol% chlorosulfonic acid. This discrepancy might be explained by water in the reaction setup, as chlorosulfonic acid is known to react with water to yield sulfuric acid and hydrogen chloride. Residual water could have been expected given the use of commercially available compounds without additional purification and should not be of concern, since it is good practice to properly characterize individual batches of sulfated polysaccharides (e.g., using the elemental analysis mentioned above) before relating macroscopic properties to the composition of the tested samples.
Control vs. sulfated hydrogels
The previous evaluation of the mechanical properties of the poly(AAM-co-AA)/alginate hybrid hydrogel helped identify formulation 2A as the most suitable candidate for cartilage tissue engineering. The versatility to display great toughness during compression, combined with great extensibility, makes this formulation the preferred one. Hence, formulation 2A was used to assess the effect of the sulfation on the mechanical properties of the hybrid hydrogel network.Tensile strength comparison - Effect of sulfation
The sulfated hybrid hydrogels reach an average tensile modulus of 75 ± 28 kPa, a slight decrease relative to the average tensile modulus of the control hybrid hydrogels (97 ± 14 kPa) (Fig. 5C). This decrease in average tensile modulus could be explained due to electrostatic repulsion and steric hindrance during crosslinking. The negatively charged sulfate groups may electrostatically repulse the alginate chains, thereby preventing ionic crosslinking. Additionally, the large sulfate moieties on the chains may prevent efficient crosslinking via steric hindrance.22,24 However, statistical analysis shows that this decrease is not statistically significant (p > 0.05), indicating the similarity in the mechanical properties of the two hydrogel compositions.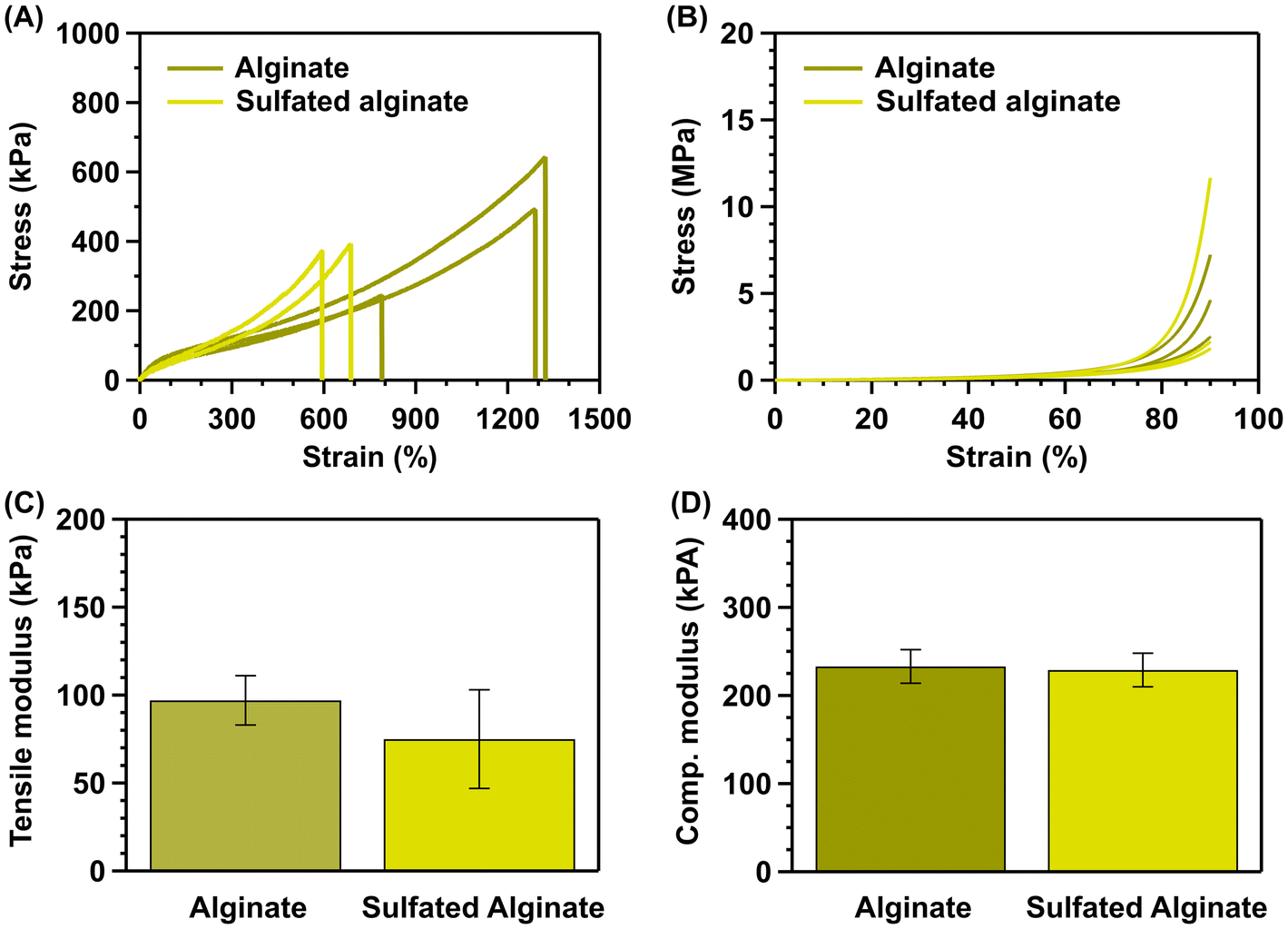 | ||
| Fig. 5 Strength and modulus comparison of the control vs. sulfated alginate hydrogels, synthesized via the one-step method. (A) Tensile stress–strain curves of the control and sulfated alginate 2A hydrogels. (B) Compressive stress–strain curves of the control and sulfated alginate 2A hydrogels. (C) Tensile modulus of the control and sulfated alginate 2A hydrogels. (D) Compressive (comp.) modulus of the control and sulfated alginate 2A hydrogels. | ||
Despite having similar tensile moduli to the one-step method, the extensibility of the sulfated hydrogel network is generally lower. The sulfated hydrogels reach a maximum strain of ∼700%, compared to the maximum strain of ∼1300% before modification (Fig. 5A). This decrease in maximum strain can be most likely be attributed to the decrease in MW after sulfation with chlorosulfonic acid (Fig. 4D). However, this decrease in extensibility is not expected to limit the applicability of the sulfated hydrogels for cartilage tissue engineering. Extensibility of 700% is still substantially larger than that of articular cartilage (up to 1.4 times).5 These elastic properties are also superior to other hydrogel systems, mimicking the function of chondroitin sulfate. Ma et al. reported a loss modulus G′, which reflects the elastic properties of ∼30 kPa for an alginate/chondroitin sulfate hybrid hydrogel.44 Shah et al. reported a loss modulus G′ of ∼4.8 kPa for a chondroitin sulfate grafted alginate-Poloxamer-407 (F127) hybrid hydrogel.45 Zare et al. reported tensile moduli ranging from 5–24 kPa for a KNG-loaded poly(lactic-co-glycolic acid) nanoparticle impregnated alginate/sulfated alginate polycaprolactone nanofiber composite hydrogel.46
Compressive strength comparison - Effect of sulfation
The sulfated hybrid hydrogels reach an average compressive modulus of 229 ± 19 kPa. A marginal decrease was observed when compared to the control hybrid hydrogels with a modulus of 244 ± 58 kPa (Fig. 5D). This decrease may be attributed to electrostatic repulsion and steric hindrance of the sulfate groups.22,24 Nevertheless, the modulus lies within the range of native cartilage (0.23–0.85 MPa).5,7Moreover, the median maximum stress exhibited by the sulfated hydrogels is 2.2 MPa (range [1.8–11.7 MPa]), slightly lower than the non-sulfated hydrogels (4.6 MPa; range [2.5–7.2 MPa]); the rather large variability likely arises from the non-elastic nature of the upper compression range (Fig. 5B). Nevertheless, the compressive performance of these materials is superior to other hydrogel systems, mimicking the function of chondroitin sulfate. The KNG-loaded poly(lactic-co-glycolic acid) nanoparticle-impregnated alginate/sulfated alginate polycaprolactone nanofiber composite hydrogel reported by Zare et al. reached a maximum stress of 6–15 kPa.46 Mhanna et al. report a compressive modulus of 2.4 ± 0.57 kPa for a pure sulfated alginate hydrogel.22 Goto et al. report a compressive modulus of 22.5 ± 6.3 kPa for a phenol-grafted sulfated alginate hydrogel.47 These reported results clearly show the benefits of the hybrid hydrogel network formulation.
Cyclical mechanical properties & energy dissipation
In addition to the mechanical characterization methods above, a preliminary cyclical compressive test was performed to further compare the mechanical strength of the control and sulfated hybrid hydrogels. The cyclic compressive test was performed in triplicate for 20 cycles with a downward strain speed of 5 mm min−1 up to 90% strain, followed by an upward strain speed of 10 mm min−1 down to 0% strain. Notably, the 90% strain greatly exceeds the strain levels expected in joint environments. Coburn et al. reported compressive strains up to 7.5% for single-leg hops.48Absorbed energy
The stress–strain curves (cycles 1, 10, and 20) (Fig. 6A and C) show that the control and sulfated hydrogels effectively dissipate energy, as suggested by the pronounced hysteresis. Maximum stress increases with cycle number in control hydrogels, indicating stiffening of the scaffold as the strain is released and reapplied. Moreover, a considerable decrease in absorbed energy occurs from the first cycle (28 ± 1.2 MJ m−3) to the second (19 ± 1.4 MJ m−3), consistent with permanent network damage, after which the energy remains relatively constant (19 ± 1.5 MJ m−3) from the second cycle onwards (Fig. 6B). However, unlike control hydrogels, pronounced strain stiffening or diminished absorbed energy is not observed for the sulfated hydrogels (Fig. 6D). The energy dissipation appears to be altered since no notable decrease in energy dissipation is observed from cycles 1 to 2. The hysteresis changes from 17.6 to 15.5 MJ m−3 from cycles 1 to 2 (Fig. 6D). The reduction (Δ2.1 MJ m−3) is marginal compared to the non-sulfated hydrogels (Δ8.3 MJ m−3), indicating that the sulfated hydrogel scaffold suffers substantially less permanent damage than the control hydrogels. The dissipated energy remains relatively constant over all 20 cycles at ∼15.7 ± 3.6 MJ m−3 (Fig. 6D). This is similar to the energy dissipation value (19.1 ± 1.5 MJ m−3) of the control hydrogels (Fig. 6B). | ||
| Fig. 6 Cyclical mechanical strength and energy dissipation comparison of the control vs. sulfated alginate 2A hydrogel, synthesized via the one-step method. (A) Cyclical stress–strain curves of the control 2A hydrogel (cycles 1, 10, and 20). (B) Change in hysteresis (dark yellow) and max stress (black) of the control 2A hydrogel. (C) Cyclical stress–strain curves of the sulfated 2A hydrogel (cycles 1, 10, and 20). (D) Change in hysteresis (yellow) and max stress (black) of the sulfated 2A hydrogel. | ||
The energy dissipation displayed in our study appears larger than in other reported hydrogel systems. The KNG-loaded poly(lactic-co-glycolic acid) nanoparticle-impregnated alginate/sulfated alginate polycaprolactone nanofiber composite hydrogel had a toughness of 2235 J m−3, while hysteresis values for tensile testing were reported to range from 588 to 2160 kJ m−3 for alginate/polyacrylamide hydrogels crosslinked with various ions.41,46 However, it should be noted that the values found in the literature were obtained under different experimental conditions. Notable differences between our gels and prior work found in the literature include the water content (75 wt% vs. 86 wt%), synthesis method (one-step vs. two-step), and testing mode (compressive testing vs. tensile testing).
The energy dissipation displayed by the sulfated hydrogel scaffolds is extremely promising for cartilage tissue engineering. Especially since loading and unloading were performed up to 90% strain each cycle, which is well above the functional range of in vivo cartilage deformation (∼7% strain).49 Moreover, the native cartilage tissue of the knee experiences both compression (superior–inferior) and shear (anterior–posterior) forces between the femur and tibia.39,40,50 Consequently, the cartilage does not fully absorb the energy by distributing the load equally to the subchondral bone plate, muscles, and tendons, dissipating the energy.39,40,50
Maximum stress
Energy absorption is accompanied by an increase in the maximum stress reached in the control hydrogels. The maximum stress rises from 5.0 MPa (range [4–5.1 MPa]) to 6.7 MPa (range [5.6–6.7 MPa]) (Fig. 6B), indicating a stiffening of the network as the strain is reapplied beyond the first cycle. On the other hand, the constant energy dissipation for the sulfated hydrogels is accompanied by relatively constant maximum stress. The maximum stress remains essentially constant over 20 cycles (Fig. 6D). Hence, the sulfated hydrogels do not seem to display the same intercycle strain stiffening behavior as their non-sulfated counterpart.This change in behavior might be due to the macroscopic water expulsion effect, which is the basis of the strain-stiffening behavior of the non-sulfated hydrogels. Sulfates are among the most hydrophilic anions and are notoriously difficult to dehydrate, limiting the drying effect during continuous loading and unloading of the hydrogel.51 Moreover, on the microscopic level, deformation reorganizes the network, increasing the number of active chains and building non-linear tension, which gives rise to the stiffening of the network.52–57 These macroscopic and microscopic mechanisms thus explain the increasing maximum stress for control hydrogels, which starts plateauing from cycle 16 onward. This indicates that over time, the amount of water exuded and the number of participating polymer chains reaches a maximum and that the toughening of the scaffold is finite.
Conclusions
The synthesis and sulfation of poly(acrylamide-co-acrylic acid)/alginate hydrogels demonstrates that the synthesis protocol and the hydrogel formulation significantly affect the mechanical properties. A one-step in situ crosslinking method results in hydrogels with superior properties compared to a two-step sequential crosslinking method. Furthermore, the results show increases in hydrogel stiffness are coupled to the wt% of the rigid, densely crosslinked alginate network, and this increase in tensile stiffness is conversely tied to an expected decreasing trend in extensibility. The compressive stiffness increases with increasing wt% of the alginate network. It was found that a formulation using 2 wt% alginate results in optimal properties, combining both high compressive and tensile stiffness with considerable extensibility. The composition of the second network showed limited influence; 80:20 or 90:10 ratio of acrylamide:acrylic acid monomers did not appreciably affect the mechanical properties. Therefore, a higher amount of acrylic acid was chosen as the most optimal formulation given future biofunctionalization via the carboxylic acid groups present. Incidentally, the compressive strength of this formulation was within the range of articular cartilage (0.23–0.85 MPa).5,7 This particular formulation also displayed strain stiffening in cyclical compression testing, showing promise for articular cartilage tissue engineering.
Moreover, sulfated alginate could mimic the functions of chondroitin sulfate, a crucial component in tissue development, by serving as co-receptors of growth factors through electrostatic interactions.19,22–24 The sulfation was successful using chlorosulfonic acid, resulting in a DS of 42.5%. The sulfate groups did not drastically deteriorate the mechanical properties; only a decreased extensibility was observed. On the other hand, cyclical testing showed that the sulfated hydrogel scaffolds suffer less permanent damage than the control hydrogels. Therefore, the mechanical properties are deemed potentially suitable for cartilage tissue engineering applications. Here, it is pertinent to acknowledge the shortcomings of these particular gel formulations, wherein the presence of poly(acrylic acid) leads to profuse swelling. Excessive swelling prohibits testing cell viability and also causes large changes in dimensions and mechanical performance. Future work will, therefore, look into the inclusion of either more hydrophobic monomers (e.g., butyl acrylate or 2-ethylhexyl acrylate) or monomers that impede swelling via additional supramolecular interactions between the polymer chains (N-acryloyl glycinamide).38,58
Although assessing the bioactivity of these hydrogels was outside of the scope of this present work, past studies have shown that sulfated hydrogel scaffolds can effectively sequester and slowly release growth factors such as TGF-β1.24 Studying the retention and release profile of growth factors from our hybrid hydrogels will be a focus of future study. The most important outcome of this study relates to the retained performance after sulfation of the alginate, employing the hybrid network approach that has proven so appealing in terms of response to mechanical deformation. Additionally, the effect of the hydrogels’ mechanical properties on encapsulated chondrocytes can be studied by looking into the upregulation of key biomarkers, including collagen types I and II, aggrecan, and c-Jun.59,60
To summarize, this work provides the basic fundamental insights required to further enhance the mechanical properties of the proposed poly(acrylamide-co-acrylic acid)/sulfated alginate hybrid hydrogel system while introducing a potentially bioactive moiety. Our results suggest the potential utility of these hydrogels as cartilage models and support their further investigation in vitro.
Author contributions
Sander Driesen: investigation, formal analysis, visualization, writing original draft, writing – review & editing; Valentino Atella: investigation, formal analysis, visualization, writing – review & editing; Kristi Kiick: supervision, writing – review & editing; Louis Pitet: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing; Geert-Jan Graulus: funding acquisition, project administration, resources, supervision, writing – review & editing.Data availability
Data for this article are available under the Open Science Framework at https://doi.org/10.17605/OSF.IO/MEVBW.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the Research Foundation Flanders (FWO) under contracts 1S19023N and G080020N, as well as the Special Research Fund (BOF) under contract BOF21OWB16. The authors are grateful to Huguette Penxten, Elsy Thijssen, Wouter Marchal, Arthur Helsen, Koen Van Vinckenroye, and Gunter Reekmans for their technical support.References
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CAS.
- S. H. Aswathy, U. Narendrakumar and I. Manjubala, Heliyon, 2020, 6, e03719 CAS.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Mater. Sci. Eng., C, 2019, 12, 3323 CAS.
- X. Li, Q. Sun, Q. Li, N. Kawazoe and G. Chen, Front. Chem., 2018, 6, 499 CrossRef CAS PubMed.
- M. Hafezi, S. Nouri Khorasani, M. Zare, R. Esmaeely Neisiany and P. Davoodi, Polymers, 2021, 13, 4199 CrossRef CAS PubMed.
- L. Zhou, P. Guo, M. D'Este, W. Tong, J. Xu, H. Yao, M. J. Stoddart, G. J. V. M. van Osch, K. K.-W. Ho, Z. Li and L. Qin, Engineering, 2022, 13, 71–90 CrossRef.
- R. K. Korhonen, M. S. Laasanen, J. Töyräs, J. Rieppo, J. Hirvonen, H. J. Helminen and J. S. Jurvelin, J. Biomech., 2002, 35, 903–909 CrossRef CAS PubMed.
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC.
- X. Huang, J. Li, J. Luo, Q. Gao, A. Mao and J. Li, Mater. Today Commun., 2021, 29, 102757 CrossRef CAS.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 RSC.
- M. Tonbul, M. Adas, T. Bekmezci and A. D. Kara, Case Rep. Orthop., 2014, 2014, 150709 Search PubMed.
- G. Sennakesavan, M. Mostakhdemin, L. K. Dkhar, A. Seyfoddin and S. J. Fatihhi, Polym. Degrad. Stab., 2020, 180, 109308 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- S. Houben and L. M. Pitet, React. Funct. Polym., 2023, 191, 105676 CrossRef CAS.
- J. Tan, Y. Luo, Y. Guo, Y. Zhou, X. Liao, D. Li, X. Lai and Y. Liu, Int. J. Biol. Macromol., 2023, 239, 124275 CrossRef CAS PubMed.
- W. Liu, H. Madry and M. Cucchiarini, Int. J. Mol. Sci., 2022, 23, 1147 CrossRef CAS PubMed.
- O. Smidsrød and G. Skjåk-Braek, Trends Biotechnol., 1990, 8, 71–78 CrossRef PubMed.
- C. Vyas, H. Mishbak, G. Cooper, C. Peach, R. F. Pereira and P. Bartolo, Biomanuf. Rev., 2020, 5, 2 CrossRef.
- C. Klecker and L. S. Nair, in Biology and Engineering of Stem Cell Niches, ed. A. Vishwakarma and J. M. Karp, Academic Press, Boston, 2017, pp. 195–213. DOI:10.1016/B978-0-12-802734-9.00013-5.
- Y. Henrotin, M. Mathy, C. Sanchez and C. Lambert, Ther. Adv. Musculoskelet. Dis., 2010, 2, 335–348 CrossRef CAS PubMed.
- S. A. Baeurle, M. G. Kiselev, E. S. Makarova and E. A. Nogovitsin, Polymer, 2009, 50, 1805–1813 CrossRef CAS.
- R. Mhanna, A. Kashyap, G. Palazzolo, Q. Vallmajo-Martin, J. Becher, S. Möller, M. Schnabelrauch and M. Zenobi-Wong, Tissue Eng., Part A, 2013, 20, 1454–1464 CrossRef PubMed.
- Ø. Arlov and G. Skjåk-Bræk, Molecules, 2017, 22, 778 CrossRef PubMed.
- P. Baei, H. Daemi, F. Mostafaei, F. Azam Sayahpour, H. Baharvand and M. Baghaban Eslaminejad, Chem. Eng. J., 2021, 418, 129277 CrossRef CAS.
- M. Gionet-Gonzales, A. Casella, D. Diloretto, C. Ginnell, K. H. Griffin, A. Bigot and J. K. Leach, Adv. Healthc. Mater., 2021, 10, e2101048 CrossRef PubMed.
- H. Ronghua, D. Yumin and Y. Jianhong, Carbohydr. Polym., 2003, 52, 19–24 CrossRef.
- L. Fan, L. Jiang, Y. Xu, Y. Zhou, Y. Shen, W. Xie, Z. Long and J. Zhou, Carbohydr. Polym., 2011, 83, 1797–1803 CAS.
- O. Arlov, F. L. Aachmann, A. Sundan, T. Espevik and G. Skjak-Braek, Biomacromolecules, 2014, 15, 2744–2750 CAS.
- D.-W. Lim, H. S. Whang, K.-J. Yoon and S.-W. Ko, J. Appl. Polym., 2001, 79, 1423–1430 CAS.
- P. Guo, Y. Yuan and F. Chi, Mater. Sci. Eng., C, 2014, 42, 622–628 CAS.
- J. Yi, K.-C. T. Nguyen, W. Wang, W. Yang, M. Pan, E. Lou, P. W. Major, L. H. Le and H. Zeng, J. Colloid Interface Sci., 2020, 578, 598–607 CAS.
- C. Xu, X. Zhang, S. Liu, X. Zhao, C. Geng, L. Wang and Y. Xia, ACS Appl. Mater. Interfaces, 2021, 13, 25383–25391 CrossRef CAS PubMed.
- J. Li, W. R. K. Illeperuma, Z. Suo and J. J. Vlassak, ACS Macro Lett., 2014, 3, 520–523 CrossRef CAS PubMed.
- J. Wang, J. Wei, S. Su, J. Qiu and S. Wang, J. Mater. Sci., 2015, 50, 5458–5465 CrossRef CAS.
- M. Arjmandi and M. Ramezani, J. Mech. Behav. Biomed. Mater., 2019, 95, 196–204 CrossRef CAS PubMed.
- Z. Cao, Y. Zhang, K. Luo, Y. Wu, H. Gao, J. Cheng, C. Liu, G. Tao, Q. Guan and L. Zhang, J. Renewable Mater., 2021, 9, 1447–1462 CAS.
- Z. Chen, J. Tang, N. Zhang, Y. Chen, Y. Chen, H. Li and H. Liu, Colloids Surf., A, 2022, 633, 127867 CrossRef CAS.
- S. Houben, A. A. Aldana, A.-S. Huysecom, W. Mpinganzima, R. Cardinaels, M. B. Baker and L. M. Pitet, ACS Appl. Polym. Mater., 2023, 5, 1819–1827 CrossRef CAS.
- D. D. Chan, L. Cai, K. D. Butz, S. B. Trippel, E. A. Nauman and C. P. Neu, Sci. Rep., 2016, 6, 19220 CrossRef CAS PubMed.
- Z. Abusara, M. Von Kossel and W. Herzog, PLoS One, 2016, 11, e0147547 CrossRef PubMed.
- C. H. Yang, M. X. Wang, H. Haider, J. H. Yang, J.-Y. Sun, Y. M. Chen, J. Zhou and Z. Suo, ACS Appl. Mater. Interfaces, 2013, 5, 10418–10422 CrossRef CAS PubMed.
- M. X. Wang, C. H. Yang, Z. Q. Liu, J. Zhou, F. Xu, Z. Suo, J. H. Yang and Y. M. Chen, Macromol. Rapid Commun., 2015, 36, 465–471 CrossRef CAS PubMed.
- H. Daemi, M. Mashayekhi and M. Pezeshki Modaress, Carbohydr. Polym., 2018, 198, 481–485 CrossRef CAS PubMed.
- F. Ma, X. Pang and B. Tang, Carbohydr. Polym., 2019, 206, 229–237 CrossRef CAS PubMed.
- S. A. Shah, M. Sohail, S. A. Khan and M. Kousar, Mater. Sci. Eng., C, 2021, 126, 112169 CrossRef CAS PubMed.
- P. Zare, M. Pezeshki-Modaress, S. M. Davachi, P. Zare, F. Yazdian, S. Simorgh, H. Ghanbari, H. Rashedi and Z. Bagher, Carbohydr. Polym., 2021, 266, 118123 CrossRef CAS PubMed.
- R. Goto, M. Nakahata and S. Sakai, Gels, 2022, 8, 818 CrossRef CAS PubMed.
- S. L. Coburn, K. M. Crossley, J. L. Kemp, S. J. Warden, T. J. West, A. M. Bruder, B. F. Mentiplay and A. G. Culvenor, Osteoarthritis Cartilage, 2023, 31, 144–157 CrossRef CAS PubMed.
- F. Eckstein, B. Lemberger, C. Gratzke, M. Hudelmaier, C. Glaser, K. H. Englmeier and M. Reiser, Ann. Rheum. Dis., 2005, 64, 291–295 CrossRef CAS PubMed.
- F. Eckstein, M. Hudelmaier and R. Putz, J. Anat., 2006, 208, 491–512 CAS.
- N. A. Tzioumis, D. A. Cullen, K. A. Jolliffe and N. G. White, Angew. Chem., Int. Ed., 2023, 62, e202218360 CAS.
- Y. Séréro, V. Jacobsen, J. F. Berret and R. May, Macromolecules, 2000, 33, 1841–1847 Search PubMed.
- G. Marrucci, S. Bhargava and S. L. Cooper, Macromolecules, 1993, 26, 6483–6488 CrossRef CAS.
- S. Suzuki, T. Uneyama, T. Inoue and H. Watanabe, Macromolecules, 2012, 45, 888–898 CAS.
- S. X. Ma and S. L. Cooper, Macromolecules, 2001, 34, 3294–3301 CAS.
- G. Huang, H. Zhang, Y. Liu, H. Chang, H. Zhang, H. Song, D. Xu and T. Shi, Macromolecules, 2017, 50, 2124–2135 CAS.
- S. Q. Wang, Macromolecules, 1992, 25, 7003–7010 CAS.
- A. A. Abraham and A. K. Sen, J. Appl. Polym. Sci., 2010, 117, 2795–2802 CAS.
- H. P. Lee, L. Gu, D. J. Mooney, M. E. Levenston and O. Chaudhuri, Nat. Mater., 2017, 16, 1243–1251 CAS.
- J. B. Fitzgerald, M. Jin, D. Dean, D. J. Wood, M. H. Zheng and A. J. Grodzinsky, J. Biol. Chem., 2004, 279, 19502–19511 CAS.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |