
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional nanostructured composite lithium soap fibers for constructing high-performance lithium metal anode interfacial layers†
Ying
Luo
a,
Shaozhen
Huang
ab,
Jiahua
Liao
a,
Zhibin
Wu
 *ab and
Libao
Chen
*ab
*ab and
Libao
Chen
*ab
aState Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, PR China. E-mail: zhibinwu@csu.edu.cn; lbchen@csu.edu.cn
bFoshan Lifriend New Energy Co. LTD, Foshan, 528244, PR China
First published on 16th April 2025
Abstract
Lithium metal batteries are regarded as promising energy storage devices due to their high theoretical capacity, low density, and low electrode potential. However, the high reactivity of lithium leads to side reactions with the electrolyte and dendritic growth which hinder their commercial application. In this study, a lithium metal anode material with a three-dimensional nanostructured interface was prepared through interfacial modification engineering. The linear flexible molecular chains of the chelating agent, which crosslink with lithium stearate, result in a highly entangled, strong, and flexible three-dimensional porous structure of composite lithium soap fibers. The strong electrostatic interaction between the terminal RCOO− groups and Li+ attracts Li+ ions into the three-dimensional matrix, constructing a lithiophilic protective layer at the micron and nanoscale. This enhances the stability of the SEI layer and the Li+ transport rate. The large specific surface area and abundant pores provide more reaction sites for lithium deposition and stripping. The unique mesh-like molecular structure, with high liquid transport performance, regulates the distribution of the electrolyte and homogenizes the lithium ion flux, effectively suppressing dendritic growth. The Li@DA symmetrical coin cells exhibits a long-term cycling performance of over 6800 hours, and the full battery maintains a high capacity retention of 92.04% after 530 cycles at a 2C rate with a high loading of 12 mg cm−2. This three-dimensional nanostructured interfacial layer holds great potential for advancing the development of high-performance lithium batteries.
Introduction
Since the Sony Corporation launched lithium-ion batteries (LIBs) in 1990, they have been widely used in portable devices. However, limited by the low specific capacity of graphite anodes (372 mA h g−1), LIBs are gradually unable to meet the increasing demand for high energy density storage.1–3 Lithium metal batteries are regarded as an ideal choice for next-generation batteries due to their high theoretical capacity (3860 mA h g−1), low density (0.534 g cm−3), and low electrode potential (−3.04 V vs. SHE).4,5 Lithium has a small atomic radius and only one electron in its outermost shell. Under the shielding effect of the atomic nucleus and inner electrons, lithium atoms can easily lose electrons to form ions. This high reactivity leads to the instability of the solid electrolyte interphase (SEI) layer.6 During the charge–discharge process, the intercalation and deintercalation of Li+ ions cause mechanical stress and volume changes within the electrode material. Since lithium metal anodes undergo “host-free” deposition, the resulting structural collapse and the uneven lithium ion flux at the fractured sites lead to the rampant growth of lithium dendrites, which can penetrate the separator and pose a safety hazard to the battery.7 Therefore, designing a high-performance lithium metal anode interfacial structure layer with uniform lithium ion flux, a high Li+ transport rate, and structural stability is a key focus of current research.In response to the failure mechanisms of lithium metal anodes—unstable SEI films, volume changes, and lithium dendrite growth—researchers have conducted extensive studies. Solid-state electrolytes can effectively prevent the continuous reaction between lithium and liquid electrolytes, but face issues such as thick protective layers, low ionic conductivity, and high preparation costs. Interface engineering has been proven to enhance the stability of the lithium interface to some extent. By coating or constructing special structures on the surface of lithium metal, the growth of lithium dendrites can be effectively suppressed.4 Through designing thinner and lower mass fraction protective layers, the artificial construction of SEI layers can achieve synergistic effects of high lithium ion conductivity, isolation of direct contact between the electrolyte and lithium, inhibition of lithium dendrite growth, and mitigation of electrode volume expansion. These layers meet the demands of low cost and ease of large-scale preparation, thereby significantly improving the performance of lithium metal batteries. Long et al.8 prepared a porous lithium siloxene (LSIS) interfacial framework in situ on the lithium metal anode interface through a mechanical friction reaction between silica and lithium metal. This mixed ionic/electronic conductive interfacial layer significantly reduces interfacial impedance and enhances lithium ion mobility and the uniformity of the electric field distribution. Wu et al.9 designed a polyimide covalent organic framework (COF) anchored on the lithium anode interface to construct an artificial SEI layer for uniform lithium deposition and inhibition of lithium dendrite growth. The Li–COF‖LFP full battery, paired with a commercial LiFePO4 cathode, retained a high capacity of 91% after 150 cycles. However, these interfacial layers still have certain limitations. Inorganic frameworks struggle to accommodate the volume expansion during electrode charging and discharging, while organic frameworks, despite their flexibility, face challenges related to cost and complex preparation processes, hindering their commercial application.
Composite lithium soap fibers (CLSF) are synthesized from three types of chelating agents and named after their initials. The composite lithium soap fibers synthesized using azelaic acid, sebacic acid, and dodecanedioic acid as chelating agents are respectively named AA, SA, and DA based on their initials. These fibers are highly entangled together, with the linear flexible molecular chains of the chelating agents crosslinked with saturated lithium stearate, resulting in a structure with high strength and flexibility.10–12 The composite lithium soap fibers exhibit a multilayer self-helical morphology, and are successively assembled together to form a complex three-dimensional nano/micro porous structure. This structure provides more robust mechanical support. The composite lithium soap fibers have excellent physicochemical properties, showing uniform distribution and good morphology, with a melting point close to 400 °C, indicating extremely high thermal stability. They possess abundant pore structures and a large specific surface area, with that of the composite lithium soap fibers DA reaching 22.55 m2 g−1. The pore structures mainly exist in the form of micropores and mesopores, effectively enhancing the interaction of the adsorbate (electrolyte) with the lithium metal interfacial layer. With a safe and convenient mechananical rolling process, the lithium foil is uniformly stressed overall. The unique three-dimensional structure of the composite lithium soap fibers acts as a surface stress buffer layer, which further facilitates mechanical processing. By mechanically rolling the fibers onto the lithium anode interface, the fibrous framework is grafted to stabilize the electrode structure and provide more reaction sites for lithium plating/stripping. The carboxyl functional groups at the ends of the composite lithium soap fibers anchored on the lithium metal interface have a strong electrostatic interaction with Li+ ions, attracting them into the three-dimensional matrix and forming a lithiophilic protective layer at the micron and nanoscale. The unique mesh-like molecular structure not only provides more reaction sites but also endows the protective layer with high liquid transport performance.13,14 This structure effectively regulates the distribution of the electrolyte, making the lithium ion flux more uniform and further enhancing the stability and cycling performance of the lithium anode, thus promoting the development of high-performance lithium metal anodes.
Results and discussion
Fig. 1a illustrates the preparation process of composite lithium soap fibers (CLSF) and composite lithium anodes (Li@CLSF). Fig. 1b details the chemical synthesis mechanism of CLSF. Three organic acids with different chain lengths, all based on carbon chains were used as chelating agents: Azelaic acid, sebacic acid, and dodecanedioic acid. These acids were dispersed in a mixed solvent of ethanol and deionized water. 1,2-Hydroxystearic acid was heated to dissolve in deionized water, and then mixed uniformly with the acids at a certain temperature. LiOH·H2O was added to allow the flexible linear molecular chains of the chelating agents to crosslink with saturated lithium stearate, forming the composite lithium soap fibers.10,11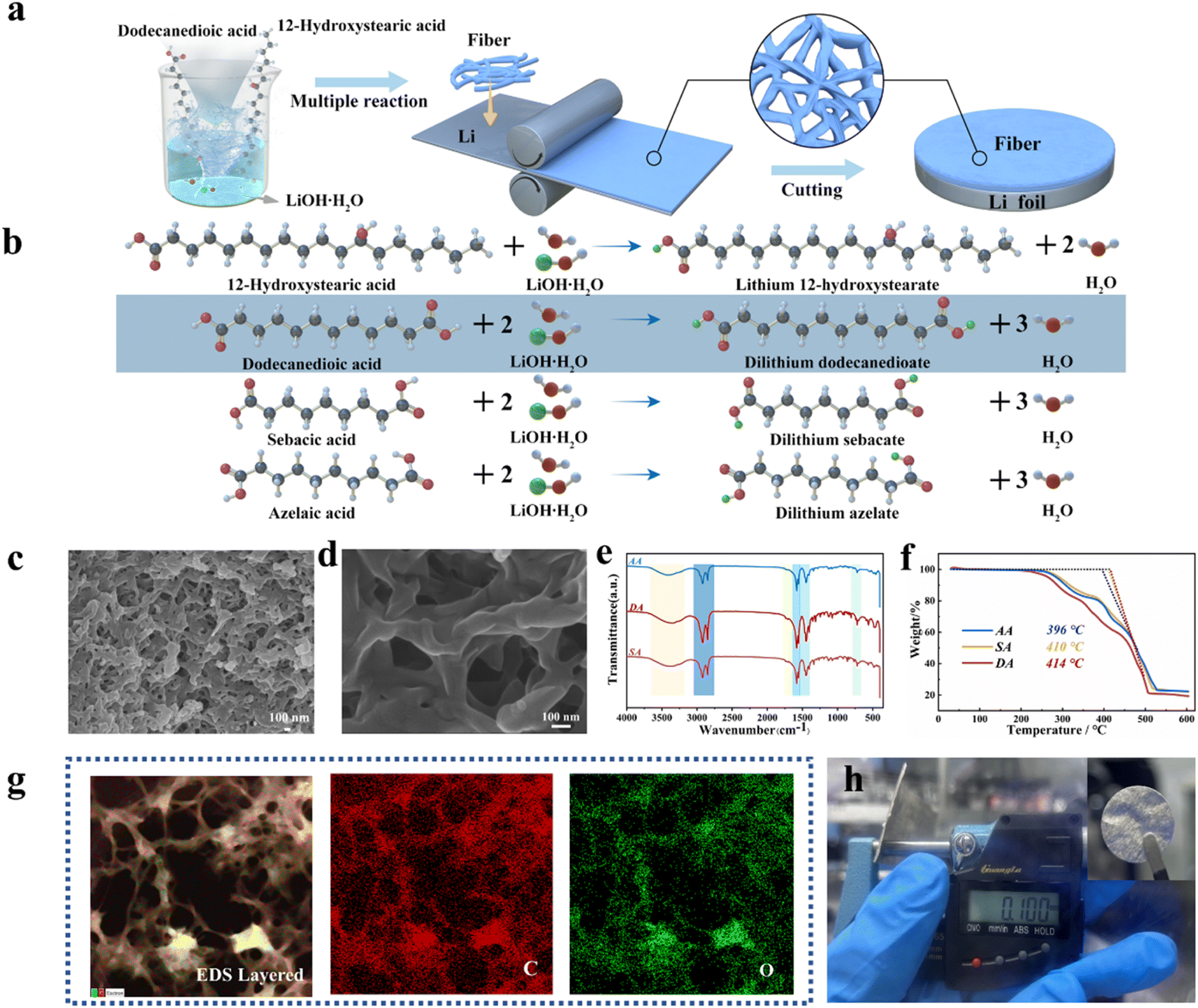 | ||
Fig. 1 (a) Schematic illustration of the preparation of composite lithium soap fibers (CLSF) and the composite lithium anode (Li@CLSF). (b) Schematic illustration of the chemical synthesis reaction mechanism of composite lithium soap fiberd. SEM images of composite lithium soap fiber DA at a scale of 100 nm under different magnifications: (c) 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and (d) 100000. (e) FT-IR spectra of the three types of composite lithium soap fibers. (f) Thermo-gravimetric analysis (TGA) curves of the three types of composite lithium soap fibers. (g) EDS layered image of DA. (h) The thickness of the Li@CLSF composite lithium metal anode, with an optical image of the electrode in the upper right corner. 000 and (d) 100000. (e) FT-IR spectra of the three types of composite lithium soap fibers. (f) Thermo-gravimetric analysis (TGA) curves of the three types of composite lithium soap fibers. (g) EDS layered image of DA. (h) The thickness of the Li@CLSF composite lithium metal anode, with an optical image of the electrode in the upper right corner. | ||
The fibers were then grafted onto the lithium metal interface via mechanical rolling. The original thickness of the lithium foil is ∼180 μm. After mechanical rolling, the thickness of the composite lithium anode can be reduced to 100 μm. This significant reduction in thickness is mainly attributed to the unique structure of the composite lithium soap fibers. The flexible linear structure of the chelating agent molecules in the composite lithium soap fibers is cross-linked with saturated lithium stearate, forming a high-strength and flexible fiber network. This network structure acts as a stress buffer during mechanical processing, allowing the lithium foil to be uniformly thinned without breaking or tearing.10,11,15 On the other hand, the composite lithium soap fibers have good compatibility with lithium metal. When mixed with rolling oil, the composite lithium soap fibers form a lithium-based grease, which effectively reduces friction and wear during mechanical rolling.4,16,17 This interaction not only helps in the uniform thinning of the lithium foil but also ensures the structural stability of the composite lithium anode after mechanical processing. After rolling, the lithium metal was cut into pieces to fabricate the anode material.4,13Fig. 1c and d show the scanning electron microscopy (SEM) images of the dodecanedioic acid-based CLSF (DA) at different magnifications. At 30000 magnification, the porous structure of the composite fibers can be observed. At 100000 magnification, the nanoscale structure of the fibers becomes clearer, showing uniform distribution and good morphology.
After preliminary characterization of the microstructures of the three types of CLSF, Fourier-transform infrared spectroscopy (FT-IR) was performed (Fig. 1e). The presence of O–H bonds, C–H stretching, scissoring, and bending vibrations, as well as vibrations of the CH backbone and COOR groups, was confirmed. This indicates that Li replaces the hydrogen in the carboxyl group, not the hydroxyl group, consistent with the proposed reaction equations and product structures. This confirms the successful synthesis of lithium 1,2-hydroxystearate and lithium organic acid salts. The reaction equations for CLSF can be summarized as follows:10–12
| CH3(CH2)5CH(OH)(CH2)10COOH + LiOH·H2O → CH3(CH2)5CH(OH)(CH2)10COOLi + 2H2O |
| HOOC(CH2)7COOH + 2LiOH·H2O → LiOOC(CH2)7COOLi + 3H2O |
| HOOC(CH2)8COOH + 2LiOH·H2O → LiOOC(CH2)8COOLi + 3H2O |
| HOOC(CH2)10COOH + 2LiOH·H2O → LiOOC(CH2)10COOLi + 3H2O |
To analyze the stability of composite lithium soap fibers, thermogravimetric analysis (TGA) was performed (Fig. 1f). The melting temperatures of AA, SA, and DA were 396 °C, 410 °C, and 414 °C, respectively, indicating good thermal stability.18 Energy-dispersive X-ray spectroscopy (EDS) (Fig. 1g) shows that DA exhibits a three-dimensional nanostructured network, with C and O elements uniformly distributed within the fibers. The three types of composite lithium soap fibers were dissolved in mineral oil, uniformly and thoroughly stirred, and then rolled onto lithium metal. The resulting composite lithium metal anodes were cut into pieces with a thickness of 100 μm (Fig. 1h). The optical image of the electrode piece Li@DA is shown in the upper right corner (Li@DA refers to the lithium anode prepared using dodecanedioic acid as the chelating agent to synthesize composite lithium soap fibers).
To further understand the physicochemical properties and differences of the composite lithium soap fibers, nitrogen adsorption–desorption tests were conducted on the three types of composite lithium soap fibers (AA, SA, and DA) at 77.3 K (Fig. 2a), with the inset showing the pore size distribution curves. The isotherms reveal a combination of Type II, IV characteristics. At low relative pressures (P/P0), the isotherms closely resemble Type II isotherms, which is due to the weak interaction between the adsorbate and the adsorbent in pores smaller than 2 nm (with the electrolyte acting as the adsorbate and the composite lithium metal interfacial layer as the adsorbent). The subsequent gradual curvature of the isotherms indicates the overlap of monolayer coverage and the onset of multilayer adsorption.19 In the range of 0.8 < P/P0 < 1.0, corresponding to the presence of large pores, hysteresis loops (H3 type) are observed, indicating capillary condensation in mesopores and macropores, with the electrolyte acting as the adsorbate on the lithium metal interfacial layer. Upon examining the inset illustrations of the isotherms, it is evident that the pore size distribution of DA exhibits a peak at 3.61 nm, compared to the composite lithium soap fibers AA and SA. This suggests that, in contrast to the other two types of fibers, the pores in DA are more predominantly micropores and mesopores. The bar chart on the far right of Fig. 2a compares the BET specific surface area and the BJH desorption average pore size among the three types of composite lithium soap fibers. Combining the information from the bar chart and the distribution curve in Fig. 2a, it can be concluded that the pore volumes of the three fiber networks are similar, while DA has the largest specific surface area, reaching 22.55 m2 g−1. Compared to the other two types, DA exhibits a higher proportion of microporous and mesoporous structures. Scanning electron microscopy (SEM) was used to characterize the surface morphology and coating thickness of the composite lithium anode. Fig. 2b shows the surface and cross-sectional views of the Li@DA electrode, clearly demonstrating that the DA composite lithium soap fibers were successfully grafted onto the lithium metal surface, with a coating thickness of ∼4 μm.
 | ||
| Fig. 2 (a) Nitrogen adsorption isotherms of the three types of composite lithium soap fibers at 77.3 K, with insets showing their pore size distribution curves. The bar chart on the right displays the BET surface area and BJH desorption average pore diameter (4V/A) of the three types of composite lithium soap fibers. (b) Scanning electron microscope (SEM) images of the surface and cross-section of the Li@DA electrode. (c) Tafel curves of symmetric cells using bare Li and the three types of composite lithium soap fibers as lithium anodes. (d) Electrochemical impedance spectroscopy (EIS) spectra of symmetric cells using bare Li and the three types of composite lithium soap fibers, before and after polarization, for the estimation of the lithium ion transference number. The bar chart on the right shows the lithium ion transference numbers for bare Li and the three types of composite lithium soap fibers. | ||
To evaluate the electrochemical properties of composite lithium anodes, Tafel tests were performed on symmetric cells assembled using lithium anodes prepared from the three types of composite lithium soap fibers (Fig. 2c). The exchange current densities of these electrodes were 0.056 mA cm−2, 0.057 mA cm−2, and 2.4 mA cm−2, respectively, while that of the bare Li electrode was only 0.046 mA cm−2. These results indicate that the composite soap fibers exhibit superior charge transfer kinetics, with Li@DA showing particularly significant improvements. Fig. 2d presents the lithium ion transference numbers for lithium metal batteries prepared with the three composite lithium soap fibers. Among them, the transference number of the Li@DA lithium metal battery was the highest, reaching 0.71, which represents a 37% increase compared to that of bare Li. This value is highly desirable. Wettability tests were conducted on bare Li and Li@DA electrodes by adding a fixed volume of ether-based electrolyte (LS-009). As shown in Fig. S1,† the contact angles of bare Li and Li@DA electrodes were 26.5° and 14.0°, respectively. Li@DA demonstrated a faster wetting rate for the electrolyte and higher affinity for lithium. This can be attributed to the three-dimensional nanostructured network on the surface of the Li@DA electrode, which effectively reduces the interfacial tension between the electrolyte and the electrode, thereby facilitating rapid diffusion and uniform wetting of the electrolyte.14 These electrochemical data indicate that Li@DA exhibits high ionic conductivity. Combining these results with the physicochemical characterization, we speculate that the pore size distribution and specific surface area are the primary factors influencing lithium ion transport. DA has the largest specific surface area, which significantly enhances the exposure of active reaction sites. The presence of micropores and mesopores further endows the interfacial layer with high liquid transport properties, effectively regulating the distribution of the electrolyte and making the lithium ion flux more uniform.
To further investigate the effects of the Li@DA electrode material on lithium plating and stripping behavior during cycling, symmetric cells were assembled and subjected to deposition at different capacities at a current density of 0.25 mA cm−2. After disassembling the cells, the electrodes were cleaned with DME, and the morphology of the electrode materials was observed using scanning electron microscopy (SEM). After delithiation for 6 hours at a current density of 0.25 mA cm−2, the stripping surfaces of bare Li and Li@DA were examined. Fig. 3a and b show that the surfaces of Li@DA were smooth and uniform, with the grafted nanostructured fibers remaining intact on the lithium metal interface. In contrast, Fig. 3c and d reveal significant pores and cracks on the surfaces of bare Li electrodes. On the plating side, lithium deposition on Li@DA was dense and uniform (Fig. 3e and f), while bare Li exhibited dendritic aggregation and irregular deposition (Fig. 3g and h). When the electrodes were first delithiated for 6 hours and then plated for another 6 hours, Li@DA maintained its dense deposition and smooth stripping surface morphology (Fig. S2†). In contrast, bare Li continued to exhibit irregular dendritic growth. After the same amount of stripping and plating, the stripping surface of bare Li did not return to its original state, with pores persisting on the interface (Fig. S3†). These pores further led to non-uniform lithium ion flux, causing dendrites and dead lithium to form continuously during cycling. When the deposition capacity was further increased, Li@DA maintained its excellent plating and stripping morphology, with the nanostructured fibers still firmly anchored on the interface (Fig. S4 and S5†), unlike bare Li, which exhibited irregular deposition and electrode pores. In situ optical microscopy measurements provided a more intuitive reflection of the above phenomena (Fig. 3i). At a current density of 2 mA cm−2, bare Li showed dendritic growth after 20 minutes, and the dendrites continued to grow and enlarge over 60 minutes. Such dendrites can easily penetrate the separator, causing battery short-circuiting and even thermal runaway, which poses a severe safety threat to the battery. In contrast, Li@DA maintained a stable plating state for up to 60 minutes. By stabilizing the solvent interface and homogenizing the lithium ion flux, Li@DA achieved a “dendrite-free” state, confirming its significant advantage in suppressing lithium dendrite formation.
 | ||
| Fig. 3 SEM images of the deposition and stripping of bare Li and Li@DA symmetric cells after delithiation at 0.25 mA cm−2 for 6 hours: (a–d) Li@DA; (e–h) bare Li. (i) In situ optical microscopy measurements of bare Li and Li@DA at 2 mA cm−2. | ||
To verify the impact of volume change on the Li@DA electrode during cycling, symmetric cells were assembled using bare Li and Li@DA electrodes with a thickness of 100 μm. These cells were tested at a current density of 0.25 mA cm−2 to observe the volume change of the lithium battery anode after stripping 3 mA h cm−2 of lithium and after stripping followed by plating 3 mA h cm−2 of lithium. The theoretical formula for the volume change of lithium metal is as follows:20,21
| ΔV = Q/(ρ × C)w |
To gain a deeper understanding of the interaction mechanism between lithium and composite lithium soap fibers, X-ray photoelectron spectroscopy (XPS) was employed to analyze the chemical composition of the interface after the reaction between composite lithium soap fiber DA and lithium. Both bare Li and Li@DA electrodes were etched for 60 seconds. In the C 1s spectra, the bare Li electrode exhibited peaks corresponding to Li2CO3, Li–C, and C–H bonds. In contrast, after the reaction of DA with lithium at the interface, additional peaks for C–C, LiCx, and RCOOLi were observed (Fig. 4a–f and S7†). The presence of C–C is attributed to the carbon chain backbone of the composite lithium soap fibers, while the LiCx peak arises from the interaction between the carbon chain and lithium during the mechanical rolling process (Fig. S8†). The carboxyl groups (–COOH) in the DA framework, due to the electronegativity of oxygen, can form significant electrostatic interactions with positively charged lithium ions (Li+). During the charging and discharging process, the terminal RCOO− groups can effectively attract lithium ions to the lithiophilic centers within the three-dimensional matrix, where they are reduced to lithium metal through charge transfer. This stable electrostatic attraction guides the deposition and stripping of lithium ions on the electrode surface, inhibits dendritic lithium growth, mitigates lithium volume expansion, and dynamically regulates the interface, thereby constructing an organic interfacial protective layer with excellent lithiophilicity at the micron and nanoscale. Fig. 4g illustrates the content of C, O, and Li elements in bare Li and Li@DA electrodes at different sputtering times. From 0 to 60 seconds, the C 1s peak in the bare Li electrode gradually decreased and disappeared, while the C 1s peak in Li@DA remained relatively stable, confirming the formation of the interfacial modification layer in Li@DA. Fig. 4h shows the compositional ratio of the interfacial layer in bare Li and Li@DA electrodes.
 | ||
| Fig. 4 The C 1s X-ray photoelectron spectroscopy (XPS) spectra: (a–c) Li@DA; (d–f) bare Li. (g) Full-scan XPS results of bare Li and Li@DA electrodes. (h) Element ratios of bare Li and Li@DA electrodes at different sputtering times. (i) Voltage profiles of bare Li and Li@DA symmetric cells at a current density of 1 mA cm−2 with a fixed capacity of 1 mA h cm−2. (j) Voltage profiles at different time periods. (k) Voltage profiles of bare Li and Li@DA symmetric cells at a current density of 3 mA cm−2 with a fixed capacity of 3 mA h cm−2. (l) Galvanostatic plating/stripping profiles of bare Li and Li@DA in symmetric cells at increasing current densities from 0.25 to 3 mA cm−2 at a fixed capacity of 0.5 mA h cm−2. | ||
Symmetric cells assembled with bare Li and Li@DA were subjected to galvanostatic charge–discharge tests at various current densities to evaluate their cycling stability and polarization voltage. At a current density of 1.0 mA cm−2 and a capacity of 1.0 mA h cm−2, cyclic testing revealed that the Li@DA electrode exhibited a significantly more stable voltage profile compared to the bare Li electrode throughout the test period, with a stable cycling time exceeding 5000 hours—more than eight times that of the bare Li electrode (Fig. 4i and j). When the current density was increased to 3.0 mA cm−2, the Li@DA symmetric cell demonstrated an impressive long-term cycling performance of over 6800 hours, far surpassing that of the bare Li electrode and achieving a remarkable improvement in the lifespan. Fig. 4l illustrates the polarization voltage behavior of Li@DA at increasing current densities of 0.25, 0.5, 1, 2, 3, and 0.5 mA cm−2 at a fixed capacity of 0.5 mA h cm−2. The results show that the polarization voltage of Li@DA gradually stabilizes with increasing current density and becomes increasingly advantageous at higher current densities. This stable polarization voltage characteristic indicates that the Li@DA electrode maintains good electrochemical performance across different current densities. These findings collectively demonstrate that the three-dimensional network structure formed by the self-helical lithium soap fibers of DA, with its large specific surface area and abundant pore structure,14 significantly increases the number of reactive sites. This not only reduces the nucleation overpotential but also promotes uniform lithium metal deposition, effectively enhancing the cycling stability of the lithium metal electrode.
To further evaluate the commercial applicability of Li@DA, researchers selected LiFePO4 (LFP) as the cathode to assemble full cells and conducted electrochemical performance tests. Fig. 5a and b show the cycling performance of bare Li and Li@DA full cells at a high loading of 12 mg cm−2 at a 2C rate. It can be observed that the Li@DA‖LFP cell showed a high capacity retention of 92.04% after 530 cycles, while the bare Li‖LFP cell showed a capacity retention of only 33.34% after 200 cycles. In terms of rate performance, Li@DA also demonstrated superior characteristics, as shown in Fig. 5c. At varying rates of 0.2–0.5–1–2–4–0.2C, the Li@DA electrode exhibited higher discharge specific capacities compared to the bare Li electrode. Fig. 5d and e display the charge–discharge curves of bare Li‖LFP and Li@DA‖LFP at different rates, with Li@DA maintaining stable charge–discharge plateaus throughout the testing period. The significant improvement in rate performance is attributed to the unique interfacial structure of Li@DA. The interfacial layer has a high Li+ diffusion coefficient and low charge transfer resistance. Additionally, the carbon chain structure within Li@DA interacts significantly with lithium ions through electrostatic forces, optimizing the lithium ion transport pathways. This three-dimensional nanostructured buffer layer enables the Li@DA anode to exhibit excellent cycling and rate performance, making it a promising candidate for high-performance lithium metal batteries.
 | ||
| Fig. 5 (a) Cycling performance of bare Li‖LFP and Li@DA‖LFP cells at a 2C rate. (b) The charge–discharge profiles of cells at 2C after cycling of Li@DA. (c) Cycling performance of bare Li‖LFP and Li@DA‖LFP cells at different rates. The charge–discharge profiles at different rates: (d) bare Li‖LFP; (e) Li@DA‖LFP. | ||
Conclusions
In summary, a lithium metal anode material with a three-dimensional nanostructured interface was successfully prepared through interfacial modification engineering. This interface layer, composed of lithium stearate crosslinked by flexible molecular chains and lithiophilic soap fibers, forms a highly entangled, strong, and flexible three-dimensional porous structure. By mechanically rolling the composite lithium soap fibers onto the lithium metal surface, the strong electrostatic interaction between the terminal RCOO− groups of the fibers and Li+ ions attracts Li+ into the three-dimensional matrix, constructing a lithiophilic protective layer at the micron and nanoscale. This enhances the stability of the SEI layer and the Li+ transport rate. The large specific surface area and abundant pores provide more reaction sites for lithium deposition and stripping. The unique mesh-like molecular structure, with its high liquid transport performance, regulates the distribution of the electrolyte and homogenizes the lithium ion flux, thereby stabilizing the electrode structure and providing more reaction sites for lithium deposition and stripping. The Li@DA symmetrical cell demonstrated an impressive long-term cycling performance of over 6800 hours. When paired with LFP in a full cell, Li@DA showed a high capacity retention of 92.04% after 530 cycles at a 2C rate with a high loading of 12 mg cm−2. The three-dimensional nanostructured composite lithium soap fiber interfacial layer developed in this study offers a new strategy for high-performance lithium metal anodes and holds great potential for application in high-energy-density battery systems.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ying Luo: experimental design, data analysis, data processing, thesis writing; Shaozhen Huang: data analysis, data processing; Jiahua Liao: data analysis; Zhibin Wu and Libao Chen: project conceptualization, data analysis, financial support, scientific discussion.Conflicts of interest
The authors declare no conflicts of interest. They have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was financially supported by the Natural Science Foundation of Hunan Province (2024JJ4058), the National Natural Science Foundation of China (U24A20501 and 52471166) and the Innovation-Driven Project of Central South University (1053320230998).References
- Y. X. Zuo, B. A. Li, N. Jiang, W. S. Chu, H. Zhang, R. Q. Zou and D. G. Xia, Adv. Mater., 2018, 30, 5 Search PubMed.
- S. Q. Zhang, N. S. Andreas, R. H. Li, N. Zhang, C. C. Sun, D. Lu, T. Gao, L. X. Chen and X. L. Fan, Energy Storage Mater., 2022, 48, 44–73 Search PubMed.
- C. Li, X. Jiang, H. Qi, D. Chen, T. You, S. Huang, H. Yu, Y. Huang, M. Rao, G. Li, B. Xu, Y. Chen and L. Chen, Energy Storage Mater., 2025, 75, 104012 CrossRef.
- S. Huang, K. Long, Y. Chen, T. Naren, P. Qing, X. Ji, W. Wei, Z. Wu and L. Chen, Nano-Micro Lett., 2023, 15, 235 Search PubMed.
- W. Y. Chen, R. Salvatierra, J. T. Li, D. X. Luong, J. L. Beckham, V. D. Li, N. La, J. A. Xu and J. M. Tour, Adv. Mater., 2022, 34, 12 Search PubMed.
- Z. Wang, Z. He, Z. Wang, K. Long, J. Yang, S. Huang, Z. Wu, L. Mei and L. Chen, Chem. Sci., 2025, 16, 3571–3579 Search PubMed.
- W. J. Ren, X. C. Shang, Y. Lin, H. Ren, L. C. Zhang, H. Su, Q. Li, L. J. Zhi, M. B. Wu and Z. T. Li, Adv. Energy Mater., 2024, 2405284 Search PubMed.
- K. Long, S. Huang, H. Wang, Z. Jin, A. Wang, Z. Wang, P. Qing, Z. Liu, L. Chen, L. Mei and W. Wang, Energy Storage Mater., 2023, 58, 142–154 Search PubMed.
- X. Wu, S. Zhang, X. Xu, F. Wen, H. Wang, H. Chen, X. Fan and N. Huang, Angew. Chem. Int. Ed., 2024, 136, e202319355 Search PubMed.
- H. Yan, M. Cai, S. Song, Y. Huang, X. Fan and M. Zhu, Prog. Org. Coat., 2022, 163, 106604 CrossRef CAS.
- G. Ren, C. Zhou, S. Wang, X. Fan, Y. Han and G. Jin, Tribol. Int., 2022, 175, 107826 Search PubMed.
- N. Xu, W. Li, M. Zhang, G. Zhao and X. Wang, RSC Adv., 2015, 5, 54202–54210 RSC.
- Y. X. Ren, L. Zeng, H. R. Jiang, W. Q. Ruan, Q. Chen and T. S. Zhao, Nat. Commun., 2019, 10, 3249 CrossRef CAS PubMed.
- Z. Su, J. Zhang, J. Jin, S. Yang and G. Li, Chem. Eng. J., 2022, 430, 132865 Search PubMed.
- J. Ren, K. L. Gong, G. Q. Zhao, X. H. Wu and X. B. Wang, Tribol. Lett., 2021, 69, 166 CrossRef.
- M. Liu, B. Liu, P. Zhao, X. Xiong, H. Jing, C. Liu, J. Shu, Z. Wu, X. Xia, W. Lei and Q. Hao, Chem. Eng. J., 2025, 503, 158640 Search PubMed.
- N. Xu, W. Li, M. Zhang and X. Wang, J. Ind. Eng. Chem., 2017, 46, 157–164 Search PubMed.
- C. Zhou, G. Ren, X. Fan and Y. Lv, J. Ind. Eng. Chem., 2022, 111, 51–63 Search PubMed.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem. Int. Ed., 2019, 58, 849–853 Search PubMed.
- X. Liu, K. Long, P. Qing, S. Huang, P. Xiao, C. Ling, Z. Wu and L. Chen, Sci. China Mater., 2023, 66, 4349–4356 CrossRef CAS.
- K. Yoo, S. Banerjee and P. Dutta, J. Power Sources, 2014, 258, 340–350 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01994j |
| This journal is © The Royal Society of Chemistry 2025 |