
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of allosteric sites and ligand-induced modulation in the dopamine receptor through large-scale alchemical mutation scan†
Lisa
Schmidt
 * and
Bert L.
de Groot
* and
Bert L.
de Groot
Max Planck Institute for Multidisciplinary Sciences, Department of Theoretical and Computational Biophysics, Group of Computational Biomolecular Dynamics, Goettingen, Germany. E-mail: bgroot@gwdg.de
First published on 22nd April 2025
Abstract
G protein coupled receptors, particularly class A GPCRs are arguably the most important class of membrane receptors and preferred targets for drug development. Despite extensive research on how ligands modulate the receptor response, discovering new, highly specific ligands remains challenging. However, finding residues outside the conserved microswitches that affect the active–inactive state equilibrium and are specific for a certain receptor, can be beneficial for the design of ligands with higher receptor selectivity. Focusing on the human dopamine receptor 2 (DRD2), we uncover crucial residues for the activation modulation using alchemical non-equilibrium free energy calculations. Our findings match with literature on activation microswitches and experimental studies, while also uncovering novel important residues. Further, we analyzed mutation-induced changes in residue contact networks and found that modulating these networks can lead to a stabilization of the respective opposite state, an effect that could as well be achieved by well-engineered (small) ligands. This way we provide insights into the mechanism of action of the well-known drugs risperidone and bromocriptine and showcase on these two examples how our data can be used for the design of new ligands.
1 Introduction
G protein-coupled receptors (GPCRs), particularly class A GPCRs, represent the largest group of membrane receptors in eukaryotes. They are not only highly abundant but also exceptionally diverse in their ability to modulate cellular responses to extracellular signals, such as ligand binding.1–5 Among these, the aminergic GPCRs, and in particular the human dopamine receptor 2 (DRD2), play a crucial role in regulating physiological behavior.6–9Structurally, GPCRs consist of a seven-transmembrane (7TM) helix bundle, which can be divided into three regions based on membrane topology: (i) the extracellular cleft (ECC), which contains ligand-binding sites, including the orthosteric binding site (OBS) and extracellular loops; (ii) the intracellular cleft (ICC), which houses G protein binding sites and intracellular loops; and (iii) the transmembrane (TM) region, which connects the ECC and ICC and is essential for signal transduction.1,3,9–11
GPCR activation is a highly coordinated allosteric process, in which local conformational changes of individual residues propagate through the receptor, leading to macroscopic conformational and functional transitions.3,12–18 A key hallmark of activation is the outward movement of transmembrane helix 6 (TM6), which opens the ICC for G protein binding.2,3,13,19
Several microswitches, consisting of critical residues and residue clusters, play pivotal roles in the activation process by altering intramolecular interactions through side-chain rotamer switching.1–3,6,10,13,20,21 Among the most functionally significant microswitches are the CWxP motif, the PIF motif, the sodium activation pocket, the NPxxY motif and the DRY motif, which collectively regulate GPCR activation and signaling.2,3,8,13,19–23
Active and inactive receptor structures differ not only in the conformation of TM6 but also in the organization of their ligand-binding pockets.1,5,8,9,24 Consequently, ligand binding to a specific pocket stabilizes the corresponding conformational state. Since ligands can be engineered to induce contact network rearrangements similar to those caused by mutations, understanding how mutations influence the active–inactive equilibrium is critical—not only for elucidating their role in disease mechanisms but also for guiding structure-based ligand design.1,3,8–10,12,14,21,23,25
To assess whether mutations impact the active–inactive state equilibrium, we compute the relative free energy differences of mutation (ΔΔG) between the active and inactive states using non-equilibrium alchemical free energy simulations with pmx.26–30 In our study, we investigate 78 different residues within the receptor domain and calculate ΔΔG values for mutations to eight different amino acids, thus covering a broad chemical spectrum: small non-polar (alanine, glycine), small polar (serine, asparagine), large non-polar (leucine), large polar (glutamine), and aromatic (phenylalanine, tyrosine).
We analyze overall trends in mutant behavior alongside structural differences between the active and inactive states. Following the definition of allostery from Weinkam et al., we classify mutation sites as allosteric if they affect the active–inactive equilibrium.17,18 Furthermore, we show that our approach aligns with previously identified activation-modulating sites, while also uncovering previously unrecognized residues that are important for the active–inactive equilibrium. Because these novel allosteric mutation sites are not conserved yet in accordance with experimental data, we propose that they may serve as potential sites for receptor subtype selectivity, making them promising targets for structure-based ligand design.
The diversity of our mutational scan enables us to investigate the role of residue sidechain properties in stabilizing either the active or inactive state. Our findings provide a mutational database for future investigations into DRD2 activation mechanisms, as well as insights into disease-associated receptor malfunctions. Additionally, we analyze how mutations alter residue interaction networks, an effect that could similarly be induced by small, highly specific ligands, providing a foundation for ligand design studies.
Finally, we compare mutation-induced residue interaction changes with residue positions in experimentally resolved receptor–ligand complexes of the active (bromocriptine-bound, PDB 7jvr31) and inactive (risperidone-bound, PDB 6cm4 (ref. 24)) receptor states. These examples demonstrate how our database can be leveraged to understand ligand function and aid structure-based drug design.
2 Methods
2.1 Structure selection and system setup
There are 6 structures of DRD2 (ref. 24 and 31–35) in the protein database (RCSB PDB8,36). Out of these, a structure for the inactive (I) and the active (A) state of the receptor at the highest available resolution were chosen, namely 6cm4(I)24 and 7jvr(A)31 with 2.9 Å and 2.8 Å resolution respectively. Since DRD2 is a transmembrane protein, the OPM server37 was used to orient the complex in the lipid bilayer.Non-wt residues in 6cm4 (ref. 24) reverted to the wild type (A3.40,122I, A6.37,375L, A6.41,379L) with the mutate tool of PyMOL.
Missing residues in 6cm4 at position 139–144 were added to the structure by copying the coordinates from 7jvr and the unresolved parts of C and N-terminus were deleted in both structures. The TM5 and TM6 helices are connected by a large unstructured loop (ICL3), that is not completely resolved due to its high flexibility. This loop was completely deleted (V5.72,223-Q6.27,365), leading to a separation of the full receptor into two subunits (S1 TM1-5, res N1.33,35-R5.71,221; S2 TM6-7 res Q6.27,365-L8.58,441).
Both the inactive and the active receptor were capped with ACE (acetylated N-terminus) and CT3 (methylated C-terminus) on the N- and C-terminal, respectively, on each of the two subunits with the CHARMM-GUI online server38 to remove charge at the terminal ends of the amino acid chain. The simulation system, including the lipid bilayer, water and ions were constructed using the online server CHARMM-GUI38–40 with the CHARMM36m force field.41 POPC (1-palmitoyl-2-oleoyl-phosphatidylcholine)39 and TIP3P42 were chosen for the lipid and the water model respectively. Sodium and chloride ions were added to the system by distance placement method up to a concentration of 150 mM to neutralize the total charge of the system and achieve a NaCl concentration at physiological level.
2.2 System minimization and equilibration of the wild type receptor
All simulation steps were carried out using GROMACS (2022.3)43–45 and the CHARMM36m force field41 with the TIP3P water model42 at 310.5 K. The system was energy minimized by steepest decent until machine precision and equilibrated in six steps with gradually decreasing restraints on atom positions and molecular dihedrals. Coulomb and van der Waals potentials were calculated with a cut-off length of 1.2 nm for the short range interaction and particle-mesh Ewald46 for the long range interactions. To ensure a smooth switch between short and long range interaction potential functions the force-switch vdW-modifier and the potential-shift Coulomb-modifier were used. The LINCS algorithm47 was applied to constrain covalent hydrogen-bonds involving hydrogen atoms (dt = 2 fs).In the first two equilibration steps (NVT) the Berendsen thermostat48 with a coupling time of 1 ps at 310.5 K was used for temperature coupling. For the following equilibration steps (NPT) the Berendsen thermostat and barostat48 with a coupling time of 5 ps was used for semiisotropic pressure coupling. This was followed by a 2000 ns production run (NPT) with Nose–Hoover thermostat (coupling time 1 ps) and semiisotropic Parrinello–Rahman barostat (coupling time 5 ps) for temperature and pressure coupling respectively. For systems with position restraints on all Cα atoms of the protein backbone and all Na+, position restraints were applied in all simulation steps with a force constant in x, y and z of FCx,y,z = 1000 kJ mol−1 nm−2.
2.3 Setup for alchemical transitions with PMX
For all simulations that applied alchemical transitions PMX27,28 was used to introduce mutations, create hybrid structures and topologies following the protocol from Aldeghi et al.26The mutated system was energy minimized by steepest decent and equilibrated for 500 ps at constant temperature (310.5 K) and pressure (1 bar) using the Langevin thermostat49 and Berendsen barostat48 for temperature and pressure coupling. This was followed by a 50 ns equilibrium simulation run with Langevin thermostat and Parrinello–Rahman barostat.
The last 20 ns of the equilibrium run were used to extract 200 start frames for the nonequilibrium transition reactions, with a spacing of 100 ps between each frame. The nonequilibrium simulations were run for 100 ps with Langevin thermostat for temperature and Parrinello–Rahman barostat for pressure coupling.
For all wt systems λ was set to 0, while for all mutant systems λ was set to 1. A cut-off of 1.0 nm and particle-mesh Ewald46 was applied for the short range and long range interactions for the calculation of Coulomb and van der Waals potentials, respectively. For the alchemical transitions soft-core potentials were used for the non-bonded interactions.50 Covalent bonds involving hydrogen atoms were constrained with the LINCS algorithm47 (dt = 2 fs). To avoid transitions between the active and the inactive state, position restrains were applied to all Cα atoms of the receptor backbone and all Na+ (FCx,y,z = 1000 kJ mol−1 nm−2) for all simulation steps. The Na+ were restrained outside of the receptor, in a way that no interactions with the receptor were possible.
2.4 Calculation and analysis of relative free energy changes and errors
Using PMX the work values from the nonequilibrium transitions were extracted and BAR was used for calculation of the free energy difference for both the active and the inactive state. The obtained values for ΔGmutationactive and ΔGmutationinactive were used to compute the relative free energy change between the systems.| ΔΔGmutationactivation = ΔGmutationactive − ΔGmutationinactive | (1) |
As already reported by Gapsys et al.51 and also observed in our tests with replicas the pmx bootstrap error is underestimating the standard error obtained by multiple repetition. Because we wanted to run a large mutation scan and were more interested in ΔΔG trends than absolute values, we decided to use the pmx bootstrap error without replicas for our error estimation of the ΔΔG.
3 Results and discussion
3.1 Comparison of the active and the inactive state simulations
We simulated the receptor starting from the experimental structures in the active (PDB 7jvr31) and the inactive (PDB 6cm4)24 state for 2000 ns without ligands or the G protein. Analysing the first two principal components (PC1, PC2; ∼60% of the total variance) of both state simulations we found that DRD2 moved away from the respective starting configurations towards a seemingly new state (possibly intermediate).13 Especially the active state showed large movements in PC1 which correspond to a closing of the intracellular cleft and almost complete transition to the inactive state structure (see Fig. 1). Since we simulated the receptor without ligands we did expect that the active state configuration would be more unstable in our simulations. This matches experimental and computational findings, that observed an unstable active receptor state without a bound G protein and/or agonist.13,19,21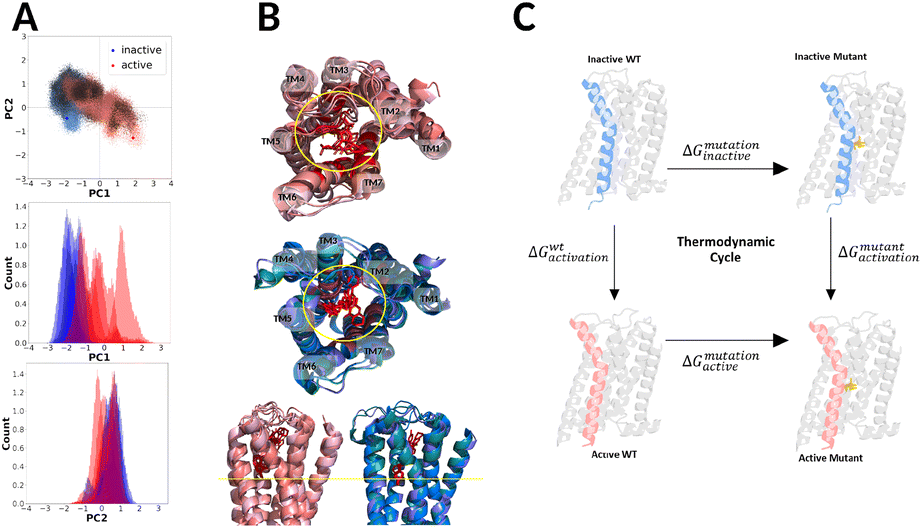 | ||
| Fig. 1 PCA (A), structural alignment of pdb structures for DRD2 (B) and the thermodynamic cycle of the activation and mutation of the dopamine receptor (C). The PCA (A) (active state simulations red, inactive state simulations blue) analysis shows different behaviour in inactive and active state. Alignment of DRD2 pdb structures shows different ligand positions for active (red) and inactive (blue) state (B). The ligand and residues reported to interact with the ligand are colored in red. Antagonists reach deeper into the cavity ((B), bottom). The thermodynamic cycle used is this study is shown in (C). | ||
We used force distribution analysis (FDA)52 to analyze the communication of residues within the receptor. Here we noticed large changes in the active and the inactive inter-residue interactions especially in the extracellular cleft (ECC). In the active state we detected attractive forces between residues in ECL2 and ECL1 (mainly W23.50,100) as well as the extracellular top of TM1, that were not observed in the inactive state. Here ECL2 is acting like a lid thereby not only partially covering the ECC but also bringing TM5 and TM2 closer together32 (see ESI Fig. 1†). However, in the inactive state ECL1 shows more attraction towards the extracellular ends of TM6 and TM7 as well as ECL3, than in the active state. Because of this changed communication in the ECC the cavity openings are slightly different in the active and the inactive state. While the ECC is less deep in the active state and covered by the ECL2, the inactive cavity protrudes deeper into the receptor.24,31–35
This change in ECC opening and communication between the active and the inactive state might also explain the different interactions of agonists and antagonists as reported in the experimental pdb structures of ligand bound D2 receptors.24,31–35 While most conventional antagonists (stabilizing inactive state) bind deeper in the ECC24,33 and form contacts to TM2, TM3, TM5, TM6 and TM7, agonists mainly form contacts to TM3, TM5, TM6 and TM7.31,34,35
3.2 Mutation scan over the whole receptor uncovers important residues for the stabilization of the active/inactive state
To gain a deeper understanding of the role of different residues in stabilizing the active and inactive states, we performed a mutation scan and calculated the relative mutation free energy differences using the pmx Python library27,28 with the GROMACS simulation software (version 2022).43–45 The thermodynamic effects of mutations on protein state stabilization can be studied by calculating the relative free energy of mutation between the wt state and the mutated state (Fig. 1, horizontal arrows, eqn (1)). This mutation relative free energy was calculated by running alchemical non-equilibrium free energy calculations with pmx.26–28 Then, to examine whether a mutation stabilizes one state over the other, the relative free energy difference of mutation activation can be calculated from the difference of the ΔG between the two states (Fig. 1). This way we show directly whether a mutation favors the active over the inactive state. To prevent our receptor from spontaneous transitions between the active and inactive states during the free energy calculation setup we used position restraints on all Cα backbone atoms. Since Na+ has been shown to have a stabilizing effect on the inactive state3,53–55 we also restrained all Na+ ions outside the receptor. This enabled us to examine the effects of mutations on sidechain interactions within the receptor in detail.We were mostly interested in finding mutants with a high impact on the state equilibrium and therefore only considered mutations that show a ΔΔG of more than ±10 kJ mol−1. Residues with a ΔΔG of smaller than −10 kJ mol−1 (stabilization of the active over the inactive state) for multiple mutations were defined as strongly mutation activating, while those that showed a ΔΔG of larger than +10 kJ mol−1 (stabilizing inactive over active state) for multiple mutations were termed strongly mutation inactivated. For residues with ΔΔG values smaller than −10 kJ mol−1 and larger than +10 kJ mol−1 depending on the mutation we use the term ambivalent residues.
Since the FDA analysis showed larger changes in the ECC region, we started our scan in this region (see ESI Section 1†) but quickly went for a larger scan, which included 78 different residues within the whole receptor domain that we mutated to 8 different amino acids. The chosen residues include known microswitches,2,3,8,20 highly conserved residues in class A GPCRs (see ESI Sections 2 and 3) and mutants that showed effects in experimental/clinical studies from the GPCR database (GPCRdb).56
Out of the 78 sampled residues 40 residues showed high ΔΔG values for more than one mutant. Most of these residues were either strongly mutation inactivated (18) or showed ambivalent ΔΔG values (13). Only 9 residues were strongly mutation activated. The higher occurrence of inactivating mutations matches with experimental and clinical studies that characterize many point mutations as inactivating or showing higher affinity for antagonists9,24,56,57 (Fig. 2).
 | ||
| Fig. 2 Results of the ΔΔG scan. The mutation matrix with the ΔΔG trends (trends: mutation inactivating in blue, mutation activating in red, ambivalent in yellow) is shown in (A). The percentual distribution of ΔΔG trends of the scanned residues is shown in (B). The amount of conserved residues ((C) left green) and their ΔΔG trends ((C) right). (D) Shows the distribution of multiple high ΔΔG residues (green) depending on their position relative to the center. The separation in inner, middle and outer circle correspond to the separation in inward, tangential and outward oriented residues. (E) Shows the residue ΔΔG trend distribution of sampled residues in the outside pocketome. In (F) the z-axis dependence (membrane normal) of the percentage of multiple high ΔΔG residues is plotted (see also ESI†). The 6 layers of the DRD2 correspond to a slicing of the receptor along the x, y-axis and starting at the ICC (1) and ending at the ECC (6). The first panel shows the total distribution of multiple high ΔΔG values (green) while the distributions for the mutation inactivating (2nd panel, blue), the ambivalent (3rd panel, yellow) and the mutation activating residues (4th panel, red) are plotted in the following panels. | ||
3.3 Comparison to literature data shows good agreement
Our predictions aligned well with mutant data from the GPCRdb,9,24,56,57 where changes in agonist/antagonist binding served as the readout. To be able to compare this data with our predictions we defined an increase in agonist binding upon mutation as activating, an increase in antagonist binding as inactivating. Of the 34 residues with available data, 28 agreed with reported trends, while 6 showed opposing trends to our predictions (see ESI Section 4†). Further, we aligned sequences from different class A GPCRs (see ESI Section 3†) and examined if residues with high ΔΔG values are conserved. Among the 78 sampled residues, 16 were highly conserved within this GPCR family. Of these, 13 displayed multiple high ΔΔG values, mostly stabilizing the inactive state (5) or showing ambivalent ΔΔG (6). Only 2 residues showed a strongly mutation activating trend. Interestingly, C6.47,385 and W6.48,386, both strongly conserved in class A GPCRs and part of the CWxP motif, did not show multiple high ΔΔG values, despite their significance as microswitches.3,8,20To examine whether our scan was able to find other microswitch residues, we compared our data to the literature on known microswitches in the class A GPCR family.2,3,8,20 We detected multiple high ΔΔG values for sampled residues from almost all reported microswitches, including the PIF motif (F6.44,382, I3.40,122 both strong mutation inactivated), the Na+-pocket (S3.39,121, N7.49,422 both strong mutation inactivated, N7.45,418 ambivalent ΔΔG values), the NPxxY motif (I3.46,128 ambivalent ΔΔG values, Y7.53,426 strong mutation activated) and the DRY motif (Y3.51,133 strong mutation activated). The only motif where we did not detect any residues with multiple high ΔΔG is the CWxP motif.
Both the PIF motif and the Na+-pocket are triggered early in the activation process2,3 and found closer to the extracellular side of the receptor. Conversely, the NPxxY and DRY motifs play a role later in the activation process (according to the ligand-first theory) and are located closer to the intracellular side.3 Notably, we observe a similar separation of sampled microswitch residues into strong mutation inactivated (early microswitches) and strong mutation activated (late microswitches). This suggests that changes in the interaction pattern of the late, intracellular microswitches lead faster to activation, possibly by weakening interactions near the ICC region and G protein binding sites. In contrast, changes in interaction patterns of earlier microswitches often stabilize the inactive state, likely by weakening interactions in the deeper ECC. This agrees with the observation that the ICC is more open in the active and more closed in the inactive state.3,10,21,24,31 The separation of microswitch residues in more mutation activated and more mutation inactivated matches with a distribution analysis of high ddG residues along the membrane normal were we detected an increase of mutation activated residues at the ICC, while mutation inactivated and ambivalent residues are found more often at the central receptor transmembrane region (Fig. 2F, ESI Section 2†).
The CWxP motif is the first motif that is triggered in the activation process.3,12,19,58 Of course it is possible that the sampled residues from this motif that are not displaying multiple high ΔΔG are false negatives. However, the CWxP motif is important in a very early stage of the activation process and is especially important in ligand binding and Na+ interaction.3,53 We did not probe for ligand or Na+ binding effects in this scan, which might be a possible explanation for why we failed to detect any multiple high ΔΔG values for the CWxP residues here. Notably, an alanine scan conducted in the presence of the antagonist risperidone bound to the inactive state in the experimental binding pose (PDB 6cm4)24 shows significant signals for the W6.48,386A and C6.47,385A mutants, both part of the CWxP motif (see ESI Section 6†).
3.4 Residues with high ΔΔG values also show larger changes in the chemical environment built by the surrounding residues
We compared the experimental crystal structures of active (PDB 7jvr31) and inactive (PDB 6cm4)24 state of DRD2 by structure alignment using the MDAnalyis toolkit.59,60 To investigate whether the extent of structural change influences the likelihood of a residue displaying multiple high ΔΔG values, we calculated the distances between the Cα atoms in the aligned structures and compared these to our sampled residues. However, we found no correlation between the Cα distances and the occurrence of multiple high ΔΔG residues.As established, residues within the intracellular part of TM6 undergo significant alterations in their surrounding environment upon activation.3,20 To explore if similar environmental differences occur elsewhere in the receptor, we extracted possible interaction partners within a 4 Å radius per residue and compared them in the active and the inactive state crystal structures (DBASS, distance based alignment of structures and sequences). While most significant changes were observed in TM6, as anticipated, a few residues in other receptor regions also showed distinct environments between the active and inactive states. Notably, environmental changes were observed in every transmembrane helix, with TM7 showing the highest number of alterations per secondary element. Most of these residues displayed high ΔΔG values upon mutation, which emphasizes the correlation between protein-residue environment, contact patterns, and stabilization effects induced by mutation. Only four residues showed large environmental changes without multiple high ΔΔG values (see ESI Fig. 5†).
3.5 Residues with large mutation effects can be found at allosteric ligand binding pockets in the outer receptor region
When analyzing the residue positions in the receptor structures, we not only assessed their orientation relative to the membrane and differences in Cα between the active and inactive state, but also their positioning relative to the receptor center. Residues with the Cα pointing towards the receptor center were classified as “inside”, those with Cα pointing to the membrane were classified as “outside” and residues whose Cα was on the side of the transmembrane helices, pointing neither directly inward or outward were defined as “tangential”. Interestingly, among the sampled residues, 14 out of 26 inside residues exhibited multiple high ΔΔG values, compared to only 3 out of 18 outside residues and 9 out of 15 tangential residues. This suggests that mutations particularly affect residues forming large contact networks within or tangential to the receptor center. Additionally, we observed 4 residues whose location relative to the receptor center differed between the active and inactive states. As expected, almost all (3) of these residues displayed multiple high ΔΔG values upon mutation, which supports the change of contact pattern hypothesis.Recently, the pocketome on the outer regions of various GPCRs has gained more attention, particularly since these pockets can be targeted by small ligands to allosterically influence the receptor response.61 Comparing our findings to known pockets of class A GPCRs, we identified 23 out of 41 sampled residues with multiple high ΔΔG (7 strongly mutation activated, 8 strongly mutation inactivated and 8 ambivalent residues) located at the pockets. Most residues were found in OS5 as well as KS4, KS5, KS6 and KS7 at TM3 and TM5 (see Fig. 3 and ESI Section 5†). These findings again highlight the importance of these pockets for the regulation of the active–inactive state equilibrium of GPCRs. Further, they show that modulating interaction patterns in these pockets can have a big impact on the state-equilibrium in GPCRs.
 | ||
| Fig. 3 Results of the pmx scan (A), the microswitch residues sampled in the mutation scan (B) and the novel state-equilibrium affecting residues (C) mapped to the structures. Residues with multiple high ΔΔG values are colored in red (strong mutation activating), blue (strong mutation inactivating) and yellow (ambivalent ΔΔG values) in (A and C). The residues from the microswitches CWxP, PIF/PIW, sodium activation pocket, NPxxY and DRY, that were sampled in the mutation scan are colored as indicated in (B). | ||
3.6 Mutational scan uncovers previously unknown activation modulating sites
As mentioned before, some of our multi-high residues are not conserved and do not belong to the previously described microswitch residues. However, mutation of these residues shows a clear effect on the active–inactive state equilibrium (see Fig. 3). Out of these 22 residues experimental data is available for 10 residues and we detect a clear match with the experiment for 7 residues. For the 12 remaining residues we were not able to find experimental data for comparison (see ESI Section 5†). Among these, 4 residues displayed strong mutation-induced activation, 5 showed strong mutation-induced inactivation, and 3 exhibited ambivalent trends. These residues are mainly located at the transmembrane regions of the receptor and oriented towards the center of the receptor (inside) or tangential (see ESI Fig. 5†). Moreover, 7 of these residues are located within outside binding pockets (M6.36,374 KS8, V6.43,381 KS7, M1.55,57 OS2, M3.35,117 KS3, T3.52,134 KS5, M4.45,155 KS5, N175 KS5) and might be important allosteric regulation sites. Since these 22 residues are not conserved among class A GPCRs and do not belong to known microswitches but still show good agreement with experimental data (if available) they might be important for DRD2 receptor subtype selectivity.3.7 Mutation scan reveals altered residue contact patterns that can be used for rational ligand design studies
To understand the effects of the point mutations on the sidechain interactions within both the active and the inactive state further, we extracted the sidechain interactions of the mutated residue with other residues over time in both states. Then, we summed all contacts per residue over time and calculated the difference between the active and the inactive contacts per mutant. In the following, we grouped the mutant summed contacts by ΔΔG (and chemical features if necessary) and extracted the most visited contacts per group and state (see Fig. 4A for a schematic overview). Comparing the resulting mutant-residue contacts of different groups we were able to identify crucial interactions that lead to altered state-stabilization upon mutation. On the example of V3.29,111, I45.51,183 and I45.52,184 for bromocriptine binding as well as L2.46,76, F6.44,382, F5.51,202, S3.39,121 and N7.49,422 for risperidone binding we show how our method can be used to understand the roles of mutations for stabilization of the active or the inactive state. All mentioned ligand interactions correspond to the experimentally solved ligand binding poses of bromocriptine (PDB 7jvr31) and risperidone (PDB 6cm4)24. | ||
| Fig. 4 Schematic of the contact analysis (A) and structural comparison of the V3.29,111 environment in active (B and D) and inactive (C) state. (B and C) Show the environment of V3.29,111 (dark red) without ligand, clearly visible is the smaller V3.29,111-ECL2 (I45.51,183 and I45.52,184 in dark salmon) pocket in the inactive state (C). In the active state I45.51,183 is rotated away from V3.29,111, leaving smaller hydrophobic pocket between V3.29,111 and ECL2. This position is stabilized by V3.29,111 and I45.51,183 contacts to the unpolar methylene group (orange) of bromocriptine (light orange, (D)). | ||
Residue V3.29,111 is not highly conserved in class A GPCRs (see ESI Fig. 1†) but is oriented towards the center of the receptor on the inside extracellular part of TM3 (see ESI Fig. 3†). Here we detect a stabilization of the active relative to the inactive state especially for the Q, N, (S) and L mutants. Comparing the inactive and active state structures and using the DBASS method alongside the analysis of inter-residue profiles of the mutant simulations, we observe that the environment surrounding V3.29,111 is predominantly shaped by I45.51,183 and I45.52,184 in ECL2 as well as L4.60,170. However, in the active state, I45.51,183 is oriented away from V3.29,111, rendering the V3.29,111-ECL2 pocket less hydrophobic and more open. This corresponds well with our predictions which show a preference for the active state over the inactive state for polar mutations (N, Q, S).
The leucine mutant stabilizes the active state by occupying more space, unlike in the inactive state where steric hindrance occurs. The aromatic mutants, that are both rather bulky, do not cause steric clashes since their rotamers are pointing out of the x3.29,111-ECL2 pocket, which explains the insignificant change in ΔΔG. These findings also match with the I45.51,183 mutations, where all mutants, especially larger amino acids like N, Q, L, F, and Y, favor the active state over the inactive state.
Our goal was to leverage this insight for rational ligand design. An agonist should stabilize the V3.29,111-ECL2 conformation by rotating I45.51,183 away from V3.29,111. This could be achieved by introducing a non-polar group that interacts with V3.29,111 and stabilizes the rotated position of I45.51,183 without distorting the other non-polar residues in the vicinity. In fact, this is achieved in bromoergocryptine and bromocryptine,31,34 two crystallized agonists. Here, interactions of both V3.29,111 and I45.51,183 with the non-polar methylene group are formed (Fig. 4D orange) which stabilize the active state in this position.
Residue L2.46,76, which is highly conserved among class A GPCRs and is implicated in the activation microswitch system,20 exhibits ambivalent ΔΔG values. Interestingly, aromatic mutants stabilize the inactive state, while mutants N, Q, G, and A favor the active state. Comparing contact patterns of 76 mutants reveals increased interactions with N7.49,422 and F6.44,382 in the active and inactive states, respectively. Aromatic mutants exhibit stronger π–π contacts with F6.44,382 in the inactive state due to closer proximity of TM7 and TM3. Polar mutants enhance contacts with S3.39,121 and N7.49,422, which form a hydrophilic patch in the active state. For steric reasons L2.46,76Y does not fit into the more tight active hydrophilic patch. On the other hand, L2.46,76A and L2.46,76 G, which are smaller in size, preferentially stabilize the active state. Even though L2.46,76 does not directly interact with external ligands24,31 like risperidone or bromocriptine, we can still use the information from our scan for ligand design. We know that the inactive state is stabilized by the L2.46,76F/Y–F6.44,382 interaction (Fig. 5). F6.44,382 is also known to be part of a hydrophobic extended binding pocket in the inactive state,24 which is stabilized by the benzisoxazole group of risperidone24 in the inactive state. In the active state, however, F6.44,382 and F5.51,202 form a π–π interaction (T-stack) which is energetically favourable. So, to be able to stabilize the inactive state configuration which does not have the F6.44,382–F5.51,202 T-stack the position of F6.44,382 needs to be stabilized. This can be achieved by the interaction from the L2.46,76F/Y mutants as well as the interaction with risperidone.
 | ||
| Fig. 5 L2.46,76 (dark red) environment formed by N7.49,422, S3.39,121, F6.44,382 (and F5.51,202 for F6.44,382 interaction) in the active (A) and inactive state ((B and C) with ligand risperidone). | ||
These findings match with our scan that shows a stabilization of the inactive state for the polar F6.44,382S, F6.44,382N and F6.44,382Q mutants, which all disturb the T-stack in the active state and therefore stabilize the inactive relative to the active state. The apolar mutants F6.44,382G and F6.44,382A also stabilize the inactive state relative to the active state, even though they disrupt the T-stacking. Here, the stabilization effect comes mainly from the interaction of F6.44,382A with L3.43,125, which is formed in the inactive state.
To summarize, we can say that disrupting the F6.44,382–F5.51,202 T-stack can lead to a stabilization of the inactive state when the position of F6.44,382 is stabilized either by ligands (see risperidone24) or by interactions from mutants like L2.46,76F. This also matches with the F5.51,202 mutants F5.51,202S/Q/G and L which all show a strong stabilization of the inactive relative to the active state. On the other side, introducing more small polar groups into the patch formed by S3.39,121 and N7.49,422 in the active state, for example by the mutants L2.46,76S/L2.46,76N/L2.46,76Q leads to a stabilization of the active state relative to the inactive state by a contraction of TM7 and TM3. Consequently mutating N7.49,422 to F, Y, L, A and G as well as the mutants S3.39,121F/Y/A/G lead to a destabilization of the hydrophilic patch and therefore a stabilization of the inactive relative to the active state.
4 Conclusions
In this study, we conducted a comprehensive in silico mutation scan of the human dopamine D2 receptor (DRD2) using non-equilibrium alchemical free energy calculations with pmx. While we successfully reproduced known trends of mutation effects on the equilibrium between the active and inactive states of the receptor,3,9,56 our work also unveiled several novel findings. Notably, we identified previously unreported residues that significantly impact the stabilization of different receptor states upon mutation. This discovery represents a major advancement, as it contributes new targets for structure-based ligand design.One of the key innovations of this study is the generation of a large mutational dataset, which not only uncovers new important residues for DRD2 activation but also demonstrates how these residues can be leveraged to explain diseases and ligand action, while providing insights for the development of new, highly specific DRD2 ligands.
We found 40 residues that exhibited multiple high ΔΔG values upon mutation to different amino acids. Out of these, 22 residues are neither conserved nor part of the known microswitches, with 12 of them being previously unexplored in experimental mutation studies. Importantly, the residues were not considered important for receptor state equilibrium in other class A GPCR studies. We propose that these residues may be specific to DRD2 receptor activation regulation, offering new targets for the design of highly receptor-specific ligands.
Additionally, we identified 7 residues located within pockets of the outside pocketome61 of class A GPCRs, primarily in KS5 (T3.52,134, N175, M4.45,155). These residues represent promising new sites for further ligand design studies targeting these unique pockets.
Our study also reveals how certain mutations in DRD2 are linked to neurological diseases. For example, the mutation trend of V3.29,111I62 can be explained by a similar effect to the V3.29,111L mutant, where the introduction of a larger, sterically demanding amino acid leads to overstabilization of the active state, potentially contributing to disease.
Finally, by combining the results from our mutation scan with structural analysis, we were able to provide new insights into how changes in the inter-residue contact network affect the allosteric modulation of the receptor. This allows us to explain the nuanced effects of DRD2 ligands like bromocriptine31 and risperidone.24 We also demonstrated how our results, using specific examples such as V3.29,111 and I45.51,183, can be applied to engineer interactions that selectively stabilize either the active or inactive state. This offers a solid foundation for future structure-based ligand design studies.
Data availability
Additional information can be found in the ESI.† All parameter files for the MD simulations (*.mdp) and all starting structures for all mutants (*.gro) are publicly available at https://doi.org/10.5281/zenodo.11635150.Author contributions
Conceptualization: BLG, LS data curation: LS formal analysis: LS funding acquisition: BLG investigation: LS methodology: BLG, LS project administration: BLG resources: BLG supervision: BLG validation: LS visualization: LS writing – original draft: LS writing – review and editing: LS, BLG.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Funding from the Max Planck Society is gratefully acknowledged. Further, we thank Peter Kolb for insightful discussions.Notes and references
- A. J. Venkatakrishnan, A. K. Ma, R. Fonseca, N. R. Latorraca, B. Kelly, R. M. Betz, C. Asawa, B. K. Kobilka and R. O. Dror, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3288–3293 CrossRef CAS PubMed.
- V. Katritch, V. Cherezov and R. C. Stevens, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 531–556 CrossRef CAS PubMed.
- Q. Zhou, D. Yang, M. Wu, Y. Guo, W. Guo, L. Zhong, X. Cai, A. Dai, W. Jang, E. I. Shakhnovich, Z.-J. Liu, R. C. Stevens, N. A. Lambert, M. M. Babu, M.-W. Wang and S. Zhao, eLife, 2019, 8, e50279 CrossRef PubMed.
- A. Radoux-Mergault, L. Oberhauser, S. Aureli, F. L. Gervasio and M. Stoeber, Sci. Adv., 2023, 9, eadf6059 CrossRef CAS PubMed.
- A. de Felice, S. Aureli and V. Limongelli, Front. Mol. Biosci., 2021, 8, 673053 CrossRef CAS PubMed.
- A. S. Hauser, M. M. Attwood, M. Rask-Andersen, H. B. Schiöth and D. E. Gloriam, Nat. Rev. Drug Discovery, 2017, 16, 829–842 CrossRef CAS PubMed.
- A. S. Hauser, S. Chavali, I. Masuho, L. J. Jahn, K. A. Martemyanov, D. E. Gloriam and M. M. Babu, Cell, 2018, 172, 41–54 Search PubMed.
- D. Yang, Q. Zhou, V. Labroska, S. Qin, S. Darbalaei, Y. Wu, E. Yuliantie, L. Xie, H. Tao, J. Cheng, Q. Liu, S. Zhao, W. Shui, Y. Jiang and M.-W. Wang, Signal Transduct. Targeted Ther., 2021, 2059–3635 CAS.
- M. Vass, S. Podlewska, I. J. P. de Esch, A. J. Bojarski, R. Leurs, A. J. Kooistra and C. de Graaf, J. Med. Chem., 2019, 62, 3784–3839 CrossRef CAS PubMed.
- A. J. Venkatakrishnan, X. Deupi, G. Lebon, F. M. Heydenreich, T. Flock, T. Miljus, S. Balaji, M. Bouvier, D. B. Veprintsev, C. G. Tate, G. F. X. Schertler and M. M. Babu, Nature, 2016, 536, 484–487 Search PubMed.
- G. Pándy-Szekeres, C. Munk, T. M. Tsonkov, S. Mordalski, K. Harpsøe, A. S. Hauser, A. J. Bojarski and D. E. Gloriam, Nucleic Acids Res., 2017, 46, D440–D446 Search PubMed.
- R. O. Dror, D. H. Arlow, P. Maragakis, T. J. Mildorf, A. C. Pan, H. Xu, D. W. Borhani and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18684–18689 CrossRef CAS PubMed.
- N. R. Latorraca, A. J. Venkatakrishnan and R. O. Dror, Chem. Rev., 2017, 117, 139–155 CrossRef CAS PubMed.
- P. Xu, S. Huang, H. Zhang, C. Mao, X. E. Zhou, X. Cheng, I. A. Simon, D.-D. Shen, H.-Y. Yen, C. V. Robinson, K. Harpsøe, B. Svensson, J. Guo, H. Jiang, D. E. Gloriam, K. Melcher, Y. Jiang, Y. Zhang and H. E. Xu, Nature, 2021, 592, 469–473 CrossRef CAS PubMed.
- X. Yuan, S. Raniolo, V. Limongelli and Y. Xu, J. Chem. Theory Comput., 2018, 14, 2761–2770 CrossRef CAS PubMed.
- R. E. Jefferson, A. Oggier, A. Füglistaler, N. Camviel, M. Hijazi, A. R. Villarreal, C. Arber and P. Barth, Nat. Commun., 2023, 14, 2875 Search PubMed.
- P. Weinkam, Y. C. Chen, J. Pons and A. Sali, J. Mol. Biol., 2013, 425, 647–661 Search PubMed.
- R. Nussinov and C.-J. Tsai, Cell, 2013, 153, 293–305 Search PubMed.
- A. Mafi, S.-K. Kim and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2110085119 Search PubMed.
- A. S. Hauser, A. J. Kooistra, C. Munk, F. M. Heydenreich, D. B. Veprintsev, M. Bouvier, M. M. Babu and D. E. Gloriam, Nat. Struct. Mol. Biol., 2021, 28, 879–888 CrossRef CAS PubMed.
- R. Nygaard, Y. Zou, R. O. Dror, T. J. Mildorf, D. H. Arlow, A. Manglik, A. C. Pan, C. W. Liu, J. J. Fung, M. P. Bokoch, F. S. Thian, T. S. Kobilka, D. E. Shaw, L. Mueller, R. S. Prosser and B. K. Kobilka, Cell, 2013, 152, 532–542 CrossRef CAS PubMed.
- R. C. Kling, N. Tschammer, H. Lanig, T. Clark and P. Gmeiner, PLoS One, 2014, 9, e100069 CrossRef PubMed.
- K.-Y. M. Chen, D. Keri and P. Barth, Nat. Chem. Biol., 2020, 16, 77–86 CrossRef CAS PubMed.
- S. Wang, T. Che, A. Levit, B. K. Shoichet, D. Wacker and B. L. Roth, Nature, 2018, 555, 269–273 CrossRef CAS PubMed.
- R. O. Dror, H. F. Green, C. Valant, D. W. Borhani, J. R. Valcourt, A. C. Pan, D. H. Arlow, M. Canals, J. R. Lane, R. Rahmani, J. B. Baell, P. M. Sexton, A. Christopoulos and D. E. Shaw, Nature, 2013, 503, 295–299 CrossRef CAS PubMed.
- M. Aldeghi, B. L. de Groot and V. Gapsys, in Accurate Calculation of Free Energy Changes upon Amino Acid Mutation, ed. T. Sikosek, Springer New York, New York, NY, 2019, pp. 19–47 Search PubMed.
- V. Gapsys, S. Michielssens, D. Seeliger and B. L. de Groot, J. Comput. Chem., 2015, 36, 348–354 CrossRef CAS PubMed.
- D. Seeliger and B. L. de Groot, Biophys. J., 2010, 98, 2309–2316 CrossRef CAS PubMed.
- G. E. Crooks, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 60, 2721–2726 CrossRef CAS PubMed.
- D. Branduardi, F. L. Gervasio and M. Parrinello, J. Chem. Phys., 2007, 126, 054103 Search PubMed.
- Y. Zhuang, P. Xu, C. Mao, L. Wang, B. Krumm, X. E. Zhou, S. Huang, H. Liu, X. Cheng, X.-P. Huang, D.-D. Shen, T. Xu, Y.-F. Liu, Y. Wang, J. Guo, Y. Jiang, H. Jiang, K. Melcher, B. L. Roth, Y. Zhang, C. Zhang and H. E. Xu, Cell, 2021, 184, 931–942 CrossRef CAS PubMed.
- D. Im, A. Inoue, T. Fujiwara, T. Nakane, Y. Yamanaka, T. Uemura, C. Mori, Y. Shiimura, K. T. Kimura, H. Asada, N. Nomura, T. Tanaka, A. Yamashita, E. Nango, K. Tono, F. M. N. Kadji, J. Aoki, S. Iwata and T. Shimamura, Nat. Commun., 2020, 11, 6442 CrossRef CAS PubMed.
- L. Fan, L. Tan, Z. Chen, J. Qi, F. Nie, Z. Luo, J. Cheng and S. Wang, Nat. Commun., 2020, 11, 1074 CrossRef CAS PubMed.
- J. Yin, K.-Y. M. Chen, M. J. Clark, M. Hijazi, P. Kumari, X.-c. Bai, R. K. Sunahara, P. Barth and D. M. Rosenbaum, Nature, 2020, 584, 125–129 CrossRef CAS PubMed.
- P. Xu, S. Huang, B. E. Krumm, Y. Zhuang, C. Mao, Y. Zhang, Y. Wang, X.-P. Huang, Y.-F. Liu, X. He, H. Li, W. Yin, Y. Jiang, Y. Zhang, B. L. Roth and H. E. Xu, Cell Research, 2023, 33, 604–616 CrossRef PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- I. D. Pogozheva, H. Joo, H. I. Mosberg and A. L. Lomize, Nucleic Acids Res., 2011, 40, D370–D376 Search PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. I. Brooks, A. D. J. MacKerell, J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- P. Bauer, B. Hess and E. Lindahl, GROMACS 2022.3 Manual, 2022, DOI:10.5281/zenodo.7037337.
- E. Lindahl, B. Hess and D. v. d. Spoel, Mol. Model., 2001, 7, 306–317 CrossRef CAS.
- D. v. d. Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 Search PubMed.
- T. A. Darden, D. M. York and L. G. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- N. Goga, A. J. Rzepiela, A. H. de Vries, S. J. Marrink and H. J. C. Berendsen, J. Chem. Theory Comput., 2012, 8, 3637–3649 Search PubMed.
- T. C. Beutler, A. E. Mark, R. C. van Schaik, P. R. Gerber and W. F. van Gunsteren, Chem. Phys. Lett., 1994, 222, 529–539 CrossRef CAS.
- V. Gapsys, A. Yildirim, M. Aldeghi, Y. Khalak, D. van der Spoel and B. L. de Groot, Commun. Chem., 2021, 4, 61 CrossRef CAS PubMed.
- B. I. Costescu and F. Gräter, BMC Biophys., 2013, 6, 5 Search PubMed.
- J. Selent, F. Sanz, M. Pastor and G. De Fabritiis, PLoS Comput. Biol., 2010, 6, 1–6 Search PubMed.
- K. A. Neve, M. G. Cumbay, K. R. Thompson, R. Yang, D. C. Buck, V. J. Watts, C. J. DuRand and M. M. Teeter, Mol. Pharmacol., 2001, 60, 373–381 CrossRef CAS PubMed.
- C. J. Draper-Joyce, R. K. Verma, M. Michino, J. Shonberg, A. Kopinathan, C. K. Herenbrink, P. J. Scammells, B. Capuano, A. M. Abramyan, D. M. Thal, J. A. Javitch, A. Christopoulos, L. Shi and J. R. Lane, Sci. Rep., 2018, 1208 CrossRef PubMed.
- C. Munk, K. Harpsøe, A. S. Hauser, V. Isberg and D. E. Gloriam, Curr. Opin. Pharmacol., 2016, 30, 51–58 CrossRef CAS PubMed.
- J. Chen, Y. L. Vishweshwaraiah and N. V. Dokholyan, Curr. Opin. Struct. Biol., 2022, 73, 102334 Search PubMed.
- R. O. Dror, A. C. Pan, D. H. Arlow, D. W. Borhani, P. Maragakis, Y. Shan, H. Xu and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13118–13123 CrossRef CAS PubMed.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, J. Domański, D. L. Dotson, S. Buchoux, I. M. Kenney and O. Beckstein, Proceedings of the 15th Python in Science Conference, 2016, pp. 98–105 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- J. B. Hedderich, M. Persechino, K. Becker, F. M. Heydenreich, T. Gutermuth, M. Bouvier, M. Bünemann and P. Kolb, Nat. Commun., 2022, 13, 2567 CrossRef CAS PubMed.
- V. Isberg, B. Vroling, R. van der Kant, K. Li, G. Vriend and D. E. Gloriam, Nucleic Acids Res., 2013, 42, D422–D425 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04723k |
| This journal is © The Royal Society of Chemistry 2025 |