
Highly efficient Ag6Si2O7/WO3 photocatalyst based on heterojunction with enhanced visible light photocatalytic activities†
Yonggang Hu,
Hong Zheng*,
Tongzhou Xu,
Ning Xu and
Hongwen Ma*
Beijing Key Laboratory of Materials Utilization of Nonmetallic Minerals and Solid Wastes, National Laboratory of Mineral Materials, School of Materials Science and Technology, China University of Geosciences, Beijing 100083, China. E-mail: zhengh@cugb.edu.cn; mahw@cugb.edu.cn; Fax: +86 10 82322974; Fax: +86 10 82323374; Tel: +86 10 82322974 Tel: +86 10 82323374
First published on 25th October 2016
Abstract
Ag6Si2O7/WO3 photocatalysts were prepared by an ultrasound-assisted precipitation method and characterized by X-ray diffraction (XRD), field emission scanning electron microscopy (FE-SEM), energy-dispersive spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), UV-vis diffuse reflectance spectra (DRS) and photoluminescence (PL) spectroscopy. The effects of Ag6Si2O7/WO3 mole ratios on photocatalytic activity of Ag6Si2O7/WO3 were investigated. The results showed that the photocatalytic degradation efficiency of methylene blue (MB) by Ag6Si2O7/WO3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) attains 97.4%, which is much higher than that (87.3%) by sole Ag6Si2O7 and that (14.0%) by sole WO3 after visible light irradiation for 30 min, and the apparent rate constant is 1.59 times that of sole Ag6Si2O7 and 44.6 times that of sole WO3. Photocatalytic activities of different photocatalysts with the same weight of visible-light-active components were compared and showed that the rate constant of Ag6Si2O7/WO3 (1:1) is 2.88 times that of mathematical sum of Ag6Si2O7 and WO3. Moreover, rhodamine B (RhB), methyl orange (MO) and 2,4-dichlorophenol were also effectively degraded. After 3 recycling runs, the photocatalytic performance of the Ag6Si2O7/WO3 (1:1) was still effectively maintained. In addition, the quenching effects of different scavengers proved that the ˙OH plays important roles in the photocatalytic reaction under visible light irradiation. The visible light photocatalytic activity enhancement of the Ag6Si2O7/WO3 (1:1) came from the efficient separation of electron–hole pairs, which resulted from the heterojunction of Ag6Si2O7/WO3. These results indicate that Ag6Si2O7/WO3 heterojunction is highly efficient photocatalyst and there is a significant potential in the degradation of organic contaminants under visible light irradiation.
:1) attains 97.4%, which is much higher than that (87.3%) by sole Ag6Si2O7 and that (14.0%) by sole WO3 after visible light irradiation for 30 min, and the apparent rate constant is 1.59 times that of sole Ag6Si2O7 and 44.6 times that of sole WO3. Photocatalytic activities of different photocatalysts with the same weight of visible-light-active components were compared and showed that the rate constant of Ag6Si2O7/WO3 (1:1) is 2.88 times that of mathematical sum of Ag6Si2O7 and WO3. Moreover, rhodamine B (RhB), methyl orange (MO) and 2,4-dichlorophenol were also effectively degraded. After 3 recycling runs, the photocatalytic performance of the Ag6Si2O7/WO3 (1:1) was still effectively maintained. In addition, the quenching effects of different scavengers proved that the ˙OH plays important roles in the photocatalytic reaction under visible light irradiation. The visible light photocatalytic activity enhancement of the Ag6Si2O7/WO3 (1:1) came from the efficient separation of electron–hole pairs, which resulted from the heterojunction of Ag6Si2O7/WO3. These results indicate that Ag6Si2O7/WO3 heterojunction is highly efficient photocatalyst and there is a significant potential in the degradation of organic contaminants under visible light irradiation.
1. Introduction
The utilization of semiconductor photocatalysts for the decomposition of organic pollutions has been regarded as an efficient method to solve environmental pollution problems. Although TiO2 has been recognized as the very important semiconductor in photocatalysis fields owing to its high efficiency, low cost, high stability and low toxicity, the large band gap of titania and massive recombination of photo-generated charge carriers limit its overall photocatalytic efficiency.1,2 Therefore, much research has focused on developing the novel photocatalysts with high photocatalytic properties under visible light irradiation,3–5 such as the series of Bi-,6–8 W-,9,10 Ag-11–15 photocatalysts. Silicates are ubiquitous materials with abundant reserves, and in the presence of transition-metal cations the SiO4 tetrahedra in silicates can be easily distorted and get polarized, so it would be possible to construct an internal polar electric field by controlling the arrangement of the polar SiO4 tetrahedra. Furthermore, the presence of several different coordination environments for transition-metal cations can enhance not only the preferential migration of photogenerated charge carriers from one metal–oxygen polyhedra to another but also the optical absorption at various wavelengths. Ag6Si2O7, which has an internal polar electric field set along the b-direction and consists of two-, three- and four-coordinate Ag+ ions leading to AgO2, AgO3, and AgO4 units, respectively, is photocatalytic nearly in the whole visible-light region (420 nm < λ < 740 nm) and exhibits a very strong photocatalytic activity.16 However, high cost and easy deactivation of silver salt limit its application. WO3 as another inexpensive material, which have many advantages, such as stable physicochemical properties, narrow band gap (about 2.7 eV) and can absorb light in the visible range.17,18 Unfortunately, the sole WO3 shows very low photocatalytic activity under visible light irradiation for the fast recombination of electron–hole pairs.19–22 Therefore, in order to reduce the costs and enhance photocatalytic activity and stability of the catalysts, Ag6Si2O7/WO3 photocatalysts were prepared and the enhanced visible light photocatalytic activities were studied.In this paper, a series of Ag6Si2O7/WO3 photocatalysts were successfully prepared by using a facile ultrasound-assisted precipitation method and then characterized by various techniques such as XRD, FE-SEM, EDS, XPS, DRS and PL. And the photocatalytic activities of the as-prepared samples were systematically evaluated by decomposition of MB, MO, RhB and 2,4-dichlorophenol under visible light irradiation. The influence of Ag6Si2O7/WO3 mole ratios on photocatalytic activities were considered and Ag6Si2O7/WO3 (1:1) was found to have much higher activity and better stability than sole Ag6Si2O7. A possible photodegradation mechanism in Ag6Si2O7/WO3 photocatalyst was discussed.
2. Experimental
2.1. Chemicals and materials
All reagents used were of analytical purity and were used without further purification. Silver nitrate (AgNO3), sodium silicate nonahydrate (Na2SiO3·9H2O), sodium tungstate dihydrate (Na2WO4·2H2O), sodium chloride (NaCl), MB, MO, RhB, 2,4-dichlorophenol, disodium ethylenediaminetetraacetate (EDTA-2Na), benzoquinone (BQ), isopropanol (IPA) and ethanol were purchased from Beijing Chemical Works. Deionized water was used in all experiments.2.2. Preparation of as-prepared samples
The Ag6Si2O7/WO3 photocatalysts were prepared by ultrasound-assisted precipitation.23 The required amounts (0.686 g) of the as-prepared WO3 powder and 0.284 g of Na2SiO3 dissolved in 70 mL deionized water were mixed and ultrasonicated for 10 min, then 30 mL 0.1 M AgNO3 was added dropwise to the above dispersion under ultrasonication. After ultrasonication for 30 min, the sample was separated from the solution by filtering, washed with deionized water and ethanol for three times, and dried at room temperature for 24 h. The obtained Ag6Si2O7/WO3 photocatalyst (the nominal mole ratio of Ag6Si2O7 to WO3 is 1:1) was noted as Ag6Si2O7/WO3 (1:1). The Ag6Si2O7/WO3 (a:b) photocatalysts (a:b is the nominal mole ratio of Ag6Si2O7 to WO3) with other mole ratios (a:b = 6:1, 3:1, 2:1, 1:2, 1:3, 1:6) were synthesized using the same process expect adding different amounts of WO3.
For comparison, Ag6Si2O7 and WO3 powder were also prepared. The Ag6Si2O7 powder was prepared by a precipitation method as reported by Lou et al.16 The WO3 powder was prepared by adding 0.825 g Na2WO4·2H2O and 0.290 g NaCl into mixed solution of 13 mL deionized water and 4 mL HCl (37%) under magnetic stirring and continuing to stir for 30 minutes. The resulting precursor suspension was transferred into a Teflon-lined stainless steel autoclave with total volume of 30 mL and maintained at 180 °C for 24 h. The solid powder was washed several times with deionized water and dried at 80 °C for 10 h.
2.3. Characterization of as-prepared samples
The X-ray diffraction (XRD) measurements were carried out on a D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 0.15418 nm), operated at 40 kV and 100 mA. SEM images were observed with a field-emission scanning electron microscope (FESEM, S-4800, Hitachi) performed at an accelerating voltage of 15.0 kV. Oxford instruments INCA X-act energy-dispersive spectroscopy (EDS) was employed to estimate the final actual mole ratio in the Ag6Si2O7 sample. The elemental analysis in the Ag6Si2O7 sample was measured with ICP-OES (inductively coupled plasma optical emission spectrometer) (Shimadzu, ICPE-9820). Chemical states of surface elements were investigated by X-ray photoelectron spectroscopy (XPS, PHI-5300, ESCA) at a pass energy of 50 eV, using Al Kα as an exciting X-ray source. All the binding energies were referenced to the C1s peak at 284.8 eV. The UV-vis diffuse reflectance spectra (DRS) of the samples were recorded in the range from 200 to 800 nm using a Hitachi UV-3100 spectrometer equipped with an integrating sphere. Photoluminescence (PL) spectra of the as-prepared samples were measured using a HITACHI F-4600 fluorescence spectrophotometer to monitor the recombination rate of electron–hole pairs.2.4. Photocatalytic evaluation
The photocatalytic activities of as-prepared samples were evaluated by the degradation of MB, MO, RhB and 2,4-dichlorophenol under simulative visible-light irradiation using a 75 W metal halide lamp with a cutoff filter (λ > 420 nm). In each experiment, 0.02 g of photocatalyst was totally dispersed into 50 mL solution of 3 × 10−5 mol L−1 initial dye concentration or 10 mg L−1 initial 2,4-dichlorophenol concentration. Before the irradiation, the photocatalyst powder and organic solution were vigorously stirred in the dark for 30 min to ensure the establishment of an adsorption–desorption equilibrium. After those, the light was turned on, and temperature of the system was controlled by a water bath (22 ± 1 °C) running through the outer casing of the glass tube to avoid light induced heating. At certain time intervals, 5 mL aliquots were sampled and centrifuged to remove the particles. The concentration of the organics was analyzed by recording the variation of the absorption-band maximum in the visible spectrum of MB (664 nm), MO (464 nm), RhB (554 nm) or UV spectrum of 2,4-dichlorophenol (284 nm) solution using a UV765 UV-vis spectrophotometer. Total organic carbon (TOC) of the illuminated solution at different time intervals was measured by the TOC analyzer (Shimadzu, L-series).3. Results and discussion
3.1. Characterization of as-synthesized samples
Fig. 1 displays the XRD patterns of the Ag6Si2O7, WO3 and Ag6Si2O7/WO3 with different mole ratios. The XRD pattern of as-prepared WO3 is in accordance with the standard card (JCPDS no. 43-1035), indicating that monoclinic WO3 was prepared. The main XRD peaks of the as-prepared Ag6Si2O7 match those of the standard Ag6Si2O7 structure (JCPDS no. 47-0704), indicating that Ag6Si2O7 was prepared, though the diffraction peaks were broad due to its small particles. Low signals on a high background are attributed to its low crystallinity. For the composites samples, besides the diffraction peaks of WO3, almost no diffraction peak of Ag6Si2O7 was observed due to much stronger diffraction peaks of WO3 than those of Ag6Si2O7. Moreover, the intensities of diffraction peaks of WO3 increase slightly with the increase of the mole ratio of WO3.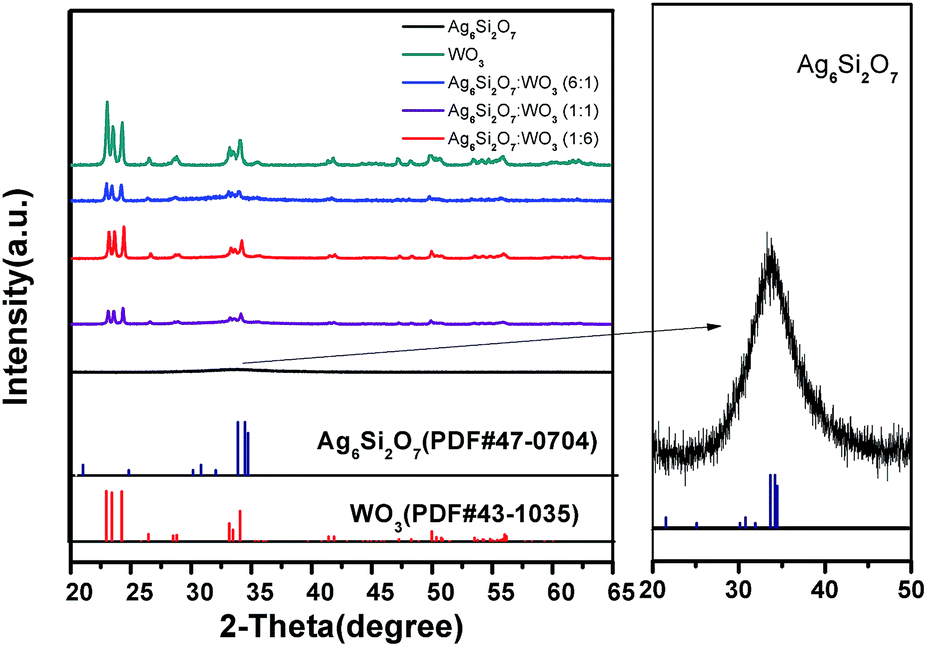 | ||
| Fig. 1 XRD patterns of as-prepared samples. | ||
Fig. 2 shows the FE-SEM images of Ag6Si2O7, WO3 and Ag6Si2O7/WO3 (1:1). The as-prepared Ag6Si2O7 is irregular in shape with size of 100 nm, which can be observed in the FE-SEM images (Fig. 2a). The FE-SEM images of WO3 particles prepared are square flake with size of 100–800 nm, larger than that of Ag6Si2O7 (Fig. 2c). Obviously we can see the square flake phase mixed with some irregular Ag6Si2O7 particles for Ag6Si2O7/WO3 (1:1) (Fig. 2d). The EDS spectrum of Ag6Si2O7 (Fig. 2b) shows the presence of only three elements, O, Si and Ag, with atomic percentages 49.84, 12.45, and 37.70%, respectively, indicating that estimated atom ratio of Ag to Si is around 3:1. The atom ratio of Ag to Si was further determined by ICP-OES to be 2.86:1 and slightly less than 3:1 due to adsorption of Si2O76− ions to the surface of Ag6Si2O7 particles, which confirms that the as-prepared samples are Ag6Si2O7.
 | ||
| Fig. 2 FESEM images of (a) Ag6Si2O7, (c) WO3, (d) Ag6Si2O7/WO3 (1:1) composite and (b) EDS of Ag6Si2O7. | ||
To investigate the optical absorption properties of the as-prepared samples, the UV-vis diffuse reflectance spectra of the WO3, Ag6Si2O7 and Ag6Si2O7/WO3 (1:1) were determined and are shown in Fig. S1.† It can be seen that an absorption edge for Ag6Si2O7 occurs at 630 nm, which is different from the absorption spectrum previously reported by Lou et al.16 The possible reason is attributed to the difference in crystallinity. Sole WO3 exhibits the fundamental absorption edge at about 460 nm, corresponding to the band gap of 2.7 eV.24 The UV-vis DRS spectrum of Ag6Si2O7/WO3 (1:1) is quite similar to WO3, but the main adsorption edge moves toward the visible light region, showing that visible light absorption was improved after combining with Ag6Si2O7 and Ag6Si2O7/WO3 (1:1) has potential ability for photocatalytic decomposition of organic contaminants under visible-light irradiation.
More detailed information regarding the chemical and bonding environment of Ag6Si2O7/WO3 (1:1) are ascertained by XPS (Fig. 3). The high resolution XPS spectrum for the Ag 3d is shown in Fig. 3a. The peaks located at 367.66 and 373.63 eV correspond to the Ag 3d5/2 and Ag 3d3/2, respectively, suggesting the presence of Ag+. No peak is observed at 369.2 or 375.8 eV, demonstrating that no Ag0 is formed during the preparation.25 There are two binding energy peaks at 529.80 and 531.85 eV corresponding to O 1s (Fig. 3b).16,18 The XPS peak of the Si 2p can be found at 102.10 eV (Fig. 3c), which is characteristic of Si4+ in the Ag6Si2O7/WO3 (1:1).16 In the high resolution spectrum of W 4f (Fig. 3d), there are two peaks at 34.70 eV and 36.90 eV which can be ascribed to W 4f7/2 and W 4f5/2, respectively. These results signify that W element is in the +6 oxidation state with the typical binding energies. It is worth noting that the binding energy values of W 4f7/2 and W 4f5/2 in the Ag6Si2O7/WO3 (1:1) are slightly lower than the XPS results provided by the literature reported for bare WO3.26 Such a shift should be attributed to the interaction between WO3 and Ag6Si2O7.27
 | ||
| Fig. 3 XPS spectra of Ag6Si2O7/WO3 (1:1) composite: (a) Ag 3d, (b) O 1s, (c) Si 2p, and (d) W 4f. | ||
3.2. Photocatalytic activity of as-prepared samples
MB was chosen as the model pollutant to evaluate the photocatalytic activities of the Ag6Si2O7/WO3 under the visible light irradiation. As shown in Fig. 4a, all the Ag6Si2O7/WO3 photocatalysts exhibit higher photocatalytic activity than sole WO3 sample. Moreover, the photocatalytic activities of Ag6Si2O7/WO3 improve generally with the increase of Ag6Si2O7 mole ratio. The Ag6Si2O7/WO3 (1:1) exhibits the most significantly enhanced photocatalytic performance. The photocatalytic degradation efficiency of MB by Ag6Si2O7/WO3 (1:1) attains 97.4%, which is much higher than that (87.3%) by sole Ag6Si2O7 and that (14.0%) by sole WO3 after visible light irradiation for 30 min. This change may be attributed to the effective interfacial charge separation resulted from the fabrication of a heterojunction. However, with the further increases of Ag6Si2O7 content, the degradation rate decreases obviously even if the photocatalytic activity of Ag6Si2O7/WO3 (2:1) is close to that of sole Ag6Si2O7. The possible reason is attributed to the fact that less WO3 is disadvantage to the formation of heterojunction, which lead to decreasing the photocatalytic activity.
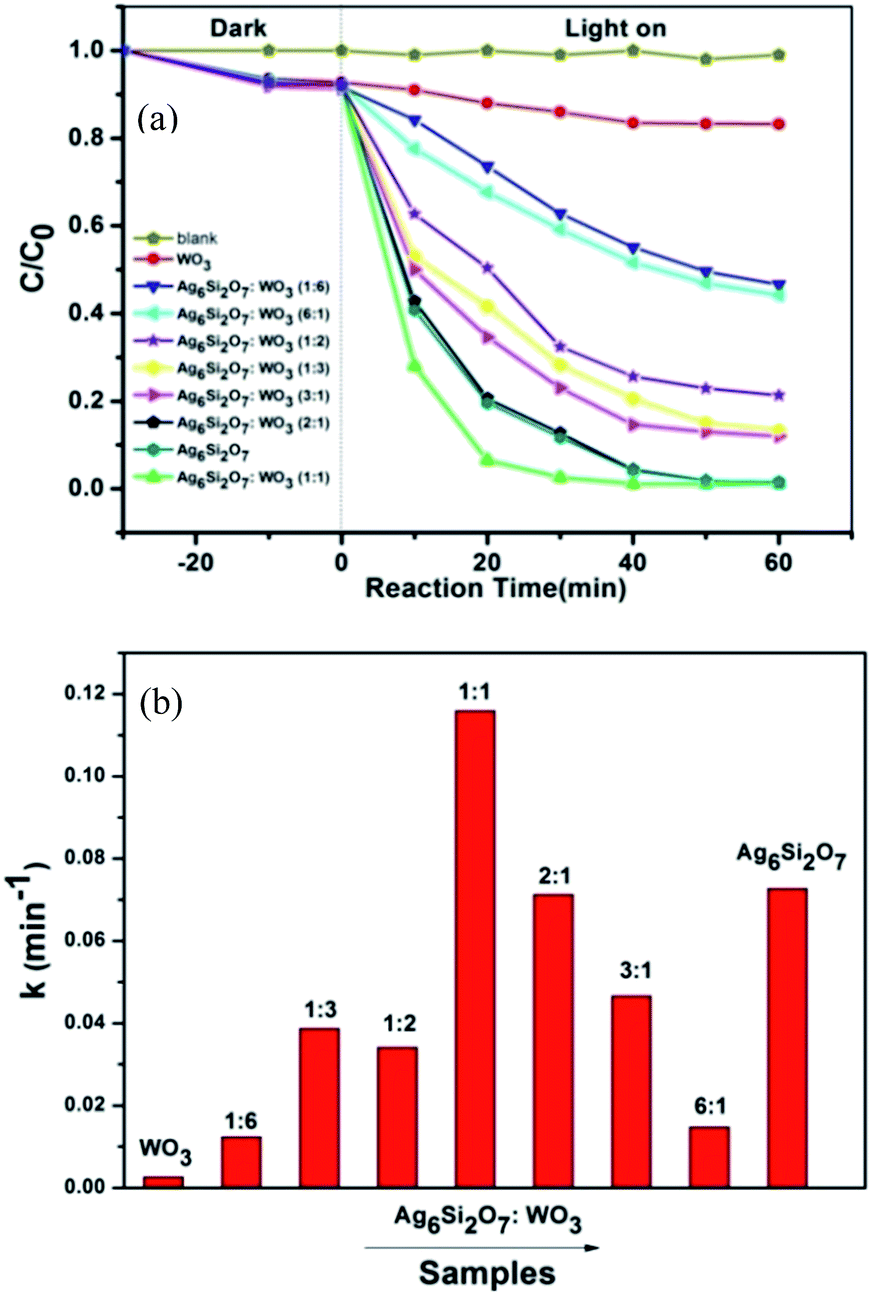 | ||
| Fig. 4 Photocatalytic degradation curves (a) and corresponding apparent rate constants (b) for the photocatalytic degradation of MB over Ag6Si2O7, WO3 and Ag6Si2O7/WO3 composites under visible light irradiation. | ||
On the basis of Langmuir–Hinshelwood (L–H) model, the photocatalytic reaction can be described as follows:
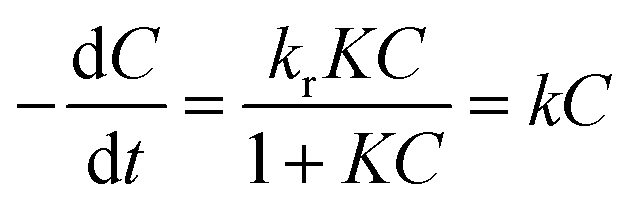 | (1) |
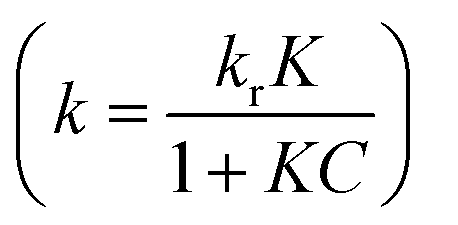 of the pseudo-first-order reaction. According to this equation, the pseudo-first-order kinetics constants for all the photocatalytic reactions were calculated from the experimental data and shown in Fig. 4b, the Ag6Si2O7/WO3 (1:1) has the highest apparent rate constant of 0.116 min−1, which was about 1.59 times as that of sole Ag6Si2O7 sample (0.073 min−1) and 44.6 times as that of sole WO3 (0.0026 min−1), suggesting that there is a significant synergistic effect between Ag6Si2O7 and WO3 for Ag6Si2O7/WO3 (1:1) under visible light irradiation.
of the pseudo-first-order reaction. According to this equation, the pseudo-first-order kinetics constants for all the photocatalytic reactions were calculated from the experimental data and shown in Fig. 4b, the Ag6Si2O7/WO3 (1:1) has the highest apparent rate constant of 0.116 min−1, which was about 1.59 times as that of sole Ag6Si2O7 sample (0.073 min−1) and 44.6 times as that of sole WO3 (0.0026 min−1), suggesting that there is a significant synergistic effect between Ag6Si2O7 and WO3 for Ag6Si2O7/WO3 (1:1) under visible light irradiation.
The decolorization and mineralization at different time intervals under visible light irradiation were compared and are shown in Fig. S2.† 48.7% of the organic carbon content of MB was degraded in 60 min of the reaction time for Ag6Si2O7/WO3 (1:1) under visible light irradiation and is still much higher than that by sole Ag6Si2O7 (27.7%) or sole WO3 (14.9%) although the mineralization process is slower than decolorization.
As we know, the dye sensitization can contribute a lot to dye degradation.28,29 To further study the photocatalytic activity of Ag6Si2O7/WO3 (1:1) under visible light, colorless 2,4-dichlorophenol was also chosen as the model pollutant. From Fig. 5 we can see that the degradation and mineralization of 2,4-dichlorophenol by Ag6Si2O7/WO3 (1:1) is still higher than those by sole WO3 or Ag6Si2O7, also suggesting that the dye sensitization is not the main contribution of dye degradation. Moreover, we didn't find the generation of AgCl on the surface of catalysts after photocatalytic degradation by XRD determination (not shown). The possible reason is attributed to the lower concentration of initial 2,4-dichlorophenol (10 mg L−1).
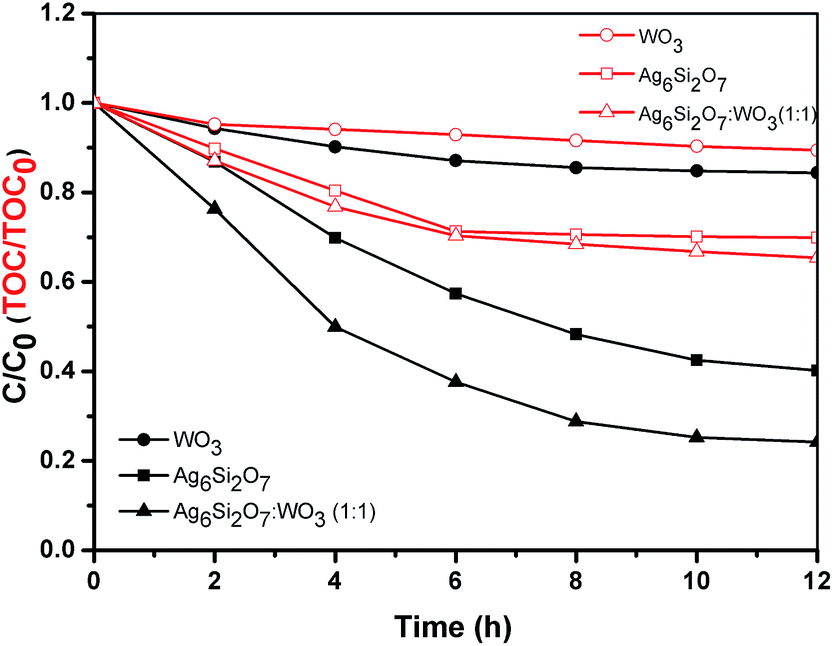 | ||
| Fig. 5 The degradation and mineralization of 2,4-dichlorophenol over Ag6Si2O7, WO3 and Ag6Si2O7/WO3 (1:1) composite under visible light irradiation. | ||
To further evaluate the visible-light photocatalytic performance of Ag6Si2O7/WO3 (1:1), photocatalytic activities of different photocatalysts with the same weight of visible-light-active components were compared, as shown in Fig. 6. It can be seen clearly that the photocatalytic activity of Ag6Si2O7/WO3 (1:1) (0.02 g) is much higher than that of mathematical sum of Ag6Si2O7 (0.0156 g) and WO3 (0.0044 g), in which they contain the same weight of visible-light active components of Ag6Si2O7 and WO3. After visible-light irradiation for 30 min the degradation efficiency of MB by Ag6Si2O7/WO3 (1:1) (0.02 g) is 97.4%, but the degradation efficiency by sum of Ag6Si2O7 (0.0156 g) and WO3 (0.0044 g) is only 73.4%. Especially, its k was 2.88 times that of sum of Ag6Si2O7 (0.0156 g) and WO3 (0.0044 g) (not shown). High photocatalytic activity of Ag6Si2O7/WO3 (1:1) can result from the decrease in recombination rate of electron–hole pairs, which can be attributed to the heterojunction between Ag6Si2O7 and WO3 formed in Ag6Si2O7/WO3 (1:1), as suggested by some literatures,30,31 the heterojunction can remarkably facilitate the separation of photoproduced electrons and holes.
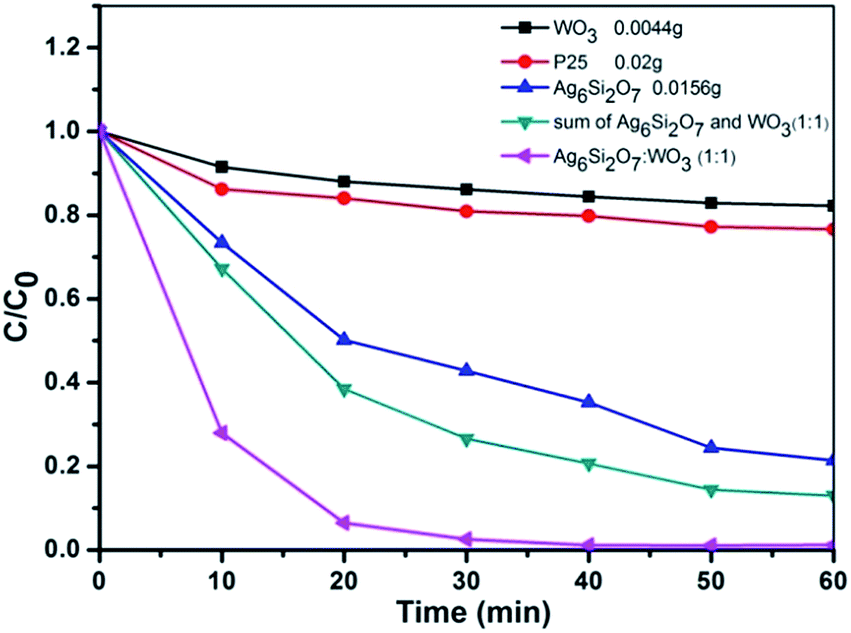 | ||
| Fig. 6 Comparison of photocatalytic activities of different photocatalysts with the same weight of visible-light-active component on the degradation of MB under visible light. | ||
To further evaluate its stability and reusability, the as-prepared Ag6Si2O7/WO3 (1:1) was repeatedly used for three recycles, and its photocatalytic performances are shown in Fig. 7. It can be observed that the photocatalytic activity of the as-prepared Ag6Si2O7/WO3 (1:1) has no apparent deactivation (10%) after three recycles and shows higher stability than sole Ag6Si2O7,16 indicating that the as-prepared Ag6Si2O7/WO3 (1:1) can serve as a stable and highly efficient photocatalyst. Good reusability of Ag6Si2O7/WO3 (1:1) for the degradation of 2,4-dichlorophenol also proves that the generation of AgCl on the surface of catalysts can be ignored.
 | ||
| Fig. 7 Cycling runs of Ag6Si2O7/WO3 (1:1) composite for the degradation of MB (a) and 2,4-dichlorophenol (b) under visible light irradiation. | ||
MO and RhB were also chosen as the model dye to evaluate the photocatalytic activity of Ag6Si2O7/WO3 (1:1). All the three dyes can be significantly photocatalytically degraded by Ag6Si2O7/WO3 (1:1) under visible light irradiation and the photocatalytic degradation efficiency decreases in the order: MB > RhB > MO (as shown in Fig. S3†). The possible reason is attributed to the molecular structure and the charge properties of the dyes that lead to different degradation efficiencies.32
3.3. Mechanism of photocatalytic activity enhancement
PL spectrum analysis was carried out to observe the separation rate of photo-induced electron–hole pairs over different samples. Lower fluorescence emission intensity implies lower electron–hole recombination rate and corresponds to higher photocatalytic activity.33–35 From Fig. 8 we can see that the Ag6Si2O7/WO3 photocatalysts exhibit the emission peaks locating at almost the same position with the sole WO3, but emission intensities decrease, suggesting that the recombination of photogenerated charge carriers was greatly inhibited in the Ag6Si2O7/WO3 photocatalysts. The emission intensity of Ag6Si2O7/WO3 (1:1) is even lower than that of sole Ag6Si2O7. The possible reason is ascribed to the heterojunction formed at interface between Ag6Si2O7 and WO3 in Ag6Si2O7/WO3 (1:1), which can prevent the recombination of photogenerated charge effectively.
 | ||
| Fig. 8 Photoluminescence spectra of Ag6Si2O7, WO3 and Ag6Si2O7/WO3 composites. | ||
It is generally accepted that the organic pollutants can be photodegraded via photocatalytic oxidation process. Therefore some active species, including hole (h+), hydroxyl (˙OH), superoxide radical (˙O2−), were examined by investigating the effects of different scavengers added on the degradation of MB by Ag6Si2O7/WO3 (1:1) in an attempt to elucidate the reaction mechanism. From Fig. S4† we can see that the photocatalytic degradation efficiency of MB was significantly depressed when the system is replenished with IPA, showing ˙OH is the main active species under visible light irradiation. It is also seen that the photocatalytic degradation of MB was almost not affected by the addition of EDTA-2Na or BQ, indicating that h+ and ˙O2− have minor effect on the degradation of MB.
On the basis of the above results, a possible mechanism for the higher visible-light photocatalytic activity of Ag6Si2O7/WO3 (1:1) than that of sole Ag6Si2O7 and WO3 was proposed (as shown in Fig. 9). Visible light can be absorbed efficiently by sole Ag6Si2O7 and WO3. Electrons in the valence bands of Ag6Si2O7 and WO3 can be excited to the conduction band and a high amount of electron–hole pairs are generated. For Ag6Si2O7/WO3 photocatalyst with an optimal mole ratio of Ag6Si2O7 to WO3, i.e. Ag6Si2O7/WO3 (1:1), Ag6Si2O7 particles deposited on the surface of WO3 particles, resulting in the formation of the heterostructure between Ag6Si2O7 and WO3. Some of the photogenerated electrons on Ag6Si2O7 particles could transfer easily to CB of WO3 through the intimate interface because the conduction edge potential of Ag6Si2O7 (0.44 eV vs. NHE) was lower than that of WO3 (0.64 eV vs. NHE). Many researchers also demonstrated that the differences in the CB edge potentials were probably a more powerful driving force, which promoted electron flow.36–38 Meanwhile, some of the photogenerated holes in the WO3 particles could transfer easily to VB of Ag6Si2O7 through the intimate interface because the valance edge potential of WO3 (3.34 eV vs. NHE) was higher than that of Ag6Si2O7 (2.02 eV vs. NHE), which promotes the effective separation of the electron–hole pairs. Since the VB levels of Ag6Si2O7 and WO3 were more positive than the potential of ˙OH/OH− (1.99 eV vs. NHE),39 as a result, the holes on the VB of both Ag6Si2O7 and WO3 could react with OH− adsorbed on the surface of the catalyst to generate ˙OH radical species. Compared with the reduction potential of oxygen E° (O2/˙O2−) (−0.046 eV vs. NHE),40 the electrons on the CB of WO3 and Ag6Si2O7 could not be taken up by O2 adsorbed on the surface of the catalyst to generate ˙O2− radical species because of more positive CB level of WO3 and Ag6Si2O7. However, since the CB levels of Ag6Si2O7 and WO3 were less positive than the potential of O2/H2O2 (0.695 eV vs. NHE for two-electron reduction), the electrons on the CB of both Ag6Si2O7 and WO3 could be consumed through a multi-electron reaction with oxygen to generated H2O2.24,41,42 And these produced H2O2 could further reacts with additional electron to produce ˙OH. Subsequently, the highly reactive ˙OH radical species participated in the photodecomposition of the MB aqueous solution.
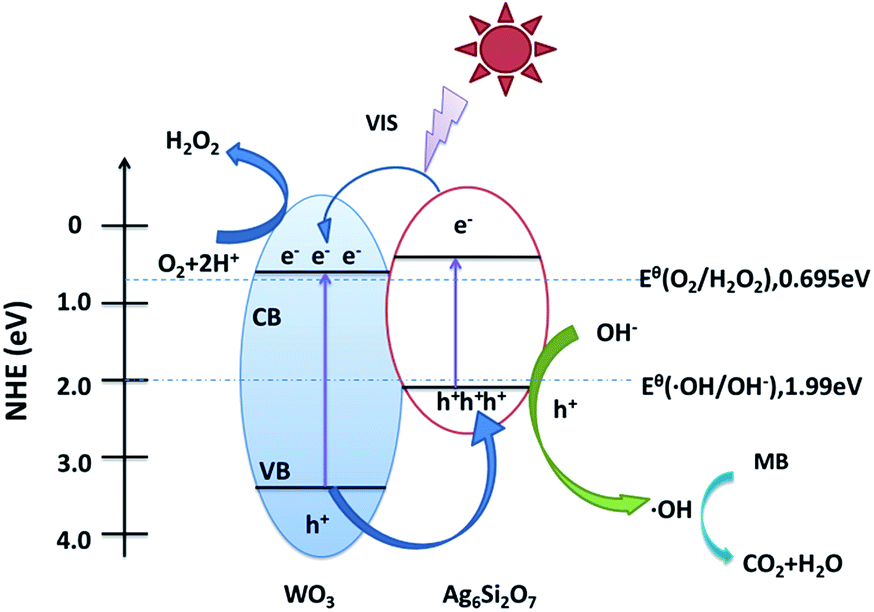 | ||
| Fig. 9 Schematic diagram of electron–hole pairs separation and the possible reaction mechanism of Ag6Si2O7/WO3 (1:1) composite under visible light irradiation. | ||
4. Conclusions
Ag6Si2O7/WO3 photocatalysts were prepared by ultrasound-assisted precipitation method and the effects of mole ratios of Ag6Si2O7/WO3 on photocatalytic activity of Ag6Si2O7/WO3 were investigated. The results showed that the photocatalytic degradation efficiency of MB by Ag6Si2O7/WO3 (1:1) attains 97.4%, which is much higher than that (87.3%) by sole Ag6Si2O7 and that (14.0%) by sole WO3 after visible light irradiation for 30 min, and the apparent rate constant of Ag6Si2O7/WO3 (1:1) is 1.59 times that of sole Ag6Si2O7 and 44.6 times that of sole WO3. RhB, MO and 2,4-dichlorophenol were also effectively degraded. After 3 recycling runs for the photodegradation of MB, the photocatalytic performance of the Ag6Si2O7/WO3 (1:1) was effectively maintained, indicating that Ag6Si2O7/WO3 heterojunction can somewhat inhibit deactivation of Ag6Si2O7. The quenching effects of different scavengers proved that the ˙OH plays important roles in the photocatalytic reaction under visible light irradiation. The highly efficient visible light photocatalytic activity of Ag6Si2O7/WO3 (1:1) came from the efficient separation of electron–hole pairs, which resulted from the heterojunction of Ag6Si2O7/WO3.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (No. 2652015261).References
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- L. Cao, Z. Gao, S. L. Suib, T. N. Obee, S. O. Hay and J. D. Freihaut, J. Catal., 2000, 196, 253 CrossRef CAS.
- M. V. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2013, 14, 13 CrossRef CAS.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625 CrossRef CAS PubMed.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286 CrossRef CAS PubMed.
- T. Saison, N. Chemin, C. Chanéac, O. Durupthy, V. Ruaux, L. Mariey, F. Maugé, P. Beaunier and J. P. Jolivet, J. Phys. Chem. C, 2011, 115, 5657 CAS.
- A. Walsh, Y. Yan, M. N. Huda, M. M. Al-Jassim and S. H. Wei, Chem. Mater., 2009, 21, 547 CrossRef CAS.
- J. Zhang, F. Shi, J. Lin, D. Chen, J. Gao, Z. Huang, X. Ding and C. Tang, Chem. Mater., 2008, 20, 2937 CrossRef CAS.
- G. R. Bamwenda and H. Arakawa, Appl. Catal., A, 2001, 210, 181 CrossRef CAS.
- H. Wang, P. Xu and T. Wang, Mater. Des., 2002, 23, 331 CrossRef CAS.
- J. Cao, B. Luo, H. Lin, B. Xu and S. Chen, J. Hazard. Mater., 2012, 217–218, 107 CrossRef CAS PubMed.
- G. Panthi, S. J. Park, T. W. Kim, H. J. Chung, S. T. Hong, M. Park and H. Y. Kim, J. Mater. Sci., 2015, 50, 4477 CrossRef CAS.
- X. Zhu, Z. Wang, B. Huang, W. Wei, Y. Dai, X. Zhang and X. Qin, APL Mater., 2015, 3, 104413 CrossRef.
- X. Ma, B. Lu, D. Li, R. Shi, C. Pan and Y. Zhu, J. Phys. Chem. C, 2011, 115, 4680 CAS.
- M. Ge, N. Zhu, Y. Zhao, J. Li and L. Liu, Ind. Eng. Chem. Res., 2012, 51, 5167 CrossRef CAS.
- Z. Lou, B. Huang, Z. Wang, X. Ma, R. Zhang, X. Zhang, X. Qin, Y. Dai and M. H. Whangbo, Chem. Mater., 2014, 26, 3873 CrossRef CAS.
- I. Grigioni, K. G. Stamplecoskie, E. Selli and P. V. Kamat, J. Phys. Chem. C, 2015, 119, 20792 CAS.
- G. H. He, C. J. Liang, Y. D. Ou, D. N. Liu, Y. P. Fang and Y. H. Xu, Mater. Res. Bull., 2013, 48, 2244 CrossRef CAS.
- J. Cao, B. Luo, H. Lin, B. Xu and S. Chen, Appl. Catal., B, 2012, 111–112, 288 CrossRef CAS.
- H. Widiyandari, A. Purwanto, R. Balgis, T. Ogi and K. Okuyama, Chem. Eng. J., 2012, 180, 323 CrossRef CAS.
- D. Li and H. Haneda, J. Photochem. Photobiol., A, 2003, 160, 203 CrossRef CAS.
- T. Nishiguchi, K. Oka, T. Matsumoto, H. Kanai, K. Utani and S. Imamura, Appl. Catal., A, 2006, 301, 66 CrossRef CAS.
- Y. Yan, H. Guan, S. Liu and R. Jiang, Ceram. Int., 2014, 40, 9095 CrossRef CAS.
- J. Zhang, K. Yu, Y. Yu, L. L. Lou, Z. Yang, J. Yang and S. Liu, J. Mol. Catal. A: Chem., 2014, 391, 12 CrossRef CAS.
- J. Cao, B. Luo, H. Lin, B. Xu and S. Chen, J. Hazard. Mater., 2012, 217, 107 CrossRef PubMed.
- L. Cheng, Y. Hou, B. Zhang, S. Yang, J. Guo, L. Wu and H. Yang, Chem. Commun., 2013, 49, 5945 RSC.
- B. Weng, J. Wu, N. Zhang and Y. Xu, Langmuir, 2014, 30, 5574 CrossRef CAS PubMed.
- L. Yu, X. Zhang, G. Li, Y. Cao, Y. Shao and D. Li, Appl. Catal., B, 2016, 187, 301 CrossRef CAS.
- J. Qin, J. Huo, P. Zhang, J. Zeng, T. Wang and H. Zeng, Nanoscale, 2016, 4, 2249 RSC.
- W. Cui, H. Wang, Y. Liang, B. Han, L. Liu and J. Hu, Chem. Eng. J., 2013, 230, 10 CrossRef CAS.
- S. Wu, H. Zheng, Y. Wu, W. Lin, T. Xu and M. Guan, Ceram. Int., 2014, 40, 14613 CrossRef CAS.
- S. Wang, F. Teng and Y. Zhao, RSC Adv., 2015, 5, 76588 RSC.
- J. Tian, Y. Sang, G. Yu, H. Jiang, X. Mu and H. Liu, Adv. Mater., 2013, 25, 5075 CrossRef CAS PubMed.
- K. Nagaveni, M. S. Hegde and G. Madras, J. Phys. Chem. B, 2004, 108, 20204 CrossRef CAS.
- Y. Lu, Y. Lin, T. Xie, S. Shi, H. Fan and D. Wang, Nanoscale, 2012, 4, 6393 RSC.
- R. Hao, G. Wang, H. Tang, L. Sun, C. Xu and D. Han, Appl. Catal., B, 2016, 187, 478 CrossRef.
- Y. X. Yang, Y. N. Guo, F. Y. Liu, X. Yuan, Y. H. Guo, S. Q. Zhang, W. Guo and M. X. Huo, Appl. Catal., B, 2013, 142, 828 CrossRef.
- Y. P. Zang, L. P. Li, Y. S. Xu, Y. Zuo and G. S. Li, J. Mater. Chem. A, 2014, 2, 15774 CAS.
- Y. M. He, L. H. Zhang, B. T. Teng and M. H. Fan, Environ. Sci. Technol., 2015, 49, 649 CrossRef CAS PubMed.
- B. Chai, T. Y. Peng, J. Mao, K. Li and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 16745 RSC.
- J. Kim, C. W. Lee and W. Choi, Environ. Sci. Technol., 2010, 44, 6849 CrossRef CAS PubMed.
- Y. Chang, K. Yu, C. Zhang, R. Li, P. Zhao, L. L. Lou and S. Liu, Appl. Catal., B, 2015, 176, 363 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23591c |
| This journal is © The Royal Society of Chemistry 2016 |