Production of γ-valerolactone from biomass-derived compounds using formic acid as a hydrogen source over supported metal catalysts in water solvent†
Pham Anh Son,
Shun Nishimura and
Kohki Ebitani*
School of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: ebitani@jaist.ac.jp; Fax: +81 761 51 1149; Tel: +81 761 51 1610
First published on 31st January 2014
Abstract
γ-Valerolactone (GVL) is a key intermediate for production of fuels and chemicals. In this research, GVL is synthesized from biomass-derived compounds using formic acid (FA) as a hydrogen source over various supported metal catalysts which are prepared by a simple impregnation or co-precipitation method. Under optimum conditions, levulinic acid (LA) is almost converted to GVL by Ru/C, Ru/SBA, Au/ZrC and Au/ZrO2 catalysts with above 90% yield in water solvent. Especially, the Au/ZrO2 showed excellent activity and recyclability; the Au/ZrO2 catalyst can decompose completely FA to CO2 and H2, which gives high yield of GVL (ca. 97%) from hydrogenation of LA, and can retain its activity for at least 5 recycle runs. GVL is also obtained from one-pot dehydration/hydrogenation reaction of fructose in water solvent. In this reaction, FA plays two roles: an acid catalyst for dehydration of fructose to LA, and a hydrogen source for hydrogenation of the obtained LA over supported metal catalysts. The Au/ZrO2 is the best catalyst for dehydration/hydrogenation reaction with overall GVL yield of 48% and can be reused several times.
1. Introduction
The rapid developments of industry and transportation all over the world have led to a drastic increase in the demand for fuels. Currently, over 84% of this demand is based on the burning of fossil fuels – non-renewable resources (oil 34%, gas 28% and coal 22%), but these resources are finite and becoming more expensive. Furthermore, the combustion of fossil fuels for the production of heat and power is associated with a worldwide increase in greenhouse gas levels which is considered the main cause of climate change.1–4Diminishing fossil resource reserves and degradation of the environment are strong driving forces for the search for sustainable and renewable resources.4 Various alternative energy sources have been developed, such as hydroelectric energy, wind power, geothermal energy, solar energy, and so on. Since the application of those energy sources might take longer than they are expected, the development of efficient processes to convert biomass resource into liquid fuels and valuable chemicals is a key research area in the next few decades.5–11 Biomass is an abundant and renewable alternative resource that is the best candidate to replace fossil fuels for the sustainable production of energy1–3,12,13 as well as chemicals.12,14–17
Some of the most important characteristics for an expected ideal platform chemical include the possibility to use it for production of both energy and carbon-based products, renewable, easy and safe to store and move in large quantities, low melting point, high boiling and flash points, low or non-toxicity, and easy for bio-degradation.6 Because of its versatile properties, γ-valerolactone (GVL) is considered as one of the most promising platform molecules that satisfy above requirements. GVL can be converted into liquid fuels, fuel additives,18,19 green solvents,12,20 food additives, and intermediates for chemicals and pharmaceutical industries.2,21,22
One of the most effective methods that can be used for production of GVL is the catalytic hydrogenation of levulinic acid (LA) which can be obtained from the hydrolysis followed by dehydration of carbohydrate compounds in acidic media.23,24 The hydrogenation of LA in vapor phase takes place at an atmospheric pressure of hydrogen and gives high yield of GVL.25–27 Nevertheless, vapor phase reaction needs huge amount of energy for vaporization of reactants. Hydrogenation of LA in liquid phase is more common; however, the reaction usually requires high pressured H2 gas (1.2–5.5 MPa). In liquid phase, hydrogenation reaction of LA can occur in the presence of either homogeneous or heterogeneous catalysts. The homogeneous catalysts are usually complexes of Ru and Ir.28–31 These homogeneous catalysts have high activities but are difficult to separate from the reaction mixture.
Many supported metal catalysts have been developed for liquid phase hydrogenation of LA under high pressured H2.32–34 In these works, various metals such as Ir, Rh, Pd, Ru, Re, Ni, etc. on different supports such as carbon and metal oxides have been screened. The results showed Ru catalyst supported on carbon gave the highest yield of GVL; however, a weak point of Ru/C is its recyclability. The reaction of carbohydrate compounds usually generates huge amount of undesired solid known as humins that deactivate the catalysts. The reuse of catalyst, therefore, needs calcination at high temperature of catalyst to combust humins. Under this condition, the Ru/C is destroyed completely.
Up to now, most studies have been used hydrogen gas that is a non-sustainable source for hydrogenation of LA, there have been few researches focusing on the utilization of the alternative hydrogen source. These are some advantages when using formic acid (FA) as the hydrogen resource compared with hydrogen gas because FA is cheaper, safer, giving higher atomic efficiency, and needing less instrumental requirements.35–39 The first purpose of this work is to synthesize GVL from LA using FA as a hydrogen source and heterogeneous catalysts in water solvent. We found that Ru/SBA-15 had good activity as well as Ru/C. Besides Ru/SBA, Au/ZrO2 and Au/ZrC acted as acid-tolerant catalysts with excellent catalytic activities for production of GVL from LA using FA as a hydrogen donor. The second topic of this work is one-pot dehydration/hydrogenation of fructose to GVL over supported metal catalysts in water solvent. To the best of our knowledge, this is the first report on production of GVL via dehydration/hydrogenation of fructose in the absence of mineral acid catalysts.
2. Experimental
2.1. Chemicals
Metal supported catalysts, metal oxides, and salts used in present work including 5 wt% Ru/C, 5 wt% Ru/Al2O3, 5 wt% Pt/C, 5 wt% Pd/C, RuCl3·3H2O, HAuCl4·4H2O, Cu(NO3)2·3H2O, Co(NO3)2·6H2O, Fe(NO3)3·9H2O, Ni(NO3)2·6H2O, activated carbon, mordenite zeolite (hydrogen form), formic acid (FA), and naphthalene were supplied from Wako Pure Chemical Ind., Ltd. Fructose, acetone, TiO2, ZrO2, Al2O3, ZrOCl2·8H2O, and Na2CO3 were obtained from Kanto Chemical Co., Inc. Levulinic acid (LA) was bought from Tokyo Chemical Industry Co., Ltd. Tri-block co-polymer poly(ethylene oxide)-poly(propylene oxide)-poly(ethyleneoxide) (P123, (EO)20(PO)70(EO)20, M = 5800), tetraethylorthosilicate (98%, TEOS), and γ-valerolactone (GVL) were purchased from Sigma Aldrich.2.2. Catalyst preparation
Preparations of supported metal catalysts were performed by impregnation method. This method is used for preparation of Ru, Pd, Pt or Au supported on mesoporous silica (SBA-type, see ESI†), TiO2, ZrO2, Al2O3, C, mordenite zeolite (hydrogen form), and ZrC (as-prepared zirconium carbonate according to the reference,40 see ESI†). Typically, 0.2 g of supporting material and 20 mL of water were introduced into a round-bottom flask, and then the mixture was stirred (500 rpm) at room temperature. Thereafter, a desired amount of metal precursor was added, and the mixture was kept at same conditions for 2 h (in case of Au catalysts, the pH of mixture was adjusted to 10 by an ammonia solution). Subsequently, the mixture was refluxed at 140 °C for 4 h. After cooling to room temperature, the obtained solid was collected by filtration, and washed with 1 L of water followed by drying at 100 °C overnight.Metals (Ru, Au, Ag, Fe, Co, Cu, and Ni) supported on ZrO2 were prepared by co-precipitation method. Typically, a 50 mL of 0.25 M Na2CO3 solution was added slowly (1 mL min−1) into a round-bottom flask containing a 20 mL solution of 0.5 M ZrOCl2·8H2O including the desired amount of metal precursor. The obtained gel was kept at room temperature under a violent stir for 2 h, followed aging at 140 °C for 12 h. Solid was recovered by filtration, extensively washed with 2 L of distilled water, and dried at 100 °C. After ground to fine powder, the solid was calcined at 500 °C for 4 h.
2.3. Procedure of catalytic reaction and product analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The temperature program for a column is described as follows: 40 °C isothermal for 0.5 min, 20 °C min−1 to 200 °C, 7 °C min−1 to 250 °C, and isothermal at 250 °C for 2 min. Retention times of acetone, GVL, LA, and naphthalene are observed at 1.5 min, 3.7 min, 4.6 min, and 5.7 min, respectively. The amounts of fructose and FA were determined by a high performance liquid chromatograph (HPLC, Waters Co., Ltd.) equipped with an Aminex HPX-87H column (Bio-Rad Laboratories, Inc.) and a refractive index detector. 10 mM H2SO4 aq. was used as an eluent at a flow rate of 0.5 mL min−1. Both column and detector were operated at 50 °C. Typically, fructose, FA, and HMF were obtained in the HPLC chart at 12.3 min, 17.2 min, and 39.5 min, respectively. All data were based on repeated runs and the error was less than 2%. Metal content in the catalyst was measured by inductively coupled plasma-atomic emission spectroscopy (ICPS-7000 Ver.2, Shimadzu Co., Ltd.). All samples was dissolved in aqua regia before ICP measurement.
:1. The temperature program for a column is described as follows: 40 °C isothermal for 0.5 min, 20 °C min−1 to 200 °C, 7 °C min−1 to 250 °C, and isothermal at 250 °C for 2 min. Retention times of acetone, GVL, LA, and naphthalene are observed at 1.5 min, 3.7 min, 4.6 min, and 5.7 min, respectively. The amounts of fructose and FA were determined by a high performance liquid chromatograph (HPLC, Waters Co., Ltd.) equipped with an Aminex HPX-87H column (Bio-Rad Laboratories, Inc.) and a refractive index detector. 10 mM H2SO4 aq. was used as an eluent at a flow rate of 0.5 mL min−1. Both column and detector were operated at 50 °C. Typically, fructose, FA, and HMF were obtained in the HPLC chart at 12.3 min, 17.2 min, and 39.5 min, respectively. All data were based on repeated runs and the error was less than 2%. Metal content in the catalyst was measured by inductively coupled plasma-atomic emission spectroscopy (ICPS-7000 Ver.2, Shimadzu Co., Ltd.). All samples was dissolved in aqua regia before ICP measurement.3. Results and discussion
3.1. Hydrogenation of LA to GVL
The reaction to GVL by LA hydrogenation is shown in Scheme 1. In this pathway, FA is decomposed to CO2 and H2 by supported metal catalyst. Subsequently, H2 takes part in the hydrogenation of LA to generate GVL.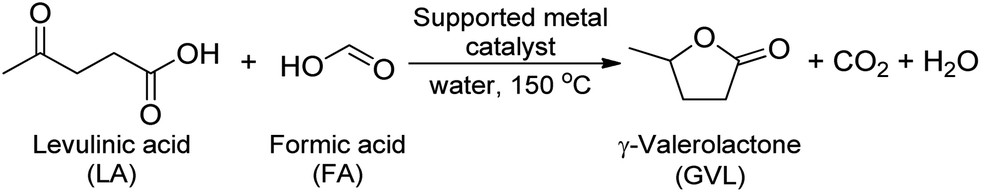 | ||
| Scheme 1 Hydrogenation of LA toward GVL using FA as a hydrogen source. | ||
Firstly, the reaction was carried out with 1/1 mole ratio of LA/FA at 150 °C using Ru-supported carbon as catalyst because Ru/C is the most common catalyst for hydrogenation of LA using H2 gas as reductant.32,34,41–43 The conversion and yield were monitored as functions of time (Fig. 1).‡ The reaction occurred quite fast and almost reached maximum performance after 5 h. Therefore, 5 h of reaction time was applied for next experiments.
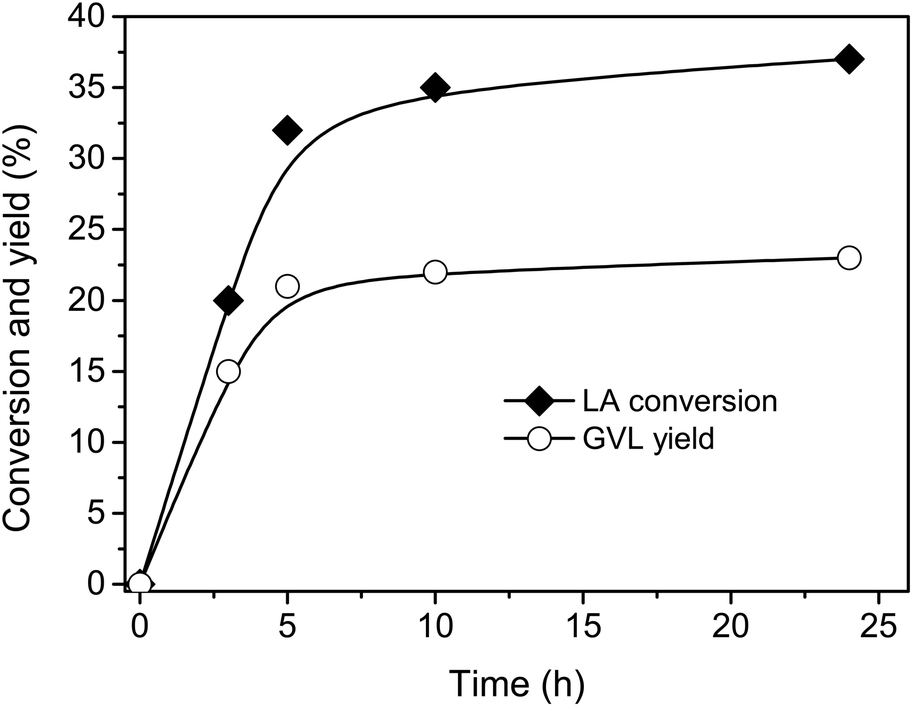 | ||
| Fig. 1 LA conversion (black diamond) and GVL yield (open circle) as a function of reaction time. Reaction conditions: LA (2 mmol), FA (2 mmol), 5 wt% Ru/C (0.02 g), water (1 mL), temperature (150 °C). | ||
In order to estimate the effect of support materials on the activity, the impregnation method was used for grafting Ru on various supports such as C, SBA-15 (ordered mesoporous SiO2), TiO2, ZrO2, and Al2O3 to afford Ru/C, Ru/SBA-15, Ru/TiO2, Ru/ZrO2, and Ru/Al2O3, respectively. The activities of these catalysts were listed in Table 1. Obtained results showed the similar activities of C and SBA-15 supported Ru catalysts (21–22% GVL yield, entries 1 and 2), while metal oxide supported Ru catalysts had much lower activities with trace amount of product (GVL yields were below 3%, entries 3–5). Because of good support for Ru catalyst, C support was also used for preparation of other catalyst such as the Pt/C, Pd/C, and Au/C; nevertheless, the activities of these catalysts were much lower than that of the Ru/C (1–2% GVL yield, entries 6–8).
| Entry | Catalyst | LA conv. (%) | GVL yield (%) | GVL sel. (%) |
|---|---|---|---|---|
| a Reaction conditions: LA (2 mmol), FA (2 mmol), 5 wt% supported metal catalyst (0.02 g), water (1 mL), temperature (150 °C), time (5 h). | ||||
| 1 | Ru/C | 29 | 21 | 73 |
| 2 | Ru/SBA-15 | 31 | 22 | 71 |
| 3 | Ru/Al2O3 | 16 | 3 | 17 |
| 4 | Ru/TiO2 | 10 | 2 | 20 |
| 5 | Ru/ZrO2 | 11 | 2 | 18 |
| 6 | Pt/C | 13 | 2 | 13 |
| 7 | Pd/C | 9 | 2 | 17 |
| 8 | Au/C | 13 | 1 | 9 |
Interestingly, Ojeda and Iglesia reported that well-dispersed Au species could selectively decompose gaseous FA.44 The obtained small Au clusters had very high activity in FA dissociation reaction and even better than some well-known catalysts such as Pt or Ru. In this report, various supported Au catalysts were also examined for hydrogenation of LA to GVL using FA as a hydrogen source (Fig. 2). Among tested catalysts, the Au/ZrO2 and Au/ZrC possessed the highest activity with 77% yield of GVL. Activities of the other metals on ZrO2 were quite low compared with that of Au (1–2% GVL yield, Table S1, ESI†). These results indicated that both of the noble metal and the nature of the support played important roles in the catalytic activity of LA hydrogenation to GVL using FA as a hydrogen source.
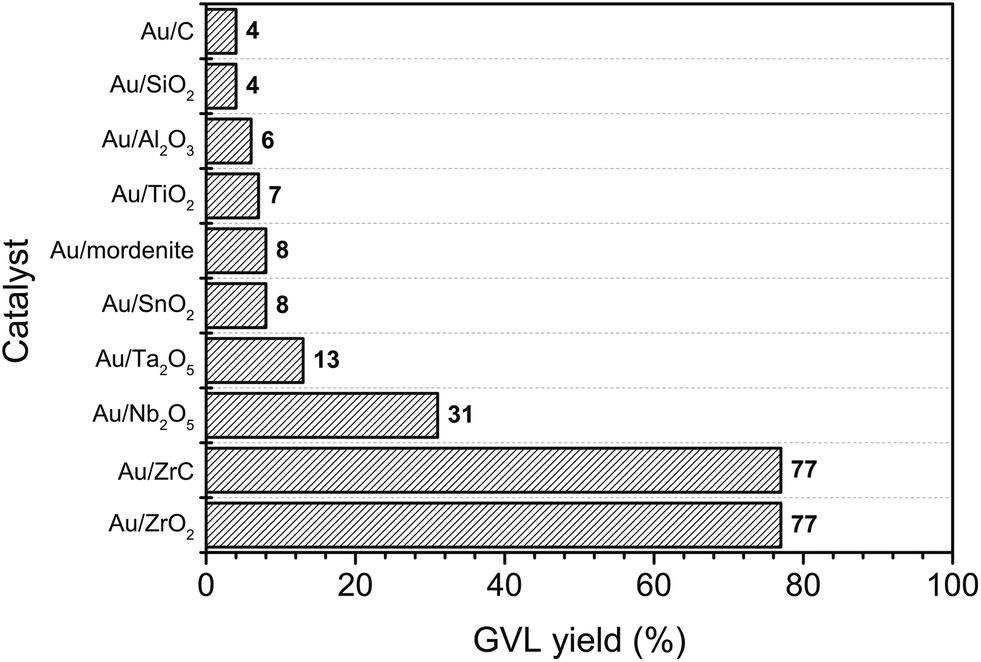 | ||
| Fig. 2 GVL yield from LA hydrogenation over 5 wt% Au catalyst on various supports. Reaction conditions: LA (2 mmol), FA (2 mmol), 5 wt% Au-supported catalyst (0.02 g), water (1 mL), temperature (150 °C), time (5 h). | ||
The influence of FA/LA mole ratio on LA conversion and GVL yield was investigated on 5 wt% Ru/C and 5 wt% Au/ZrO2 catalysts (Fig. 3). In case of 5 wt% Ru/C catalyst, the hydrogenation reaction is proportional to the initial amount of FA. With FA/LA mole ratio of 1, GVL yield was only 21%, while LA was converted completely giving 90% GVL yield when the FA/LA mole ratio changed to 3. At every FA/LA mole ratios, the Au/ZrO2 catalyst was more efficient than the Ru/C for hydrogenation of LA. 77% GVL yield was obtained over 5 wt% Au/ZrO2 at mole ratio of FA/LA = 1, while there was no significant difference in reaction performances with FA/LA mole ratios of 2 and 3 (94–97% GVL yield). Therefore, the best FA/LA mole ratio for 5 wt% Ru/C and 5 wt% Au/ZrO2 were 3 and 2, respectively. Higher activity of the Au/ZrO2 than Ru/C was due to the differences in their decomposition ability of FA (Table 2 and S2, ESI†); FA was decomposed completely at all investigated amounts with the Au/ZrO2 catalyst, whereas Ru/C converted only 56%, 49%, and 43% FA when using 2 mmol, 4 mmol, and 6 mmol of FA, respectively. It seems that the Au/ZrO2 was very stable in presence of FA, but the Ru/C was deactivated gradually by FA during the reaction. Indeed, Ru/C almost lost its activity after the first run and could not be recycled, in contrast to Au/ZrO2 (vide infra).
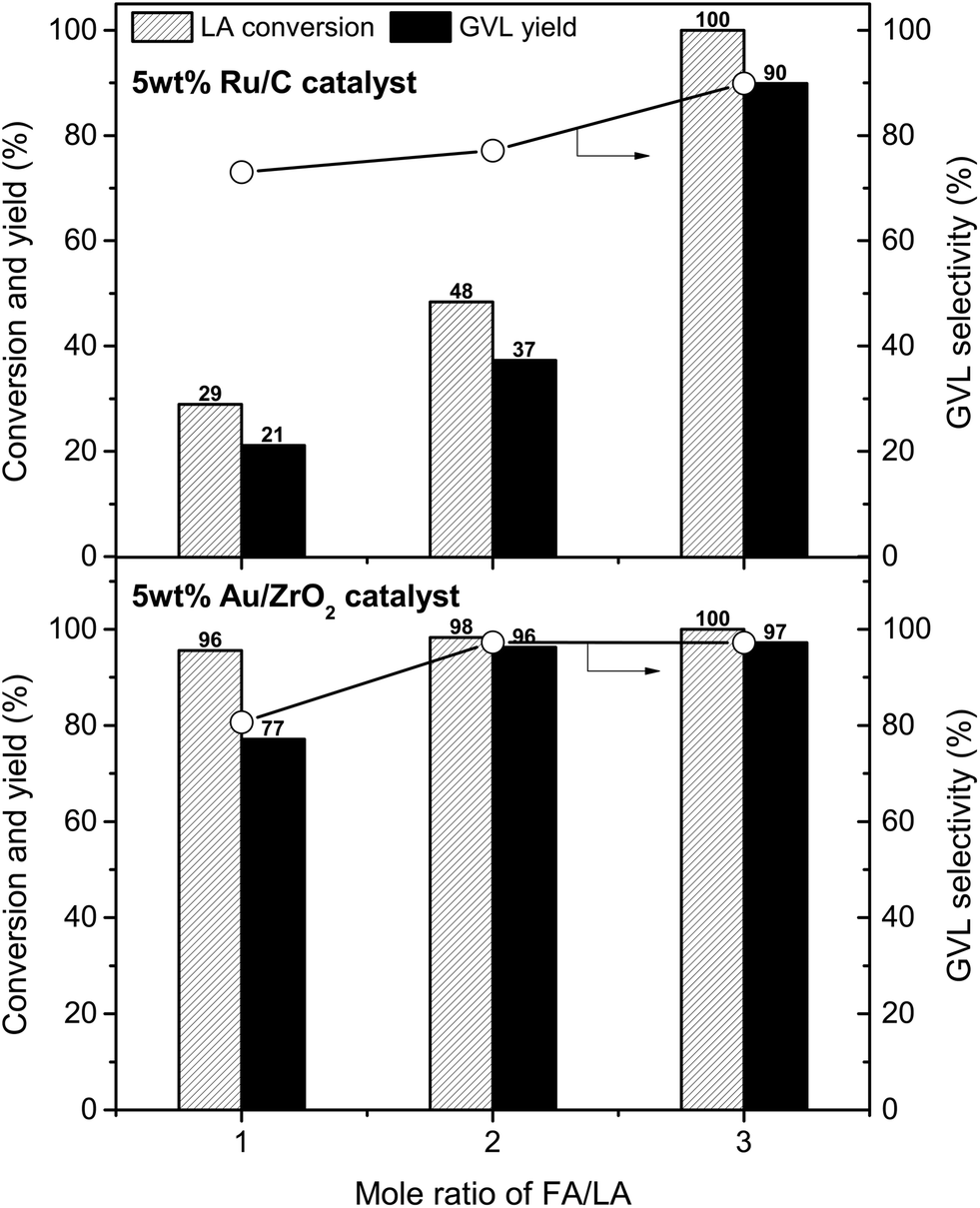 | ||
| Fig. 3 Effect of FA/LA mole ratio on the hydrogenation performance. Reaction conditions: LA (2 mmol), catalyst (0.02 g), water (1 mL), temperature (150 °C), time (5 h). | ||
| Entry | Catalyst | React. Temp. (°C) | LA conv. (%) | GVL yield (%) | GVL sel. (%) | FA Conv. (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: LA (2 mmol), FA (a 6 mmol, b 4 mmol), catalyst (0.02 g), water (1 mL), time (5 h). | ||||||
| 1 | 5 wt% Ru/Ca | 100 | 7 | 2 | 29 | 13 |
| 2 | 110 | 16 | 5 | 30 | 16 | |
| 3 | 120 | 30 | 12 | 39 | 19 | |
| 4 | 130 | 35 | 22 | 62 | 33 | |
| 5 | 140 | 50 | 32 | 63 | 37 | |
| 6 | 150 | 100 | 90 | 90 | 43 | |
| 7 | 5 wt% Au/ZrO2b | 100 | 7 | 2 | 28 | 10 |
| 8 | 110 | 19 | 11 | 56 | 24 | |
| 9 | 120 | 21 | 14 | 67 | 32 | |
| 10 | 130 | 40 | 26 | 67 | 52 | |
| 11 | 140 | 73 | 66 | 91 | 100 | |
| 12 | 150 | 100 | 97 | 97 | 100 | |
The hydrogenation of LA using FA as a hydrogen source strongly depended on reaction temperature. Two represented catalysts for this experiment were Ru/C and Au/ZrO2 (Table 2). At temperatures lower than 120 °C, GVL yield was less than 15% in both cases (entries 1–3, 7–9) but increased gradually when rising reaction temperature higher than 130 °C. At 150 °C, GVL yield suddenly achieved more than 90%. These results showed that at low temperatures (below 130 °C), FA was not decomposed significantly, but violently decomposed when reaction temperature was higher than 140 °C to release sufficient amount of hydrogen for the hydrogenation reaction. LA conversion only reached 100% when reaction temperature was elevated to 150 °C (entries 6 and 12). Therefore, the suitable temperature for decomposition of FA and consecutive hydrogenation of LA was 150 °C.
The activities of various contents of Au catalyst on ZrO2 were tested (Table S3, ESI†). The increase in percentage of Au led the rise of product yield. However, above 3 wt% of Au, the active sites on ZrO2 support became saturation leading to the unchanged GVL yield with 96–97%. The highest TON value of 630 was obtained with 3 wt% Au/ZrO2 catalyst.
In order to investigate the recycling properties of the catalyst, the Au/ZrO2 was removed from the reaction mixture by centrifugation, washed with acetone, dried at 80 °C for 2 h before subjected to further catalytic reaction. As a result, we found that the Au/ZrO2 could be recycled at least five times without significant loss of catalytic activity (Fig. 4).
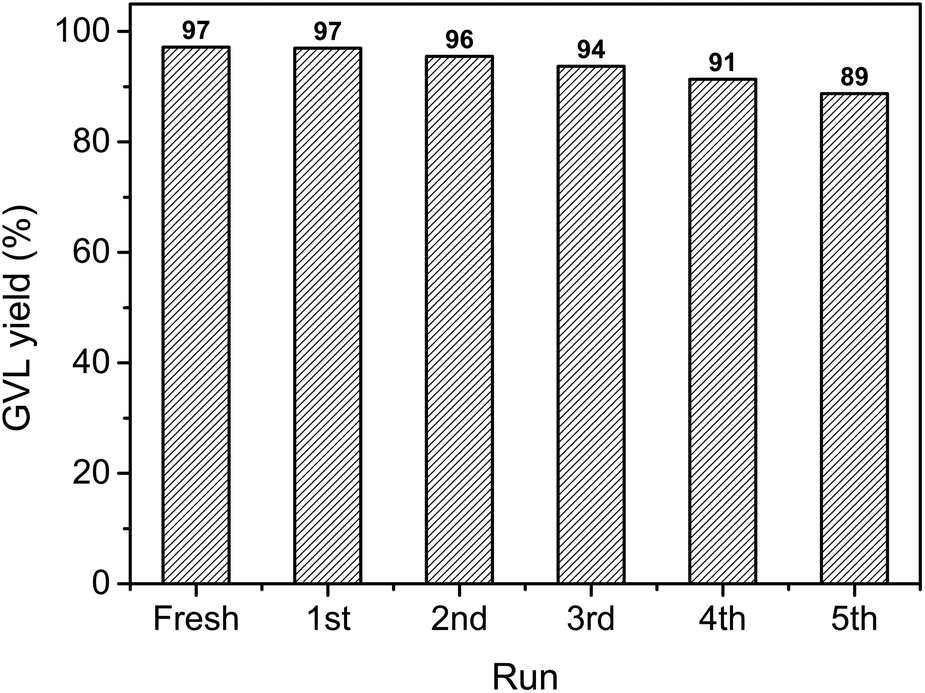 | ||
| Fig. 4 Recycling properties of 3 wt% Au/ZrO2 in the production of GVL via hydrogenation of LA with FA as hydrogen source. Reaction conditions: LA (2 mmol), FA (4 mmol), 3 wt% Au/ZrO2 catalyst (0.02 g), water (1 mL), temperature (150 °C), time (5 h). | ||
3.2. One-pot dehydration/hydrogenation reaction of fructose to GVL
The transformation of fructose to GVL undergoes two main steps: (i) acid-catalyzed dehydration of fructose yielding LA and (ii) hydrogenation of obtained LA to GVL (Scheme 2). The latter process is promoted by supported metal catalysts. As mentioned above, FA is decomposed slowly below 130 °C by supported metal catalysts. Fortunately, this temperature is suitable for dehydration of fructose to LA.45 Therefore, FA can serve as homogeneous acid catalyst for dehydration of fructose to LA at 120 °C. In next step, the temperature is elevated to 150 °C or higher to facilitate the decomposition of FA to H2, followed by hydrogenation of LA obtained in previous step to GVL. Consequently, it is expected that this multi-step reaction can be done in a one-pot manner. In this work, instead of utilization of mineral acids (H2SO4, HCl, etc.), FA was attempted to use as both an acid catalyst for dehydration of fructose step at low temperature (120 °C) and a hydrogen source for hydrogenation step that occurred over supported metal catalysts at higher temperature (150 °C).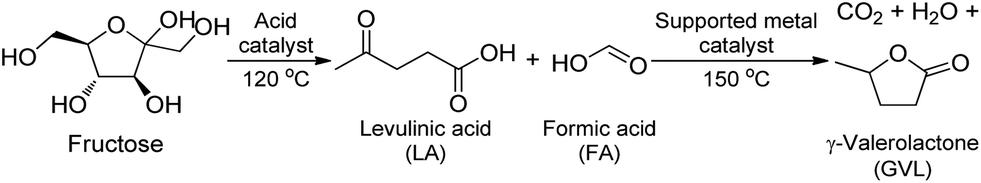 | ||
| Scheme 2 Dehydration/hydrogenation reaction of fructose to GVL using FA as both of an acid catalyst and a hydrogen source. | ||
A preliminary experiment was carried out to estimate the performance of dehydration of fructose by FA: a mixture including fructose (16 mmol) and FA (48 mmol) was heated at 120 °C for 3 h. HPLC analysis showed the complete conversion of fructose affording 50% LA yield and huge amount of undesired solid compounds, known as humins as side products in this reaction.
Four catalysts were selected for one-pot dehydration/hydrogenation of fructose included Au/ZrO2, Au/ZrC, Ru/SBA-15, and Ru/C. For direct conversion of fructose to GVL, the Au/ZrO2 exhibited an excellent catalytic activity with 48% yield of GVL (TON = 317). Result from Table 3 also showed that Au/ZrC might be another potential candidate of catalyst for this reaction, since activity of the Au/ZrC could be comparable to that of the Au/ZrO2. Ru catalysts had lower activities in this one-pot reaction (entries 3 and 4). The recyclability of Au/ZrO2 and Ru/SBA-15 catalysts were also tested. After the reaction, reaction mixture was separated by centrifugation to collect solid that contained both catalyst and humins. To remove humins and other products deposited on the catalysts, the solid was calcined at 500 °C for 4 h. Calcined Au/ZrO2 and Ru/SBA-15 were used for the next run. After 3 runs, activity of the Au/ZrO2 decreased gradually from 48 to 37%, while the Ru/SBA-15 lost its activity faster than the Au/ZrO2 catalyst (only 9% GVL yield was obtained for the 3rd run) (Fig. S1, ESI†). The Au/ZrO2 catalyst was still reused for several times though it was partly deactivated.
| Entry | Catalyst | Actual metal content (wt%)a | GVL yield (%) | TONb |
|---|---|---|---|---|
| a Reaction conditions: Fructose (2 mmol), FA (4 mmol), 3 wt%, supported metal catalyst (0.02 g), water (1 mL), temperature (150 °C), time (5 h). a Determined by ICP. b Turnover number (TON) is defined as mole of formed GVL per actual mole of supported metal. | ||||
| 1 | Au/ZrO2 | 3.02 | 48 | 317 |
| 2 | Au/ZrC | 2.81 | 47 | 307 |
| 3 | Ru/SBA-15 | 2.63 | 26 | 87 |
| 4 | Ru/C | 2.68 | 21 | 71 |
4. Conclusion
γ-Valerolactone (GVL) can be obtained from hydrogenation of levulinic acid (LA) using formic acid (FA) as hydrogen source and supported metal catalysts in water. For Ru catalyst, the best supports include carbon or mesoporous silica (SBA-15). Under optimized reaction conditions, the reaction with these catalysts can give at least 90% GVL yield. Au supported on zirconia and zirconium carbonate (denoted as Au/ZrO2 and Au/ZrC) are also found to be excellent catalysts for decomposing FA and hydrogenating LA to produce GVL with 97% yield. The Au/ZrO2 is found to be a highly stable, acid-tolerant catalyst and can be reused for 5 recycles without significant loss of its activity. In the one-pot transformation of fructose to GVL, FA acts as an acid catalyst for dehydration reaction of fructose to LA at 120 °C before participating in the hydrogenation stage as a hydrogen source at higher temperature (150 °C) to afford GVL. In this reaction, the Au/ZrO2 is also the best catalyst for one-pot synthesis of GVL from fructose in water which provides 48% yield of GVL.Acknowledgements
This work is supported by Scientific Research (C) (no. 25420825) under Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. P.A.S. is thankful to 322 Project of Ministry of Education and Training (MOET) from Vietnam Government for the scholarship.Notes and references
- J. C. Escobar, E. S. Lora, O. J. Venturini, E. E. Yanez, E. F. Castillo and O. Almazan, Renewable Sustainable Energy Rev., 2009, 13, 1275–1287 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- P. S. Nigam and A. Singh, Prog. Energy Combust. Sci., 2011, 37, 52–68 CrossRef CAS PubMed.
- International Energy Outlook, 2013, (IEO2013), http://www.eia.doe.gov/forecasts/ieo/.
- K. Yan, C. Jarvis, T. Lafleur, Y. Qiao and X. Xie, RSC Adv., 2013, 3, 25865–25871 RSC.
- I. T. Horváth, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- D. O. Otieno and B. K. Ahring, Bioresour. Technol., 2012, 112, 285–292 CrossRef CAS PubMed.
- S. Dutta, RSC Adv., 2012, 2, 12575–12593 RSC.
- M. Marzo, A. Gervasini and P. Carniti, Catal. Today, 2012, 192, 89–95 CrossRef CAS PubMed.
- I. Sádaba, S. Lima, A. A. Valente and M. López-Granados, Carbohydr. Res., 2011, 346, 2785–2791 CrossRef PubMed.
- L. Hu, G. Zhao, W. Hao, X. Tang, Y. Sun, L. Lin and S. Liu, RSC Adv., 2012, 2, 11184–11206 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- A. Takagaki, S. Nishimura and K. Ebitani, Catal. Surv. Asia, 2012, 16, 164–182 CrossRef CAS.
- J. Zhang, S. B. Wu, B. Li and H. D. Zhang, ChemCatChem, 2012, 4, 1230–1237 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- J. Q. Bond, D. M. Alonso, R. M. West and J. A. Dumesic, Langmuir, 2010, 26, 16291–16298 CrossRef CAS PubMed.
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203 CAS.
- R. Palkovits, Angew. Chem., Int. Ed., 2010, 49, 4336–4338 CrossRef CAS PubMed.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed.
- G. Novodárszki, N. Rétfalvi, G. Dibó, P. Mizsey, E. Cséfalvay and L. T. Mika, RSC Adv., 2014, 4, 2081–2088 RSC.
- Z. Sun, M. Cheng, H. Li, T. Shi, M. Yuan, X. Wang and Z. Jiang, RSC Adv., 2012, 2, 9058–9065 RSC.
- A. P. Dunlop and J. W. Madden, US Pat. 2786852, 1957.
- P. P. Upare, J. M. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J. S. Chang, J. Ind. Eng. Chem., 2011, 17, 287–292 CrossRef CAS PubMed.
- P. P. Upare, J. M. Lee, Y. K. Hwang, D. W. Hwang, J. H. Lee, S. B. Halligudi, J. S. Hwang and J. S. Chang, ChemSusChem, 2011, 4, 1749–1752 CrossRef CAS PubMed.
- M. Chalid, A. A. Broekhuis and H. J. Heeres, J. Mol. Catal. A: Chem., 2011, 341, 14–21 CrossRef CAS PubMed.
- W. Li, J. H. Xie, H. Lin and Q. L. Zhou, Green Chem., 2012, 14, 2388–2390 RSC.
- S. A. Rajeev and A. C. Larry, Chem. Phys. Lett., 2012, 541, 21–26 CrossRef PubMed.
- J. Deng, Y. Wang, T. Pan, Q. Xu, Q. X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1163–1167 CrossRef CAS PubMed.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS PubMed.
- Z. Yan, L. Lin and S. Liu, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
- M. G. Al-Shaal, W. R. H. William and R. Palkovits, Green Chem., 2012, 14, 1260–1263 RSC.
- L. Deng, J. Li, D. M. Lai, Y. Fu and Q. X. Guo, Angew. Chem., 2009, 121, 6651–6654 CrossRef.
- L. Deng, Y. Zhao, J. Li, Y. Fu, B. Liao and Q. X. Guo, ChemSusChem, 2010, 3, 1172–1175 CrossRef CAS PubMed.
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247–1255 RSC.
- D. M. Alonso, S. G. Wettstein, J. Q. Bond, T. W. Root and J. A. Dumesic, ChemSusChem, 2011, 4, 1078–1081 CrossRef CAS PubMed.
- J. Tuteja, H. Choudhary, S. Nishimura and K. Ebitani, ChemSusChem, 2014, 7, 96–100 CrossRef CAS PubMed.
- P. A. Son, S. Nishimura and K. Ebitani, React. Kinet., Mech. Catal., 2014, 111, 183–197 CrossRef CAS.
- A. M. R. Galletti, C. Antonetti, V. D. Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC.
- R. A. Bourne, J. G. Stevens, J. Ke and M. Poliakoff, Chem. Commun., 2007, 4632–4634 RSC.
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 4800–4803 CrossRef CAS PubMed.
- P. A. Son, S. Nishimura and K. Ebitani, React. Kinet., Mech. Catal., 2012, 106, 185–192 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation method, hydrogenation of LA over various metal supported ZrO2, decomposability of FA, and recylability. See DOI: 10.1039/c3ra47580h |
| ‡ The difference between conversion and yield may be caused by the formation of intermediates or unidentified compounds formed during the reaction. |
| This journal is © The Royal Society of Chemistry 2014 |