
One-dimensional assembly of TiO2 nanoparticles toward enhancing light harvesting and electron transport for application in dye-sensitized solar cells
Abstract
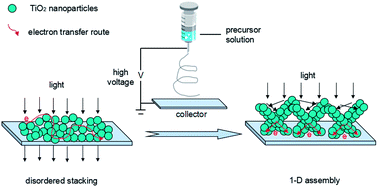
One-dimensional (1D) assembly of TiO2 nanoparticles (NPs) was successfully achieved by an improved electrospinning technique. Electrospinning precursor solution was prepared with conventional TiO2 NPs and polyvinylpyrrolidone dispersed in ethanol. The newly developed 1D assembly of nanoparticles (AS-NP) has been introduced into the photoanode in dye-sensitized solar cells (DSSCs). Compared to the traditional disordered stacking of TiO2 NPs, the AS-NPs bring in faster charge transport and longer electron lifetime, as well as a higher light scattering ability (especially in the wavelength range from 500 nm to 650 nm). It is exhibited that the AS-NPs can enhance electron transport and light scattering, while retaining the merits of NP morphology. Consequently, the efficiency of a cell based on an AS-NP/NP bilayer photoanode could be improved by about 15% in comparison to a reference cell made of absolute TiO2 NPs.
 Please wait while we load your content...
Please wait while we load your content...

