An isoindigo and dithieno[3,2-b:2′,3′-d]silole copolymer for polymer solar cells†
Romain
Stalder
a,
Caroline
Grand
a,
Jegadesan
Subbiah
b,
Franky
So
b and
John R.
Reynolds
*a
aThe George and Josephine Butler Polymer Research Laboratory, Department of Chemistry, Center for Macromolecular Science and Engineering, University of Florida, Gainesville, FL 32611, USA. E-mail: reynolds@chem.ufl.edu
bDepartment of Materials Science and Engineering, University of Florida, Gainesville, FL 32611, USA
First published on 27th October 2011
Abstract
The copolymer of isoindigo and dithieno[3,2-b:2′,3′-d]silole, P(iI-DTS), is reported as prepared by Stille coupling to yield a soluble high molecular weight material absorbing light throughout the visible spectrum up to 800 nm. With deep HOMO and LUMO energy levels (high ionization potential and electron affinity) electrochemically measured at −5.55 and −3.95 eV respectively, this new p-type polymer enabled the fabrication of high open circuit voltage polymer solar cells when blended with fullerene derivatives. By employing solvent additives, the morphology of the devices was optimized to yield power conversion efficiencies of 4%.
Introduction
In order to complement other renewable sources of energy, including different kinds of thin film technologies,1 the field of organic photovoltaics (OPVs) has pushed for the development of photonic materials capable of solution processing for large area low-cost fabrication on light-weight flexible substrates.2,3Conjugated polymers, in bulk heterojunctions (BHJs) with fullerene derivatives,4 were deemed capable of such task in the last decade as polymer solar cells (PSCs) became better understood thus more efficient: due to the limited absorption of n-type fullerene derivatives,5–7 it became evident that extending the absorption of the p-type conjugated polymer counterpart while properly tuning the energy of its frontier molecular orbitals would lead to better performing BHJ PSCs.8–10 The donor–acceptor (D–A) approach11 for the rational synthetic design of conjugated polymers enabled the precise tuning of these organic semiconductors' properties,12,13 ultimately leading to power conversion efficiencies (PCEs) now exceeding 7% for PSCs.14–17The use of isoindigo (iI) as a new acceptor in conjugated systems taking advantage of the D–A approach for OPVs was first developed in our group in iI-based small molecules.18 We, and others, followed this with D–A conjugated polymers19–24 reaching 3% efficiency in BHJ solar cells with PCBM,24 and hole mobilities of 0.79 cm2V−1s−1 in air-stable p-type OFETs.21 During the preparation of this Communication, Andersson et al. reported an isoindigo-based copolymer (having a different repeat unit structure) affording 6.3% efficiency, which is the highest for materials based on this acceptor.25 We recently reported the synthesis of the homopolymer of isoindigo—an all–acceptor conjugated polymer—and its use in all–polymer BHJ solar cells with P3HT, yielding efficiencies approaching 0.5%.26 Remarkably, the LUMO energy levels of all iI-based conjugated materials reported so far are confined to the −3.7 to −3.9 eV range. With bandgaps between 1.9 and 1.5 eV depending on the aromatic units conjugated with isoindigo, the HOMO energy levels are between −5.5 and −5.9 eV. Such high ionization potentials (deep HOMOs) for D–A polymers lead to devices with high open circuit voltages (Voc) for BHJs with fullerene derivatives, and with scalable high yielding synthesis, along with extended absorption towards the near-IR and high hole mobilities, these polymers are strong candidates for high efficiency low-cost solar cells.
In an effort to tailor the structure of iI-based D–A polymers for optimized PSCs, we report the synthesis of the copolymer of isoindigo and dithieno[3,2-b:2′,3′-d]silole, P(iI-DTS), along with its optical and electrochemical properties. This high molecular weight soluble polymer, absorbing light up to 800 nm with a HOMO energy level at −5.55 eV, was used in PSCs in BHJ with PC70BM. Employing solvent additives during active layer processing allowed us to increase the power conversion efficiency up to 4%, mainly due to improved BHJ morphology as indicated by atomic force and transmission electron microscopy.
Results and discussion
Polymer synthesis and characterization
In the design of an iI-based donor–acceptor copolymer as a p-type material for BHJ solar cells, the electron-rich comonomer should be chosen such that suitable backbone planarity, hole delocalization/mobility and solubility are achieved when copolymerized with dibromoisoindigo. The simplest electron-rich moiety serving this purpose is one thiophene ring, which was used by Wang and co-workers in an iI-based conjugated polymer achieving 3% solar cell efficiency.24 Extending the donor unit length to two thiophene rings likely increases the delocalization of positive charge carriers along the backbone thereby enhancing the p-type character of the iI-based polymer, as demonstrated by Lei and co-workers.21 In copolymers based on different acceptors than isoindigo, the presence of a bridging atom such as carbon (cyclopentadithiophene, CPDT) or silicon (dithienosilole, DTS) between two thiophene rings has been shown to further planarize the electron-rich unit, while providing an alkylation site for solubility purposes. The silicon bridge of DTS is advantageous as the alkyl chains stemming from silicon are able to remain in-plane to a greater extent than in CPDT, resulting in a more planar backbone.27,28 Therefore, we suspected DTS to be an electron-rich unit best suited for high efficiency D–A copolymers based on isoindigo as a conjugated acceptor. For solubility purposes, we functionalized both monomers with 2-ethylhexyl side-chains. The DTS moiety was prepared and converted to its ditin derivative following a previously reported procedure,14 and was purified by preparative HPLC in order to guarantee proper functional group stoichiometry during polymerization (see Figures S1 and S2†). We then copolymerized the 6,6′-dibromo-N,N′-(2-ethylhexyl)-isoindigo monomer 1 with 2,2′-bistrimethylstannyl-4,4′-bis-(2-ethylhexyl)-dithieno[3,2-b:2′,3′-d]silole monomer 2 under Stille coupling conditions to afford P(iI-DTS) in 94% overall yield after purification. The copolymerization was carried out using Pd2dba3 as Pd(0) source and P(o-tolyl)3 as ligand, in dry degassed toluene at 85 °C as displayed in Scheme 1.![Synthesis of the copolymer of isoindigo and dithieno[3,2-b:2′,3′-d]silole, P(iI-DTS), by Stille cross-coupling.](/image/article/2012/PY/c1py00402f/c1py00402f-s1.gif) | ||
| Scheme 1 Synthesis of the copolymer of isoindigo and dithieno[3,2-b:2′,3′-d]silole, P(iI-DTS), by Stille cross-coupling. | ||
Before quenching the reaction, 2-bromothiophene and 2-tributyltin thiophene were added in succession in the reaction medium as an attempt to replace undesired backbone chain-end groups with thiophene rings.29 After purification of the polymer in a Soxhlet extractor using methanol and hexanes, the high molecular weight fraction of P(iI-DTS) extracted with chloroform was analyzed using size exclusion chromatography in THF against polystyrene standards. The polymer from the chloroform fraction used in the following study has a number average molecular weight of 36.0 kDa and a polydispersity of 2.77, and is soluble in all chlorinated solvents and in THF and toluene. The analyzed elemental composition for C, H and N is within 0.4% of the calculated elemental composition. From the thermogravimetric analysis (Figure S5†) performed under nitrogen with a 5% weight loss set as decomposition threshold, the polymer was found to be thermally stable up to 410 °C.
Optical and electrochemical properties
The UV–vis absorption spectrum of P(iI-DTS) in solution and in thin films is shown in Fig. 1. With a peak absorption (λmax) at 720 nm and a low-energy onset of absorption (λonset) at 805 nm in the solid state, the polymer absorbs strongly in the visible towards the near-IR with an optical bandgap of 1.54 eV as calculated from λonset. At wavelengths less than 550 nm, the absorption decreases, peaking at 435 nm with 33% of the intensity of the λmax at 720 nm. Thin films of P(iI-DTS) thus look blue-green as they absorb mostly in the red region of the visible spectrum. This absorption gap at wavelengths lower than 550 nm is compensated by the absorption of PC70BM, and blend films of P(iI-DTS):PC70BM absorb broadly across the entire visible (Figure S6†).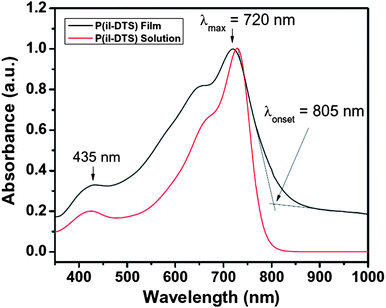 | ||
| Fig. 1 Absorption spectra of P(iI-DTS) in solution (red line) and in thin films (black line). | ||
We used cyclic voltammetry (CV) and differential pulse voltammetry (DPV) to investigate the electrochemical properties of the polymer and measure its frontier orbital energies, on thin films drop-cast onto Pt button electrodes, using 0.1M tetrabutylammonium hexafluorophosphate (TBAPF6) in acetonitrile as supporting electrolyte, as shown in Figure S7.†
Since we used a Ag/Ag+ reference electrode calibrated against Fc/Fc+, the potentials reported in the following are versusFc/Fc+. In the oxidative CV (Figure S7†), one reversible oxidation process was observed with a half-wave potential at 0.69 V. In the oxidative DPV experiment, we recorded an onset of oxidation at 0.45 V. Assuming that the energy of SCE is 4.7 eV vs. vacuum,30Fc/Fc+ being at 0.38 V vs.SCE31 sets the latter redox standard at 5.1 eV vs. vacuum, as highlighted by Bazan and co-workers recently.32 We thus calculated the HOMO energy level to be −5.55 eV. The reductive CV shows two reversible reduction processes, with half-wave potentials at −1.17 V and −1.57 V. From the onset of reduction at −1.15 V in the DPV, we calculated the LUMO level to be at −3.95 eV. The electrochemical bandgap of 1.60 eV is consistent with the optical bandgap of 1.54 eV. The high value of the LUMO at −3.95 eV is closer to that of PC70BM than the ∼0.3 eV offset recommended for efficient electron transfer, but the extended absorption of the polymer, and the high ionization potential (deep HOMO) of −5.55 eV were promising for devices with high Voc, as the latter is closely related to the offset of the LUMO of the fullerene derivative and the HOMO of the p-type polymer in BHJ solar cells.
Polymer solar cells
We first investigated a conventional cell architecture based on ITO/PEDOT:PSS/P(iI-DTS):PCBM/LiF/Al, with PC60BM and PC70BM as electron acceptors. After initial device testing, PC70BM was deemed a better acceptor than PC60BM, because of the enhanced photon absorption of the cells under AM1.5 illumination, due to the more extended absorption of PC70BM in the visible. The ratio of P(iI-DTS) to PC70BM, solvent (chloroform (CF) and chlorobenzene (CB)), solution concentration, spin-coating speed and annealing conditions were optimized. A donor/acceptor weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in CB at a concentration of 25 mg/mL spun cast at 1000 rpm and annealed at 150 °C before LiF/Al deposition gave the best efficiency. The J–V curves of the optimized BHJ obtained under AM1.5 illumination (100 mWcm−2) are shown in Fig. 2.
:4 in CB at a concentration of 25 mg/mL spun cast at 1000 rpm and annealed at 150 °C before LiF/Al deposition gave the best efficiency. The J–V curves of the optimized BHJ obtained under AM1.5 illumination (100 mWcm−2) are shown in Fig. 2.
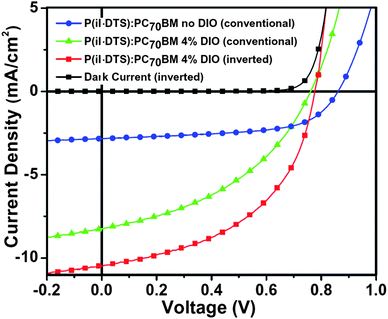 | ||
| Fig. 2 J–V curves of the P(iI-DTS):PC70BM (1:4) based BHJ solar cells with and without DIO additive, under AM1.5 solar illumination, in conventional (blue and green lines) and inverted architecture (red line). | ||
Consistent with the high HOMO energy value of the polymer, the P(iI-DTS):PC70BM cell has a high Voc of 0.86 V (blue line). Despite the high fill factor (FF) of 60%, the efficiency is limited by the low short circuit current (Jsc) of 2.82 mA/cm2. This low Jsc can be explained by the large domains observed under AFM on the order of 0.5 microns displayed in Fig. 3a and 3c, which entails a limited donor/acceptor interface in the BHJ, leading to a reduced number of excitons being dissociated in the film. Large phase separation likely decreases the carrier recombination rate, which could explain the observed high FF. In these optimized cells, the efficiency was found to be of 1.45% (Table 1).
 | ||
| Fig. 3
AFM images of the P(iI-DTS):PC70BM blend at 1:4 ratio, processed without (a) and with (b) 4% DIO additive (2 μm-side, 20 nm-height scales). TEM images of the aforementioned blend, processed without (c) and with (d) 4% DIO additive (200 nm scale bars). | ||
:4) blend
| Device processing | Jsc(mA/cm2) | Voc (V) | FF (%) | PCE (%) |
|---|---|---|---|---|
| Conventional cell without DIO | 2.82 | 0.86 | 60 | 1.45 |
| Conventional cell with DIO (4%) | 8.26 | 0.76 | 42 | 2.62 |
| Inverted cell with DIO (4%) | 10.49 | 0.77 | 50 | 4.01 |
The use of solvent additives such as octanedithiol or diiodooctane (DIO) has previously been shown to decrease the domain sizes in the BHJ of PSCs.33,34 Given the morphological limitations stated above, we monitored the effect of two solvent additives (1,8-diiodooctane (DIO), chloronaphthalene) on device performance. As shown in the AFM and TEM images in Fig. 3b and 3d, we found that the addition of 4% in volume of DIO in the spin-casting solution significantly reduces the features sizes in the active layer.
While the AFM images show a smoother surface morphology, the TEM images reveal an intricate network of donor/acceptor phases in the bulk of the film, similar to that observed in previously reported studies,14,35 with segregation scales reduced from 0.5 micron without additives to tens of nanometers with 4% DIO. Because of the reduced domain size, excitons are more likely to reach the P(iI-DTS)/PC70BM interface and generate charge carriers. As can be seen in Table 1, 4% DIO additive leads to a three-fold increase of the Jsc. To further improve carrier extraction at the electrodes, devices using the inverted architecture ITO/ZnO/P(iI-DTS):PC70BM(1:4)/MoO3/Ag were fabricated, while keeping the processing conditions for the active layer the same. As can be seen in Fig. 2 and Table 1, this architecture leads to increased device performance from 2.62% to 4.01%, since it is likely to take better advantage of a vertical phase separation present in the BHJ film.14
Conclusions
Synthesized by Stille cross-coupling, the copolymerP(iI-DTS) of isoindigo and dithienosilole is soluble at high molecular weight when functionalized with 2-ethylhexyl side chains on each conjugated unit, and absorbs light up to 800 nm in the solid state. With HOMO and LUMO energy levels at −5.55 and −3.95 eV vs. vacuum respectively, this polymer has a bandgap of 1.60 eV as measured by DPV in the solid state, which correlates well with an optical bandgap of 1.54 eV. Despite its high electron affinity (deep LUMO) close to that of fullerene derivatives, P(iI-DTS) still enables moderate PCE of 1.45% when blended with PC70BM in PSCs processed without solvent additives: the high open circuit voltage of 0.86 V is undermined by a low short circuit current. When 4% diiodooctane was used as an additive, we measured a PCE increase to 2.62%, accountable to improved film morphology for charge separation. When the device architecture was modified to enhance carrier extraction at the electrodes, the P(iI-DTS):PC70BM cells reached 4% PCE, one of the highest reported so far for isoindigo-based conjugated polymers.Acknowledgements
We gratefully acknowledge the Air Force Office of Scientific Research (FA9550-09-1-0320) and the Office of Naval Research (N-00014-11-1-0245) for financial support. R.S. acknowledges the University of Florida Alumni Awards Program for a fellowship.Notes and references
- T. D. Nielsen, C. Cruickshank, S. Foged, J. Thorsen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2010, 94, 1553–1571 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394–412 CrossRef CAS.
- C. N. Hoth, P. Schilinsky, S. A. Choulis and C. J. Brabec, Nano Lett., 2008, 8, 2806–2813 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- J. A. Mikroyannidis, A. N. Kabanakis, S. S. Sharma and G. D. Sharma, Adv. Funct. Mater., 2011, 21, 746–755 CrossRef CAS.
- A. Varotto, N. D. Treat, J. Jo, C. G. Shuttle, N. A. Batara, F. G. Brunetti, J. H. Seo, M. L. Chabinyc, C. J. Hawker, A. J. Heeger and F. Wudl, Angew. Chem., Int. Ed., 2011, 50, 5166–5169 CrossRef CAS.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371–3375 CrossRef CAS.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS.
- J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062–10065 CrossRef CAS.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R.m. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS.
- G. Zhang, Y. Fu, Z. Xie and Q. Zhang, Macromolecules, 2011, 44, 1414–1420 CAS.
- B. Liu, Y. Zou, B. Peng, B. Zhao, K. Huang, Y. He and C. Pan, Polym. Chem., 2011, 2, 1156–1162 RSC.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS.
- X. Xu, L. Li, B. Liu and Y. Zou, Appl. Phys. Lett., 2011, 98, 063303 CrossRef.
- R. Stalder, J. Mei and J. R. Reynolds, Macromolecules, 2010, 43, 8348–8352 CrossRef CAS.
- E. Wang, Z. Ma, Z. Zhang, P. Henriksson, O. Inganas, F. Zhang and M. R. Andersson, Chem. Commun., 2011, 47, 4908–4910 RSC.
- E. Wang, Z. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganas, F. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef CAS.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- H.-Y. Chen, J. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371–375 CrossRef CAS.
- M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, H.-J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS.
- J. K. Park, J. Jo, J. H. Seo, J. S. Moon, Y. D. Park, K. Lee, A. J. Heeger and G. C. Bazan, Adv. Mater., 2011, 23, 2430–2435 CrossRef CAS.
- W. N. Hansen and G. J. Hansen, Phys. Rev. A: At., Mol., Opt. Phys., 1987, 36, 1396–1402 CrossRef CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS.
- J. T. Rogers, K. Schmidt, M. F. Toney, E. J. Kramer and G. C. Bazan, Adv. Mater., 2011, 23, 2284–2288 CrossRef CAS.
- J. C. Bijleveld, V. S. Gevaerts, D. Di Nuzzo, M. Turbiez, S. G. J. Mathijssen, D. M. de Leeuw, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2010, 22, E242–E246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer HPLC traces; synthetic polymerization procedure; proton NMR; GPC and TGA analyses for the polymer; cyclic and differential pulse voltammograms; external quantum efficiency for the best device setup. See DOI: 10.1039/c1py00402f |
| This journal is © The Royal Society of Chemistry 2012 |