Macromolecular design of poly(vinyl alcohol) by RAFT polymerization
Anton A. A.
Smith
a,
Thomas
Hussmann
a,
Joseph
Elich†
a,
Almar
Postma
b,
Marie-Helene
Alves
a and
Alexander N.
Zelikin
*a
aDepartment of Chemistry, Aarhus University, Aarhus, C 8000, Denmark. E-mail: zelikin@chem.au.dk
bCSIRO—Materials Science and Engineering, Clayton, VIC, Australia
First published on 19th October 2011
Abstract
Reversible Addition–Fragmentation chain Transfer polymerization (RAFT) is employed herein to obtain the first example of poly(vinyl alcohol), PVA, with controlled molecular weight and terminal aminegroups thus presenting a flexible tool for materials design and bioconjugation. Furthermore, we demonstrate that RAFT control can be used to facilitate syndiotactic chain propagation and obtain PVA with the highest reported content of syndiotactic dyad (∼78%).
Over the past two decades, rational macromolecular design has become an indispensable tool which provides for an overall success of polymers in diverse biotechnological and biomedical applications.1,2 Among the polymers with particular success, poly(vinyl alcohol) stands out as a candidate material with over 50 years of diverse applications, FDA approval for several uses,3 and clinical applications in humans, specifically as embolic bodies.4,5 Early studies revealed that at similar molecular weights, pharmacokinetics of PVA is near identical to that of PEG,6 a benchmark polymer in biomedicine. Further to this, a bioconjugate of PVA with superoxide dismutase was among the first examples of polymer–protein conjugates with dramatically improved pharmacokinetics.7 Nevertheless, despite early successes, currently PVA does not appear in the focus of biomedical research, largely due to that polymer science has failed to accomplish fundamental aspects of macromolecular design, namely a synthesis of PVA with controlled molecular weight and narrow polydispersities. Indeed, while pharmacokinetics6 and materials properties8 of PVA are highly dependent on the polymer molecular weight, materials science and biomedicine still rely on the use of PVA samples with polydispersity indexes at and above 2.8 Furthermore, opportunities in bioconjugation with PVA also remain scant, as compared to the polymers in the spotlight of polymer therapeutics. Herein, we specifically address these shortcomings and demonstrate the utility of Reversible Addition–Fragmentation chain Transfer (RAFT) polymerization technique to gain control over PVA molecular weight and stereochemistry and obtain samples with facile means of bioconjugation. To the best of our knowledge, macromolecular design of PVA, as described below, has no prior precedents, and we expect that presented results will lead to novel opportunities in using this FDA approved polymer in (nano)biotechnology and biomedicine.
The synthesis of PVA differs from that of typical vinylic polymers in that this polymer has no true monomer and is obtained viahydrolysis of a precursor polymer, typically poly(vinyl acetate), PVAc. This was first accomplished in 1930s, and since then vinyl acetate (VAc) remains a popular monomer of choice in elucidating mechanism and kinetics of polymerization techniques, including RAFT.9 Despite this, few reports have focused on synthesis of PVA from PVAc obtained via RAFT (or other controlled radical polymerization techniques), and while solitary successful reports include the syntheses of PVA with linear10 and multi-arm architectures,11 none of these demonstrated synthesis of PVA with molecular weights controlled over a broad range.
To accomplish this, we capitalize on the features inherent with the living character of RAFT polymerization, namely control over polymer molecular weight via the ratio of monomer to RAFT agent and polymerization time. Xanthate RAFT agents have previously been shown to afford good control over polymerization of VAc with typical polydispersity indexes 1.2–1.3.12 Indeed, with the use of S-phthalimidomethyl-O-ethyl xanthate, judicious choice of polymerization parameters allowed the synthesis of PVAc with molecular weights spanning two orders of magnitude, from 3000 to 121![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, preserving good polydispersity, Đ (ref. 13) (Fig. 1). We note that GPC characterization is presented herein for PVAc, a polymer precursor for PVA, and not for PVA, to avoid complication of molecular weight analyses arising from supramolecular association of PVA, i.e.gelation. Nevertheless, all PVAc samples were successfully deacetylated to produce the target polymer, PVA (Table 1).
000 Da, preserving good polydispersity, Đ (ref. 13) (Fig. 1). We note that GPC characterization is presented herein for PVAc, a polymer precursor for PVA, and not for PVA, to avoid complication of molecular weight analyses arising from supramolecular association of PVA, i.e.gelation. Nevertheless, all PVAc samples were successfully deacetylated to produce the target polymer, PVA (Table 1).
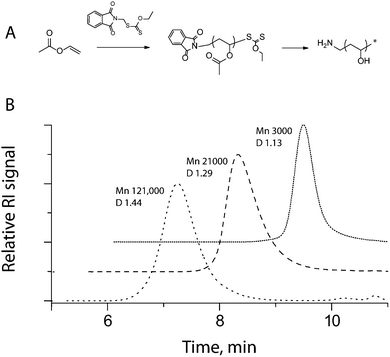 | ||
| Fig. 1 (A) Schematic illustration of the synthesis of amine-functionalized PVA via RAFT mediated polymerization of VAc, removal of the terminal phthalimideprotecting group and saponification. (B) Gel permeation chromatography elution profiles for three representative samples of PVAc obtained viaRAFT polymerization. Subsequent hydrazinolysis and saponification yielded amine-terminated PVA samples with calculated molecular weights (left-to-right) of 60, 10 and 1.5 kDa. | ||
| [M]/[RAFT] | Temp./°C | Conv. (%) | M n calc/kDa | M n GPC /kDa | Đ | R (%) | |
|---|---|---|---|---|---|---|---|
| 1 | 3900 | 60 | 50 | 84 | 60 | 1.44 | 53 |
| 2 | 3900 | 60 | 13 | 22 | 19 | 1.27 | 53 |
| 3 | 200 | 60 | 80 | 6.9 | 10 | 1.29 | 53 |
| 4 | 33 | 60 | 65 | 0.9 | 1.5 | 1.13 | (53 ± 2) |
| 5 | 33 | 50 | 53 | 0.75 | 1.0 | 1.1 | (69 ± 8) |
| 6 | 200 | 37 | 67 | 5.8 | 7.0 | 1.2 | 53 |
The second and equally important goal of the proposed design in the synthesis of PVA relates to producing samples of PVA amenable for facile bioconjugation. While existing opportunities in bioconjugation using hydroxylgroups are significantly disadvantaged compared to the classic site of conjugation (amine, thiol, etc.), incorporation of the latter as polymer terminal groups has recently been established via a choice of an appropriate RAFT agent. This notion provided an impetus for using a RAFT agent which contains a phthalimide (Phth), specifically within the so-called “R” group in the structure of RAFT agent. Phth presents a protected amine moiety with a facile deprotection using hydrazine, a strategy which has been previously realized in the context of polymer terminal groups14 to achieve e.g.fluorescent labeling of polymer samples.15 We hypothesized that, if performed on PVAc, the target polymer (PVA), unlike its pristine counterpart, would be equipped for convenient, site-specific bioconjugation. To investigate this in detail, hydrazinolysis of PVAc was conducted using varied relative amounts of hydrazine in methanol at 60 °C for 30 min (Fig. 1). Under these conditions, 50 mole equivalents of hydrazine were sufficient for a completion of reaction, as evidenced by a disappearance of Phth characteristic peaks in the NMR spectra. With longer reaction times (1 h), the relative content of hydrazine could be reduced to 20 mole equivalents, a feature which becomes increasingly important when treating oligomeric samples of PVAc. In optimizing reaction conditions for the final step to obtain the target polymer, saponification of PVAc into PVA, we found that avoiding water as a reaction medium and using methanolic NaOH was pivotal for a better polymer recovery, specifically for low molecular weight samples. The final polymer samples were analyzed viaNMR in d6-DMSO, and recorded spectra were identical to those typically reported for PVA.
We emphasize that while macromolecular design via a RAFT agent “Z” group (-OEt in the chosen RAFT agent) is appropriate for PVAc, a subsequent saponification step is expected to remove these terminal functionalities from PVA. Formation of a terminal thiol group upon removal of thioester functionality, as widely used in bioconjugation using a variety of RAFT-derived polymers,12 for PVA is also complicated by dominating side reactions.10 These two notions that make the proposed R-group design unique. Compared to rather scant opportunities in bioconjugation using PVA hydroxylgroups, we believe that terminal amine functionalities and controlled molecular weights render this polymer synthesis route highly attractive for production of PVA for bioconjugation and design of functionalized hydrogels. To provide an initial illustration to novel opportunities in materials design associated with amine-functionalization of PVA, a 4.5 kDa sample of H2N-PVA was reacted with rhodamine isothiocyanate (RITC), a model low molecular weight cargo. The resulting polymer was used to assemble surface adhered, patterned PVA hydrogels, towards their applications in guided cell adhesion and surface mediated drug delivery.16 Functionalized PVA hydrogels are unique in that both physical (matrix elasticity) and chemical (low fouling and/or pro-cell adhesion functionalization) cues are available to elicit a desired cellular response at an interface. The presented fluorescent image (Fig. 2, inset) reveals that synthesized samples of PVA, despite a low molecular weight, exhibit outstanding gelation capability and retention of model cargo, thus contributing towards a novel platform in the design of intelligent biointerfaces.17
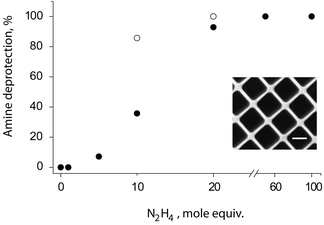 | ||
| Fig. 2 Degree of phthalimide removal from the PVAc terminus achieved by hydrazine hydrate taken at varying molar equivalents to Phthgroup in methanol at 60 °C over 30 min (filled circles) or 60 min (open circles). Inset: fluorescence microscopy image of a surface adhered PVA physical hydrogel obtained via a microtransfer molding technique and using a 4.5 kDa sample of PVA fluorescently labelled through the terminal amine functionality. Sample is imaged in PBS in the hydrated state, scale bar: 20 μm. | ||
Further to polymer molecular weight, polymer syndiotacticity is a characteristic decisive for the properties of PVA, specifically a capacity of the polymer to form physical hydrogels.18 Herein, we report the first data which suggest that the RAFT mechanism can be used to facilitate a syndiotactic chain propagation and present characterization of samples of PVA with a degree of syndiotacticity exceeding the highest values previously reported in the literature.19,20 Aiming to obtain oligomeric samples of PVA and using an increased concentration of RAFT agent (monomer-to-RAFT ratio, [M]/[RAFT] = 33), we observed that at 60 °C a high concentration of the RAFT agent resulted in an expected retardation of polymerization. Resulting PVA samples were characterized by a degree of syndiotacticity R = 53 ± 2%, Fig. 3. Surprisingly, a decrease in polymerization temperature to 50 °C resulted in a drastic change in tacticity of chain propagation and afforded PVA samples with an average degree of syndiotacticity R = 69 ± 8% (average tacticity values are quoted based on three independent experiments for each set of conditions). Notably, within the studied range, temperature alone did not affect a syndiotactic chain propagation, as evidenced by a polymerization at 37 °C at a [M]/[RAFT] ratio of ∼200, which resulted in an atactic sample of PVA (Table 1). These data strongly suggest that a high concentration of a RAFT agent, together with appropriate choice of temperature, is imperative to facilitate a syndiotactic chain propagation, a phenomenon which may have ramifications far exceeding the scope of this work.
![1H NMR spectra (d6-DMSO, 25 °C) of synthesized samples of PVA with varying degrees of syndiotacticity, R% = [rr + mr/2]/[rr + mr + rr], where r and m signify racemo and meso configuration of adjacent hydroxyls on a PVA chain, rr, mm and mr are syndiotactic, isotactic and atactic dyads. R = 53% is a value typical of atactic PVA samples; samples with R > 58% are typically considered syndiotactic; R = 74% is the highest value previously reported for PVA in the literature.19](/image/article/2012/PY/c1py00429h/c1py00429h-f3.gif) | ||
| Fig. 3 1H NMR spectra (d6-DMSO, 25 °C) of synthesized samples of PVA with varying degrees of syndiotacticity, R% = [rr + mr/2]/[rr + mr + rr], where r and m signify racemo and meso configuration of adjacent hydroxyls on a PVA chain, rr, mm and mr are syndiotactic, isotactic and atactic dyads. R = 53% is a value typical of atactic PVA samples; samples with R > 58% are typically considered syndiotactic; R = 74% is the highest value previously reported for PVA in the literature.19 | ||
Compared to other methods which afford syndiotactic PVA (bulky monomers, fluoroalcoholsolvents, etc.), the revealed approach is significantly advantaged in using a readily available monomer and a convenient polymerization temperature (as opposed to custom monomers21–23 and protocols carried out at e.g. −78 °C (ref. 24)) as well as a facile saponification step to yield PVA (in contrast to hydrolysis of e.g.vinyl pivalate25). An inherent limitation of the synthesis of s-PVA as described above, namely the relatively low molecular weight of the samples (∼1 kDa), can be overcome by employing other tools of macromolecular design, such as a comb architecture of PVA,26 which is a subject of ongoing research.
An important aspect in the recovery of s-PVA relates to a significant change in the product solubility observed with increased content of syndiotactic dyad. In a process not dissimilar to crystallization employed for low molecular weight compounds, we took advantage of this phenomenon and were able to isolate a sample of s-PVA with a syndiotacticity of 78% (overall polymer recovery of 15%). To the best of our knowledge, this is the highest value of syndiotacticity for PVA ever reported. Despite a low molecular weight, this sample exhibits a pronounced tendency to self-associate, as evidenced by a low solubility; we are now investigating solution and materials properties of this highly syndiotactic sample of PVA.
Conclusions
Despite an extensive history of applications, macromolecular design of PVA remained elusive, and the data presented above are, to the best of our knowledge, the first example of the synthesis of PVA with defined molecular weights, narrow polydispersities and facile sites for bioconjugation. The importance of this lies in that pharmacokinetic parameters of PVA (blood residence time, etc.)6 are governed by the polymer molecular weight, and while PVA showed promise in polymer therapeutics,7 the lack of well-defined methods of polymer synthesis and bioconjugation essentially arrested the utility of this polymer in biomedicine. From a different perspective, syndiotactic chain propagation in RAFT-controlled polymerization of PVAc and the synthesis of PVA with the highest ever degree of syndiotacticity, reported herein for the first time, is important for both fundamental and applied polymer sciences and opens broad possibilities for macromolecular design of PVA.Acknowledgements
This work was supported by a grant from the Lundbeck Foundation and Sapere Aude Starting Grant from the Danish Council for Independent Research, Technology and Production Sciences, Denmark.Notes and references
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347 CrossRef CAS.
- C. C. DeMerlis and D. R. Schoneker, Food Chem. Toxicol., 2003, 41, 319 CrossRef CAS.
- S. M. Tadavarthy, J. H. Moller and K. Amplatz, Am. J. Roentgenol., Radium Ther. Nucl. Med., 1975, 125, 609 CAS.
- G. S. Davidson and K. G. Terbrugge, Am. J. Neuroradiol., 1995, 16, 843 CAS.
- T. Yamaoka, Y. Tabata and Y. Ikada, J. Pharm. Sci., 1995, 84, 349 CrossRef CAS.
- Y. Kojima and H. Maeda, J. Bioact. Compat. Polym., 1993, 8, 115 CrossRef CAS.
- C. M. Hassan and N. A. Peppas, Adv. Polym. Sci., 2000, 153, 37 CrossRef CAS.
- M. H. Stenzel, L. Cummins, G. E. Roberts, T. R. Davis, P. Vana and C. Barner-Kowollik, Macromol. Chem. Phys., 2003, 204, 1160 CrossRef CAS.
- Y.-Y. Tong, Y.-Q. Dong, F.-S. Du and Z.-C. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1901 CrossRef CAS.
- M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Chem. Commun., 2004, 1546 RSC.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402 CrossRef CAS.
- For all polymerizations, VAc was purified by cryogenic distillation and polymerized in bulk. The reaction mixtures were degassed by freeze–pump–thaw cycles and incubated at a specified time and temperature, after which the synthesized polymer was recovered viaprecipitation into heptane. Removal of the phthalimideprotecting group was accomplished via a treatment using hydrazine monohydrate in methanol at 60 °C. Hereafter, aqueous or methanolic NaOH was added and the mixture was stirred overnight at room temperature. Resulting PVA was collected as a white precipitate. This procedure yielded a complete saponification in all samples tested, allowing a one-pot conversion from PVAc to PVA-NH2.
- A. Postma, T. P. Davis, R. A. Evans, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5293–5306 CrossRef CAS.
- D. M. Lynn, B. Sun, C. M. Jewell and N. J. Fredin, Langmuir, 2007, 23, 8452 CrossRef.
- Surfaced adhered, patterned physical hydrogels were assembled via a microtransfer molding technique using a poly(dimethylsiloxane) stamp and a 24 h PVA hydrogel stabilization in 0.5 M solution of sodium sulfate, as described in detail in ref. 17..
- B. E. B. Jensen, A. A. A. Smith, B. Fejerskov, A. Postma, P. Senn, E. Reimhult, M. Pla-Roca, L. Isa, D. S. Sutherland, B. Städler and A. N. Zelikin, Langmuir, 2011, 27, 10216 CrossRef CAS.
- J. H. Choi, S. W. Ko, B. C. Kim, J. Blackwell and W. S. Lyoo, Macromolecules, 2001, 34, 2964 CrossRef CAS.
- M.-H. Alves, B. E. B. Jensen, A. A. A. Smith and A. N. Zelikin, Macromol. Biosci., 2011, 11, 1293 CrossRef CAS.
- S. Murahashi, S. Nozakura, M. Sumi, H. Yuki and K. Hatada, Kobunshi Kagaku, 1966, 23, 605–612 CrossRef CAS.
- T. Nakano, K. I. Makita and Y. Okamoto, Polym. J., 1998, 30, 681–683 CrossRef CAS.
- R. Fukae, A. Tamada, N. Kawatsuki and T. Yamamoto, Polym. J., 1998, 30, 764–766 CrossRef CAS.
- W. S. Lyoo, S. S. Kim, H. D. Ghim, J. P. Kim and S. S. Lee, J. Appl. Polym. Sci., 2002, 85, 1992–2003 CrossRef CAS.
- Y. Nagara, K. Yamada, T. Nakano and Y. Okamoto, Polym. J., 2001, 33, 534 CrossRef CAS.
- T. Yamamoto, S. Yoda, H. Takase, T. Saso, O. Sangen, R. Fukae, M. Kamachi and T. Sato, Polym. J., 1991, 23, 185–188 CrossRef CAS.
- D. Quemener, M. Le Hellaye, C. Bissett, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 155 CrossRef CAS.
Footnote |
| † On leave from Cornell University, Ithaca, NY, USA |
| This journal is © The Royal Society of Chemistry 2012 |