Single site catalysts for stereoselective ring-opening polymerization of lactides
Pieter Jelle
Dijkstra
*,
Hongzhi
Du
and
Jan
Feijen
Department of Polymer Chemistry and Biomaterials, Faculty of Science and Technology, MIRA Institute for Biomedical Technology, University of Twente, P.O. Box 217, 7500, AE, Enschede, The Netherlands. E-mail: p.j.dijkstra@tnw.utwente.nl; Fax: +31 53 4892155; Tel: +31 53 4891968
First published on 8th September 2010
Abstract
Poly(lactide) (PLA) is the most well known biodegradable and biocompatible material among the aliphatic polyesters nowadays explored for biomedical, pharmaceutical and environmental applications. Different poly(lactide)s are distinguished, namely stereoregular PLLA and PDLA, atactic, heterotactic, syndiotactic and stereoblock PLAs. Because the stereochemistry of the monomeric units in the polymer chains plays a decisive role in the mechanical, physical and degradation properties of PLA materials, stereospecific catalysts to prepare different polylactide architectures are a major topic. In this review, after a general introduction on metal catalyzed ring opening polymerization, we mainly focus on single site catalyst systems inducing stereoselective polymerization of lactides.
Introduction
Polymeric materials derived from petrochemical feedstock are widely used nowadays. However, these petrochemical resources will arguably be consumed in the near future. Thus, there is an urgent need to develop new polymeric materials using renewable resources. The production and utilization of biodegradable aliphatic polyesters, such as poly(lactic acid)s (PLAs), have been developed recently to meet this requirement. The most straightforward methods to prepare PLAs are (1) the polycondensation of lactic acid and (2) the ring-opening polymerization (ROP) of lactide. It seems that polycondensation of lactic acid is the commercially most attractive route to prepare PLAs. However, condensation polymerization to form PLAs is hampered by the typical limitations of step polymerization, like the difficulty in obtaining polymers with sufficiently high molecular weights. Since esterification reactions are equilibrium processes in a condensation polymerization, it is necessary to drive the polymerization by the removal of water to achieve high degrees of polymerization. The ROP of lactide circumvents this disadvantage and is the method nowadays used to prepare PLAs.Ring-opening polymerization of lactide
Lactide, which is a cyclic dimer of lactic acid, is produced by the thermal degradation of poly(lactic acid) oligomers. A catalyst is required for the ROP of lactide and depending on the catalyst used, different mechanisms are involved, such as a cationic,1–4 anionic,5–8 or coordination–insertion mechanism.9It is very difficult to obtain PLA materials with high molecular weights by using metal complexes which act via a cationic mechanism in the ROP of lactide.10 Moreover, the ROP of lactide following an anionic mechanism usually leads to problems in controlling the molecular weight and molecular weight distribution of the PLA product, which is mainly caused by side reactions such as epimerization, chain termination, and inter-/intra-molecular transesterification reactions.11 Therefore, the study of metal complexes that catalyze lactide polymerizationvia a coordination–insertion mechanism has become an important topic.
Metal alkoxides are known to catalyze the ROP of lactidevia a coordination–insertion mechanism, which involves four steps as depicted in Fig. 1: (i) coordination of the lactide monomer to the Lewis-acid metal center, (ii) the lactide monomer inserts into the metal-alkoxide bond via nucleophilic addition, (iii) ring-opening of the lactide monomer viaacyl-oxygen cleavage and (iv) continuous insertion of lactide monomers. Finally termination of the polymerization reaction by hydrolysis of the active propagation chain is performed before isolation of the PLA material.
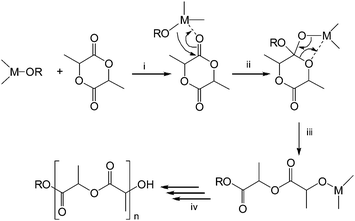 | ||
| Fig. 1 Proposed mechanism of a coordination–insertion mechanism in the ROP of lactide. | ||
Multivalent metal alkoxides
The most widely used catalyst for the industrial production of PLA materials is undoubtedly tin(II) bis(2-ethylhexanoate), usually referred to as tin(II)octanoate or Sn(Oct)2. The mechanism of the ROP of lactones and lactides catalyzed by Sn(Oct)2 has been a research subject for many years. Now it is generally accepted that protic reagents such as alcohols, or even impurities such as lactic acid present in the monomer may act as co-initiators12–14 (Fig. 2). Tin(II) alkoxides will be generated from the reaction between Sn(Oct)2 and the protic reagents in the initiation step, and act as the true active species to initiate the ROP of lactides. Aluminium alkoxides have also been proven to be efficient initiator/catalysts for the ROP of lactide.15–17 The aluminium isopropoxide, Al(OiPr)3, turned out to be remarkably less active than Sn(Oct)2. Moreover, an induction period of a few minutes is observed when applying Al(OiPr)3 as an initiator in lactide polymerization. This feature was attributed to the presence of an equilibrium between the tetramer (A4) and the trimer (A3) (Fig. 3), of which A3 was demonstrated to be more reactive than A4.18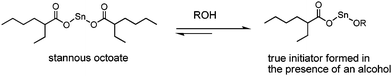 | ||
| Fig. 2 Structure for the true initiator of Sn(Oct) formed in the presence of protic reagents. | ||
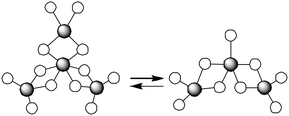 | ||
| Fig. 3 Equilibrium between the tetramer and trimer of Al(OiPr)3. Only aluminium atoms (grey) and oxygen atoms (white) are shown for clarity. | ||
Homoleptic metal alkoxide clusters were also studied in lactide polymerization. Feijen and coworkers19 have reported that Y5(µ-O)(OiPr)13, with a core structure shown in Fig. 4, has a remarkable activity in (S,S)-LA polymerization, and a non-linear relationship between the apparent propagation rate and the cluster concentration is present, which is an indication that the propagating chains aggregate in the solution. Tolman and coworkers20 reported that Fe5(µ-O)(OEt)13, having an analogous cluster structure, displayed very high rates and excellent molecular weight control in lactide polymerization.
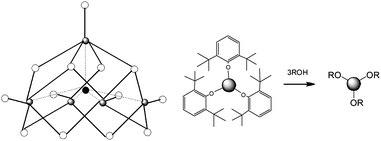 | ||
| Fig. 4 Left: core structure of Fe5(µ-O)(OEt)13 or Y5(µ-O)(OiPr)13. Metal atoms are presented by grey circles and oxygen atoms by white circles. The filled circle in the center represents the µ-5 oxygen atom connecting to all the metal atoms. All other atoms are left out for clarity. Right: In situ generation of an active yttrium alkoxide from a sterically hindered and non-active alkoxide. | ||
Although these metal alkoxides were proven to be efficient catalyst-initiators for the ROP of lactide, control of molecular weight is sometimes complicated by the clustered form of the active species. Molecular weight distributions will be broadened when more than one growing chain is connected to one metallic center. For these reasons, well-defined single-site catalysts have been designed and exploited in lactide polymerization.19
Single-site metal catalysts for stereoselective polymerization of lactide
The last two decades have witnessed a tremendous development of single-site metal catalysts in the synthesis of polyolefins21 and polyesters. These single-site catalysts have a general formula of LnMR, where M is a central metal atom surrounded by an ancillary ligand Ln. The steric and electronic properties of the ligand adjust the bonding of the metal center to the ligand, and therefore, influence the activity and stereoselectivity of the catalyst. R is the initiating group, which also affects the polymerization activity of the complexes. It is possible, by employing appropriate combinations of Ln with M and R, to generate efficient catalysts which can precisely control the polymerization rate, molecular weight, molecular weight distribution, comonomer incorporation, and even polymer stereochemistry in lactide polymerization. Spassky and coworkers22 were the first to discover that (R-SalBinap)AlOMe induced a highly stereocontrolled polymerization of rac-LA to form isotactic and crystalline PLAs with a higher melting temperature (Tm) than that of optically pure (S,S)-PLA or (R,R)-PLA. Since that time it was demonstrated that single-site metal alkoxides supported by various kinds of auxiliary ligand frameworks have unique advantages in carrying out well-controlled and in certain cases stereoselective polymerization of lactides. Because there are two stereogenic centers in one lactide molecule, different stereo-isomers of lactide are distinguished, (S,S)-LA, (R,R)-LA, and meso-LA (Fig. 5). An equivalent mixture of (S,S)-LA and (R,R)-LA is referred to as rac-LA. The alignment of R and S stereogenic centers in different modes along the polymer chain (Fig. 6) determines the mechanical and physical properties of the PLA materials. Stereoregular PLA materials can be prepared from rac- or meso-LA by using a variety of single-site metal complexes, which function via two different mechanisms (i) a chain-end-control mechanism, where the configuration of the next inserted monomer in rac-LA polymerization or the cleavage site of the monomer in meso-LA polymerization is determined by the stereogenic center in the last repeating unit along the propagating chain. If the stereogenic center in the last unit favors a meso-enchainment, isotactic PLA is obtained from rac-LA and heterotactic PLA will be obtained by using meso-LA.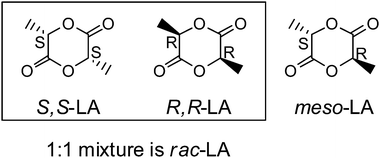 | ||
| Fig. 5 Structures of lactide stereoisomers. | ||
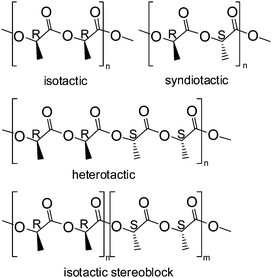 | ||
| Fig. 6 Structures of stereoregular PLA materials. | ||
However, if the stereogenic center in the last unit favors a racemic-enchainment, hetereotactic PLA will be obtained from rac-LA and syndiotactic PLA from meso-LA; (ii) an enantiomorphic site-control mechanism, where the configuration of the inserted monomer in rac-LA polymerization or the cleavage site of the monomer in meso-LA polymerization is determined by the configuration of the surrounding ligand. Thus, in the lactide polymerization following an enantiomorphic site control mechanism, only isotactic or syndiotactic PLA can be obtained from rac- or meso-LA, respectively.
Formation of isotactic/stereoblock PLAs from rac-LA
The most important breakthrough in the field of stereoselective polymerization of lactide was made by Spassky and co-workers.22 They discovered that in a rac-LA polymerization carried out at 70 °C initiated by the enantiomerically pure Schiff base aluminium methoxide (R,R)-1, the polymerization rate of (R,R)-LA is 19 times higher than that of (S,S)-LA. A living polymerization occurred as shown by the narrow molecular weight distributions of the polymers obtained. At conversions less than 50%, the polymer microstructure was predominantly isotactic, in this case (R)-PLA. At conversions higher than 60%, only (S,S)-LA remained. The reaction reached 100% conversion very slowly due to the fact that the polymerization rate of (S,S)-LA is much lower than that of (R,R)-LA. The resulting PLA had a tapered stereoblock microstructure, in which the stereoblock distribution was changing from all (R,R)-units to all (S,S)-units over the polymer chain (Fig. 7). This material exhibited a Tm of 187 °C, higher than that of the enantiopure isotactic (S)-PLA or (R)-PLA, which have Tm's in between 170 and 180 °C.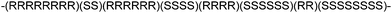 | ||
| Fig. 7 Schematic structure of tapered isotactic PLA. | ||
Coates and Ovitt23,24 discovered the presence of a bimetallic side product when preparing 1. In order to eliminate the formation of this bimetallic side product, they prepared compound 2 with an isopropoxide group connected to the central aluminium. They reported that the polymerization of rac-LA using rac-2 at 70 °C gave a stereoblock PLA material with a Tm of 179 °C. Inspecting the methine region of the homonuclear decoupled 1H NMR spectrum of the resulting polymer revealed that the PLA is not a stereocomplex formed between enantiomerically pure strands of isotactic (S,S)-PLA and (R,R)-PLA previously reported by Baker.25 Instead, the true structure of the resulting PLA is a stereoblock copolymer.
Schiff base aluminium isopropoxides (R,R)-3 and rac-3 prepared from the commercially available Jacobsen ligand were reported by Feijen and co-workers.26,27 It was demonstrated that (R,R)-3 has a moderate activity in rac-LA polymerization. Unlike (R,R)-1 which exhibits a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 preference for the polymerization of (R,R)-LA over (S,S)-LA (kRR/kSS = 20), (R,R)-3 has a strong preference for the polymerization of (S,S)-LA (kSS/kRR = 14). Rac-3 initiated and catalyzed the polymerization of rac-LA to form a stereoblock PLA material with a Pi value of 0.93 at 85% conversion of rac-LA. Notably, this excellent stereocontrol was still maintained even in a bulk polymerization. At 130 °C, the poly(rac-LA) prepared by using rac-3 has a Pi value of 0.88. So far, this is the first time that a highly isotactic PLA is obtained under bulk polymerization conditions from rac-LA. Chisholm et al.28,29 have recently re-examined the rac-LA polymerization using (R,R)-3, revealing a combined influence of some factors on the stereoselectivity of the complex, which are the chirality of the complex, the initiating group, and the nature of the polymerization solvent. Thus, it is problematic to ascribe the high stereoselectivity of (R,R)-3 either completely to a chain-end-control or to an enantiomorphic site-control mechanism.
:1 preference for the polymerization of (R,R)-LA over (S,S)-LA (kRR/kSS = 20), (R,R)-3 has a strong preference for the polymerization of (S,S)-LA (kSS/kRR = 14). Rac-3 initiated and catalyzed the polymerization of rac-LA to form a stereoblock PLA material with a Pi value of 0.93 at 85% conversion of rac-LA. Notably, this excellent stereocontrol was still maintained even in a bulk polymerization. At 130 °C, the poly(rac-LA) prepared by using rac-3 has a Pi value of 0.88. So far, this is the first time that a highly isotactic PLA is obtained under bulk polymerization conditions from rac-LA. Chisholm et al.28,29 have recently re-examined the rac-LA polymerization using (R,R)-3, revealing a combined influence of some factors on the stereoselectivity of the complex, which are the chirality of the complex, the initiating group, and the nature of the polymerization solvent. Thus, it is problematic to ascribe the high stereoselectivity of (R,R)-3 either completely to a chain-end-control or to an enantiomorphic site-control mechanism.
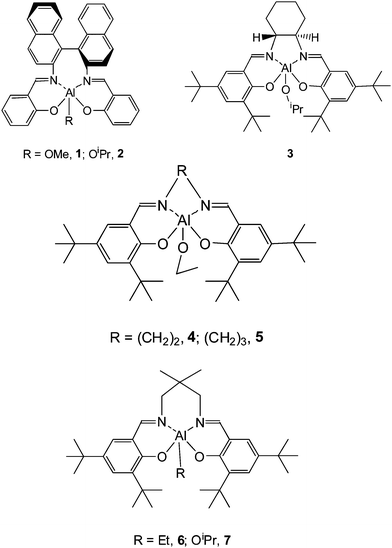
Many achiral Schiff base aluminium alkyls were proven to be efficient stereoselective catalysts for lactide polymerization in the presence of an alcohol as initiator. Nomura and coworkers30 reported on the stereoselective polymerization of rac-LA by in situ formed aluminium alkoxides from the achiral Schiff base aluminium ethyls 4 and 5 with bulky tert-butyl groups at the ortho and para positions of the phenol group. In the presence of benzyl alcohol as an initiator, complexes 4 and 5 catalyzed rac-LA polymerizationvia a chain-end-control mechanism. Complex 5 with a propylene diimine bridge furnished PLA materials with an isotacticity (Pm = 0.91) higher than that of PLA obtained with complex 4 comprising of an ethylene diimine bridge. DFT(B3LYP/6-31G*) calculations of the most stable conformation of complex 5 revealed that the propylene diimine bridge is more flexible than the ethylene diimine bridge. The more flexible propylene diimine bridge in complex 5 may allow an easy adaptation for the lactide with a specific configuration, which will increase the difference in transition state energy between lactides with different configurations, leading to an enhanced isoselectivity of 5 in rac-LA polymerization.
In 2003, Chen and coworkers31 reported on the complex 6/2-propanol as a catalyst/initiator system in rac-LA polymerization. In the presence of 2-propanol, complex 6 has a high isoselectivity in the polymerization of rac-LA generating a stereoblock PLA with a Pm value of 0.90. Thermal analysis revealed that this stereoblock PLA has a Tm of 201 °C. Kinetic data indicated that the rac-LA polymerization using complex 6/2-propanol is first-order, both in monomer and catalyst. Subsequently, the aluminium isopropoxide 7 was synthesized by the reaction between 6 and 2-propanol.32 Complex 7 was structurally determined to be monomeric with a five-coordinated central aluminium both in the solid and solution state. Polymerization data revealed that complex 7 gave the same isoselectivity and polymerization rate constant as that of 6/2-propanol, indicating that 7 is the true active species that initiates the lactide polymerization when using 6/2-propanol as a catalyst/initiator system. Further polymerizations carried out by the group of Nomuraet al.33 illustrated that compound 7 maintains a high isoselectivity in bulk polymerization of rac-LA. At 130 °C, the obtained PLA showed a high Tm of 169 °C with a Pm value of 0.84. At 150 °C, the polymerization furnished PLA materials with a Pm value of 0.82 and a Tm of 158 °C. As the polymerization temperature increased to 180 °C, the Pm and Tm values of the resultant PLA decreased to 0.80 and 155 °C, respectively. In 2007, Nomura et al. have reported on a Schiff base aluminium complex 8 with flexible but bulky tBuMe2Si substituents at the ortho and para positions of the phenol groups. This catalyst induced stereoblock PLA formation from rac-LA with a Pm value of 0.98 and a Tm of 210 °C.34 Up to now, this is the highest isotactic stereoblock PLA material which has been prepared using an achiral catalyst/initiator system in rac-LA polymerization.
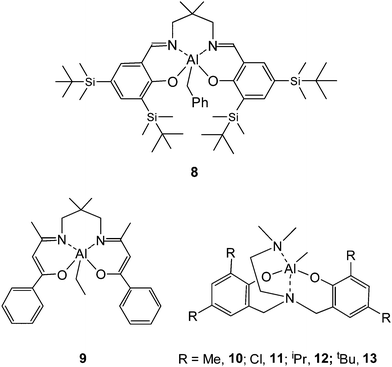
Schiff base aluminium catalysts which have been traditionally applied in lactide polymerizations are based on a bis(salicylidene) Schiff base ligand framework. Chen and co-workers35,36 reported the synthesis of a series of tetradentate enolic Schiff base aluminium ethyl complexes and their application as catalysts in rac-LA polymerization. Systematic research revealed that modifications on the auxiliary ligand exerted a dramatic influence on their catalytic performance, including activity and stereoselectivity. Lengthening the ethylene diimine bridge to a propylene diimine bridge and the presence of electron-withdrawing substituents at the 5-position in the diketone skeleton both resulted in a remarkable enhancement of stereoselectivity and polymerization rate. In the presence of 2-propanol as an initiator, complex 9 polymerized rac-LA to form isotactic enriched PLA materials with a Pm of 0.78.
A family of aluminium methyl complexes supported by tetradentate aminophenoxide ligands have been prepared by Gibson et al.37 and exploited for the ROP of rac-LA. It was found that the catalytic behavior of these complexes is highly dependent on the substituents at the ortho and para positions of the phenol group. Complexes 10 and 12 with methyl and isopropyl groups at the ortho and para positions of the phenol group furnished isotactic-biased PLA materials with Pm values of 0.73 and 0.65, respectively. However, complex 13 with tert-butyl substituents at the ortho and para positions of the phenol group led to a slight heterotactic polymerization of rac-LA, affording PLA material with a Pr value of 0.57.
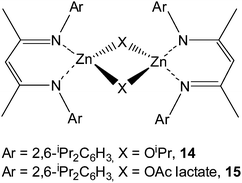
Complex 11 with chlorine substituents at the ortho and para positions of the phenol group only gave an atactic material from rac-LA.
In recent years metal free catalyst were discovered that induce stereoselective polymerization of lactides (also see next section). Most importantly, Wade et al. found that a dimeric phosphazene base polymerizes rac-LA with Pm = 0.95 at low temperatures.38
Formation of heterotactic PLAs from rac-LA
The most notable feature for β-diketiminate zinc or magnesium complexes is that they furnish highly hetereotactic PLAs in rac-LA polymerization. Coates and Ovitt39,40 reported the synthesis and characterization of the zinc isopropoxide 14 and zinc methyl lactate 15. Structural characterization revealed that both compounds are dimeric in the solid as well as in solution. Polymerization data indicated that they are both efficient initiators for lactide polymerization, producing PLAs with predictable molecular weights and narrow molecular weight distributions. Notably, complex 14 initiated the stereoselective ROP of rac-LA via a chain-end-control mechanism, yielding highly heterotactic microstructures with a Pr value up to 0.90 at room temperature and 0.94 at 0 °C.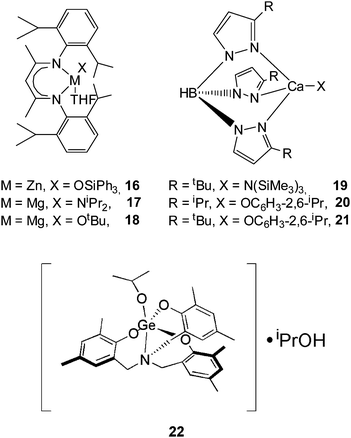
The monomeric zinc triphenylsilanoxide complex 16 supported by a β-diketiminate ligand41 with a THF molecule coordinated to the central zinc atom was shown to have the same heteroselectivity toward rac-LA in THF as that of the dinuclear zinc complex 14 in CH2Cl2. Although the magnesium complexes 17 and 18 as reported by Chisholm et al.42,43 do not show stereoselectivity toward rac-LA either in CH2Cl2 or benzene, they have a similar heteroselectivity in THF as complex 14. It seems that the coordinated THF molecule to the central metal atom plays an important role in enhancing the heteroselectivity of the β-diketiminate zinc or magnesium complexes.
Chisholm and coworkers44 have used trispindazolyl(Tp)-hydroborate ligands to coordinate calcium to form efficient initiators for lactide polymerization. The calcium amide complex 19 with bulky tert-butyl substituents on the Tp ligand was reacted with 200 equiv of rac-LA in THF. In all cases, polymerizations of rac-LA were very rapid, 90% conversion within 5 min was achieved. Complex 20 with the less bulky isopropyl substituents on the Tp ligand revealed an extremely rapid rac-LA polymerization, achieving 90% conversion in less than 1 min. Moreover, under the protection of the bulky tert-butyl substituent on the Tp ligand, complex 21 furnished a heterotactic PLA material from rac-LA with a Pr value of 0.90.
A new germanium alkoxide 22 supported by a C3-symmetric amine(trisphenolate) ligand was recently reported by Davidson et al.45 This compound has been applied in the bulk polymerization of rac-LA at 130 °C. Analysis of the microstructure of the isolated polymers revealed a strong heterotactic bias (Pr = 0.78–0.82). So far, this is the first example of a highly heteroselective polymerization of rac-LA under solvent-free conditions at a relatively high temperature.
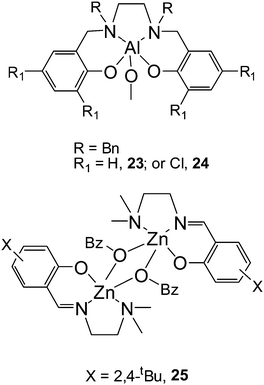
It must be emphasized that Gibson and coworkers46 have found an interesting remarkable stereocontrol of achiral aminophenoxide aluminium methyl complexes in the polymerization of rac-LA. In the presence of benzyl alcohol as an initiator, these complexes catalyzed rac-LA polymerizations in a well-controlled and living manner, affording highly isotactic PLA materials with a Pm value of 0.88 by using 23, and highly heterotactic PLAs with a Pr value of 0.96 by using 24. So far, this is the first time that aluminium complexes have been found to furnish a highly heterotactic PLA, and the first time that a dramatic switch in tacticity of the resulting PLA has been observed upon changing the substituent pattern at the ortho and para positions of the phenol group in the complexes. Preliminary kinetic data indicated that the rac-LA polymerizations catalyzed by these complexes were both first-order in monomer and catalyst.
Lin and coworkers47 synthesized a series of dinuclear zinc complexes supported by a N,N,O-tridentate Schiff base ligand framework. Polymerization data indicated that the reactivities of these complexes were dramatically affected by both the electronic and steric properties of the substituents at the ortho and para positions. Kinetic data showed that the polymerizations are both first-order in monomer and initiator. All these complexes furnish heterotactic PLA materials from rac-LA. It is worthwhile to note that a heterotactic PLA with a high Pr value up to 0.91 can be obtained at −55 °C by using complex 25.
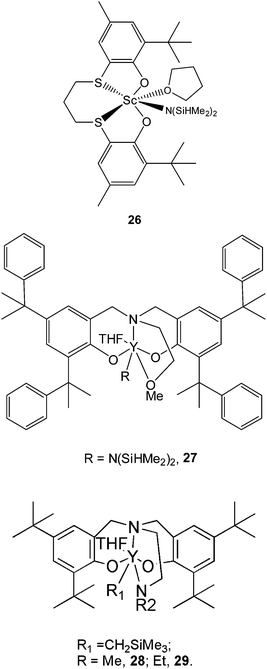
Exciting advances were made when rare earth metal complexes were applied in rac-LA polymerization. Okuda et al.48,49 synthesized a series of scandium complexes supported by 1,ω-dithiaalkanediyl-bridged bis(phenolate) ligands. These complexes showed excellent heterotactic-control in rac-LA polymerization. It was found that both THF as a solvent and bulky substituents at the ortho position of the phenol group largely improve the heterotacticity of the isolated PLAs. Moreover, polymerization data revealed that the complex with a propylene dithialkane bridge improved the heteroselectivity. At 25 °C in THF, complex 26 afforded heterotactic PLA materials from rac-LA with a Pr up to 0.96. A scandium tert-butoxy-R-lactate complex was synthesized from complex 26 and isolated as a key model complex. Structural analysis revealed that this model complex has a dimeric structure with a single ligand conformation of Λ,Λ50 in the solid state. 1H NMR spectroscopy of this model complex revealed that the dimeric structure of the complex in the solid state is retained in solution, and the R configuration of the lactate ester has selectively induced the Λ conformation of the ligand in the complex because of steric repulsion, which additionally favors the insertion of (S,S)-LA.
Carpentier et al. discovered that yttrium amido complexes supported by an amine bis(phenolate) ligand have good heteroselectivity in the polymerization of rac-LA.51–53 By introducing bulky substituents at the ortho and para positions of the phenol group and changing the donor group on the pendent chain from methoxy ether to a dimethyl substituted amine group, the heteroselectivity in rac-LA polymerization of the complex is improved. Complex 27 with bulky and conformationally flexible α,α-dimethylbenzyl groups at the ortho and para positions of the phenol group produced substantially heterotactic PLAs with a Pr up to 0.90 at 20 °C.
Cui et al.54 also reported a series of THF-solvated lanthanide (mono)alkyl complexes supported by O,N,N,O-tetradentate diamine bis(phenolate) ligands. Notably, complexes 28 and 29 displayed modest activity but high stereoselectivity in the polymerization of rac-LA to give highly heterotactic PLA materials with Pr values ranging from 0.95 to 0.99. An active oligomer connected to complex 28 prepared from rac-LA was characterized by 1H NMR. The spectrum of the active oligomer demonstrated that the ligand and the pendent nitrogen atom remain coordinated to the metal ion, and the geometry around the central metal in complex 28 did not collapse but retained its structure in solution upon monomer coordination and insertion. The spatially steric environment in the resulting propagating sites will favor the incorporation of a configurationally opposite enantiomer to lower the transition state energy.
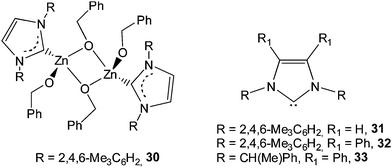
Tolman et al.55,56 discovered that the dinuclear zinc complex 30, comprising monodentate N-heterocyclic carbenes (NHCs), gave heterotactic-biased PLA materials with a Pr value of 0.60 at room temperature in rac-LA polymerization. To address whether a free carbene was participating in lactide polymerization, the free carbene 31 was exploited as a catalyst in the presence of benzyl alcohol as initiator in rac-LA polymerization. A striking difference in the tacticity of the resulting polymers obtained by using the free carbene compared to using the zinc complex 30 was discovered. Free carbene 31 produced isotactic enriched PLA with a Pm value of 0.75 from rac-LA in CH2Cl2 at −20 °C. Further studies using the more sterically hindered NHCs in lactide polymerization were carried out by Waymouth et al.57 Most notably, achiral carbene 32 produced highly isotactic PLA material with a Pm value up to 0.90 from rac-LA at −70 °C. Lactide polymerizations using chiral free carbene 33 were also investigated. The enantiomerically pure and racemic carbene 33 both furnished highly isotactic poly(rac-LA) material at −70 °C with a Pm value of 0.88.
Formation of stereoregular PLAs from meso-LA
Coates and Ovitt24,58 showed that the enantiomerically pure aluminium complex (R,R)-2 affords syndiotactic PLA from meso-LAvia an enantiomorphic site control mechanism. The polymerization data showed that when the polymerization proceeded at 70 °C in toluene for 40 h, the syndiotacticity of the resulting PLA is 0.96. Due to the high degree of syndiotacticity, the PLA produced by meso-LA polymerization using (R,R)-2 is crystalline. Following annealing at 95 °C for 60 min, this polymer exhibits a glass-transition temperature (Tg) at approximately 45 °C, and a Tm as high as 153 °C. So far this is the only example of a PLA material with high syndiotacticity. Coates and Ovitt also investigated the ROP of meso-lactide using rac-2.24 After 40 h at 70 °C, the polymerization reached 98% conversion. Although the resulting polymer has a heterotacticity of 0.80, it was amorphous and only exhibited a Tg at 43.2 °C. To explain the novel formation of the heterotactic structure from meso-LA by using rac-2, a polymer exchange mechanism (Fig. 8) was proposed, whereby each individual polymer chain effectively switches between enantiomeric aluminium centers before the insertion of the next monomer unit.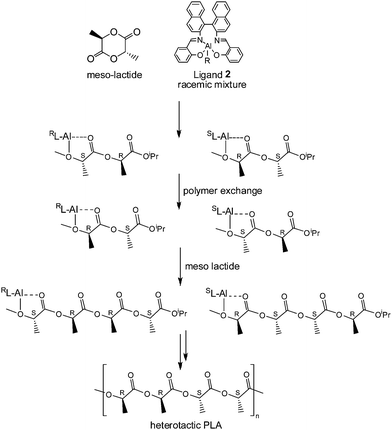 | ||
| Fig. 8 Polymer exchange mechanism for the formation of heterotactic PLA from meso-LA by using rac-2. | ||
Some other achiral metal complexes were also applied in meso-LA polymerization aiming for a chain-end-control mechanism. However, the chain-end-control of these complexes on the stereochemistry in meso-LA polymerization is very poor. Coates et al.40 reported that the dinuclear zinc isopropoxide complex 14 supported by β-diketiminate ligand affords syndiotactic PLA with a Pr value of 0.76. The yttrium amido complex 24 supported by bulky bis(phenoxy) amine ligand reported by Carpentier et al.52 also furnished a moderate syndiotactic PLA material with a Pr value of 0.75.
Applying the free carbene 32 in the polymerization of meso-Lac afforded a heterotactic-biased material with a Pr value of 0.83 at −40 °C.57
Summary and perspectives
Remarkable advances have been made in the design and synthesis of various kinds of single-site metal complexes that exert excellent stereocontrol in lactide polymerization. So far, highly isotactic and heterotactic PLA materials are formed from rac-LA, while highly syndiotactic PLA is prepared from meso-LA. The metal employed, chirality of the ligand and introduction of bulky substituents are all parameters which influence the stereocontrol. The design of new efficient and stereoselective catalyst systems will depend on e.g. the way the monomer can be conformationally positioned at the active site, stereochemistry and rigidity of the ligand, kinetics of interchange of the metal active site to polymer chains.Notes and references
- I. G. Barskaya, Y. B. Lyudvig, R. R. Shifrina and A. L. Izyumnikov, Vysokomol. Soedin., Ser. A, 1983, 25, 1283–1288 Search PubMed.
- N. Emig, H. Nguyen, H. Krautscheid, R. Reau, J. B. Cazaux and G. Bertrand, Organometallics, 1998, 17, 3599–3608 CrossRef CAS.
- D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993–9998 CrossRef CAS.
- M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071–7081 CrossRef CAS.
- C. Stere, M. Iovu, A. Boborodea, D. S. Vasilescu and I. S. Fazakas-Anca, Polym. Adv. Technol., 1998, 9, 322–325 CrossRef CAS.
- A. Bhaw-Luximon, D. Jhurry, N. Spassky, S. Pensec and J. Belleney, Polymer, 2001, 42, 9651–9656 CrossRef CAS.
- D. S. McGuinness, E. L. Marshall, V. C. Gibson and J. W. Steed, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3798–3803 CrossRef CAS.
- S. Csihony, T. T. Beaudette, A. C. Sentman, G. W. Nyce, R. M. Waymouth and J. L. Hedrick, Adv. Synth. Catal., 2004, 346, 1081–1086 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef.
- Y. Yamashit, T. Tsuda, H. Ishida, A. Uchikawa and Y. Kuriyama, Makromol. Chem., 1968, 113, 139–146 CrossRef.
- M. Bero, G. Adamus, J. Kasperczyk and H. Janeczek, Polym. Bull., 1993, 31, 9–14 CrossRef CAS.
- G. Rafler and J. Dahlmann, Acta Polym., 1992, 43, 91–95 CrossRef CAS.
- M. B. Bassi, A. B. Padias and H. K. Hall, Polym. Bull., 1990, 24, 227–232 CrossRef CAS.
- A. J. Nijenhuis, D. W. Grijpma and A. J. Pennings, Macromolecules, 1992, 25, 6419–6424 CrossRef CAS.
- P. Dubois, C. Jacobs, R. Jerome and P. Teyssie, Macromolecules, 1991, 24, 2266–2270 CrossRef CAS.
- P. Degee, P. Dubois and R. Jerome, Macromol. Symp., 1997, 123, 67–84 CAS.
- P. Degee, P. Dubois, S. Jacobsen, H. G. Fritz and R. Jerome, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2413–2420 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS.
- W. M. Stevels, M. J. K. Ankone, P. J. Dijkstra and J. Feijen, Macromolecules, 1996, 29, 6132–6138 CrossRef CAS.
- B. J. O'Keefe, S. M. Monnier, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2001, 123, 339–340 CrossRef CAS.
- G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. LeBorgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686–4692 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS.
- C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552–1553 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS.
- M. H. Chisholm, N. J. Patmore and Z. P. Zhou, Chem. Commun., 2005, 127–129 RSC.
- M. H. Chisholm, J. C. Gallucci, K. T. Quisenberry and Z. P. Zhou, Inorg. Chem., 2008, 47, 2613–2624 CrossRef CAS.
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS.
- Z. Tang, X. Chen, X. Pang, Y. Yang, X. Zhang and X. Jing, Biomacromolecules, 2004, 5, 965–970 CrossRef CAS.
- Z. Tang, X. Chen, Y. Yang, X. Pang, J. Sun, X. Zhang and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5974–5982 CrossRef CAS.
- R. Ishii, N. Nomura and T. Kondo, Polym. J., 2004, 36, 261–264 CrossRef CAS.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem.–Eur. J., 2007, 13, 4433–4451 CrossRef CAS.
- X. Pang, H. Du, X. Chen, X. Zhuang, D. Cui and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6605–6612 CrossRef CAS.
- X. Pang, H. Du, X. Chen, X. Wang and X. Jing, Chem.–Eur. J., 2008, 14, 3126–3136 CrossRef CAS.
- Z. Tang and V. C. Gibson, Eur. Polym. J., 2007, 43, 150–155 CrossRef CAS.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- G. W. Coates and T. M. Ovitt, Abstr. Pap., Jt. Conf. - Chem. Inst. Can. Am. Chem. Soc., 2000, 219, U356–U356 Search PubMed.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- M. H. Chisholm, J. C. Huffman and K. Phomphrai, J. Chem. Soc., Dalton Trans., 2001, 222–224 RSC.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785–2794 CrossRef CAS.
- M. H. Chisholm and K. Phomphrai, Inorg. Chim. Acta, 2003, 350, 121–125 CrossRef CAS.
- M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS.
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280–2283 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS.
- H. Y. Chen, H. Y. Tang and C. C. Lin, Macromolecules, 2006, 39, 3745–3752 CrossRef CAS; D. J. Darensbourg and O. Karroonnirun, Inorg. Chem., 2010, 49, 2360–2371 CrossRef CAS.
- H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818–7821 CrossRef.
- H. Ma, T. P. Spaniol and J. Okuda, Inorg. Chem., 2008, 47, 3328–3339 CrossRef CAS.
- Λ and Δ, represent the helicity of the complex structure which is chiral-at-metal.
- C. X. Cai, A. Amgoune, C. W. Lehmann and J. F. Carpentier, Chem. Commun., 2004, 330–331 RSC.
- A. Amgoune, C. M. Thomas, T. Roisnel and J. F. Carpentier, Chem.–Eur. J., 2005, 12, 169–179.
- A. Amgoune, C. M. Thomas and J. F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693–697 CrossRef CAS.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS.
- T. R. Jensen, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2004, 2504–2505 RSC.
- T. R. Jensen, C. P. Schaller, M. A. Hillmyer and W. B. Tolman, J. Organomet. Chem., 2005, 690, 5881–5891 CrossRef CAS.
- A. P. Dove, H. B. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881–2883 RSC.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072–4073 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |