Functionalized cyclic carbonates: from synthesis and metal-free catalyzed ring-opening polymerization to applications
Fabian
Suriano
a,
Olivier
Coulembier
*a,
James L.
Hedrick
b and
Philippe
Dubois
a
aCenter of Innovation and Research in MAterials and Polymers (CIRMAP), Laboratory of Polymeric and Composite Materials (LPCM), University of Mons, Place du Parc 23, 7000, Mons, Belgium. E-mail: olivier.coulembier@umons.ac.be
bIBM Almaden Research Center, 650 Harry Road, San Jose, California 95120, USA
First published on 24th September 2010
Abstract
Aliphatic polycarbonates are often referenced for their interesting applications due to their lower tendency to undergo hydrolysis, lower Tg, and their degradation in renewable resources. Their synthesis is described using a series of metal-free catalysts allowing preparation of well-defined polymers in terms of molecular weights and structures. Additionally, some highlights regarding their biomedical applications such as drug targeting are presented.
1. Introduction
Biodegradable polymers are receiving increasing attention in a wide variety of surgical, pharmaceutical and agricultural applications as well as in disposable packages.1,2 Biodegradable polymers are from various origins and structures that are mainly grouped into either natural or synthetic polymers. Natural biodegradable polymers, also referred to as biopolymers, are produced by renewable resources, i.e., plants, microorganisms and animals.3 Their synthesis generally involves enzyme-catalyzed-chain growth polymerization reactions of activated monomers, which are typically formed within cells by complex metabolic processes.4 The more widespread biopolymers such as polysaccharides (starch, cellulose, chitin and chitosan), proteins (collagen and gelatin) and polyesters (poly(β-hydroxyalkanoates)) can be obtained from microorganisms. Synthetic biodegradable polymers afford more versatility compared to natural polymers. For example, synthetic polymers may be tailored, copolymerized or produced with changes in operational conditions in order to tune specific properties or target specific needs and applications. Among the different properties able to be modified, hydrophobicity, crystallinity, degradability, solubility and thermal properties are the most investigated. Novel synthetic biodegradable polymer materials may provide considerable enhancement in medical applications due to their adaptable thermal and mechanical properties and their degradation to non-toxic by-products.5 In the frame of this highlight, we will describe the synthesis and polymerization of functional aliphatic polycarbonates and present potential applications.2. Aliphatic polycarbonates
Aliphatic polycarbonates (PCs) have been extensively used in the biomedical field due to their biocompatibility, very low toxicity and biodegradability.6 They are amorphous with a glass transition, e.g. around −20 °C for poly(trimethylene carbonate) (PTMC).7 PCs may be synthesized by polycondensation methods, which may either be carried out by phosgenation of hydroxyl compounds with KOH or pyridine as a catalyst or transesterification of diols with lower dialkyl carbonates in the presence of a basic catalyst. Similar to PLAs, polycondensation of PCs suffers from major drawbacks such as poor control over molecular parameters and limitation of molecular weights (Scheme 1).8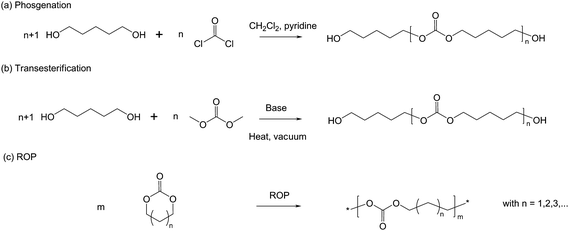 | ||
| Scheme 1 Synthetic methods for the preparation of aliphatic polycarbonates. | ||
The most efficient preparation method is the ring-opening polymerization of cyclic carbonate monomers. Cyclic carbonates may be obtained via several synthetic methods.9 The first to be reported is very similar to the preparation of lactide as it involves the depolymerization of oligomers previously synthesized by polycondensation.10,11 Transesterification has shown to lead to cyclic carbonates. For instance, trimethylene carbonate may be obtained from propane-1,3-diol and diethyl carbonate using catalytic amount of sodium ethanolate. Depending on the substituted diols, transesterification reaction may lead either to polycarbonates or the respective cyclic carbonates. While 2,2-dimethyl, 2-methyl-2-n-propyl- and 2-methyl-2-iso-amylpropane-1,3-diol gave polycarbonates in high yields using sodium methoxide as catalyst, 2,2-diethyl, 2-ethyl-2-phenylpropane-1,3-diol, pentane-2,4-diol, 2-methylpentane-2,4-diol and butane-1,3-diol resulted in cyclic carbonates.12,13Phosgene derivatives (di- and triphosgene) allow the production of cyclic monomers by coupling 2,2-disubstituted propane-1,3-diols with phosgene (Scheme 2).5e ROP of cyclic carbonates occurs viaanionic polymerization with initiators such as sec-butyllithium, sodium- and potassium naphthalene, lithium-, sodium-, and potassium alkoxides5d or via coordination–insertion with various metal compounds such as magnesium, aluminium, tin, zinc, lanthanum, neodymium, samarium, gadolinium, erbium and yttrium. Very recently, metal-free catalysts (amidines and guanidines) have proved highly efficient as well.14
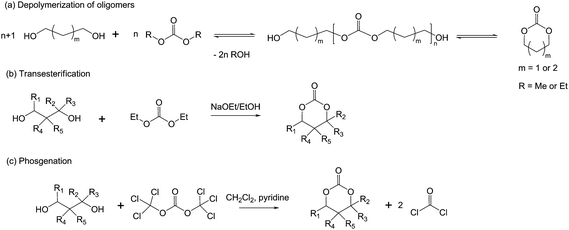 | ||
| Scheme 2 Various synthetic methods for the preparation of cyclic carbonates. | ||
As far as their degradation properties are concerned, polycarbonates are more resistant to acid-hydrolysis than polyesters. Although the carbonate group is more susceptible to alkaline hydrolysis than polyester, the rate of degradation of an aliphatic PC is lower than that of polyester which may be explained by the higher electrophilicity of the carbonate carbon atom relative to the ester counterpart. Because of this difference, the carbonyl carbon atom in the PC chain is more susceptible towards attack by hydroxide ions under basic conditions. As a consequence, polycarbonate hydrolysis does not yield carboxylic groups upon degradation unlike polyesters where the generated carboxylic groups further catalyze the hydrolysis reaction. Copolymerization of trimethylene carbonate with selected lactones can, for example, be used to prolong in vivodegradation time of resulting samples when compared to polyesters alone, which is probably due to the liberation of CO2 and hydroxyl groups formed by the chain cleavage (Scheme 3).15
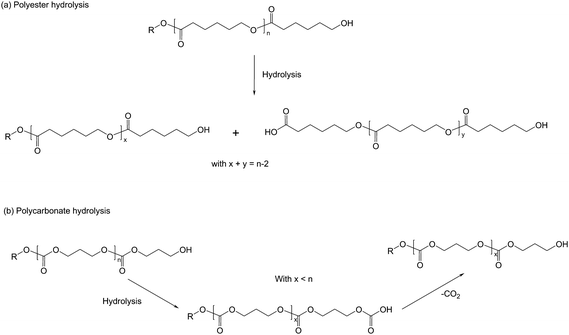 | ||
| Scheme 3 Hydrolytic degradation of (a) polyesters and (b) polycarbonates. | ||
Thus, the rate of the degradation can be finely tuned by the proper choice of polymer chain molecular weight and/or by copolymerization of different monomers. Although they may undergo hydrolysis to a lesser extent, aliphatic PCs were shown to also degrade under oxidative conditions in vivo. Oxidative degradation is intrinsically a very slow process. It can be observed in the form of surface cracking and pitting by active oxygen species.16 The increased hydrolytic stability of the aliphatic PC leads to several applications, which include their use as resins in coatings and in medical devices and implants.17,18
3. Synthesis and applications
Polymerization of aliphatic cyclic carbonates has been reported using organometallic catalysts (MAO, IBAO, Sn(Oct)2, and Al(OiPr)3) as well as enzymes.19 Recently, metal-free catalysts have also been investigated in polymerization of carbonates and others cyclic monomers such as lactones. The increasing interest in these catalysts is due to facile synthesis of polyesters and polycarbonates that is metal-free. Their use also eliminates the costly and time consuming removal of possible highly toxic heavy metals from the resulting polymers, especially relevant for some applications. A number of catalysts with a range of activities, stereocontrol and selectivity for ROP have been demonstrated. Narrowly dispersed products with predictable molecular weight and end-group fidelity have been reported.20Simple organic molecules such as 4-dimethylaminopyridine (DMAP), 4-pyrrolidinopyridine (PPY) (Scheme 4a and b), and several phosphines have shown to promote ROP of cyclic monomers in the presence of a suitable nucleophilic initiator.21–23 Many of these catalysts have the advantage of being commercially available or readily synthesized. The ROP reaction catalyzed by DMAP was proposed to occur through a monomer-activated mechanism involving a nucleophilic attack of the monomer by DMAP to generate an alkoxide/acyl pyridinium zwitterion. Subsequent proton transfer from the initiating (or propagating) alcohol, followed by acylation of the alkoxide, generates hydroxyl-terminated ring-opened monomer (Scheme 5). Polymerization with DMAP is effective in the presence of secondary or primary alcohols, while secondary alcohols undergo transesterification in a lesser manner as they are only active toward monomer and not the growing chain itself.24 Recently, Bourissou et al. proposed an anionic mechanism for the polymerization promoted by DMAP based on computational studies.25
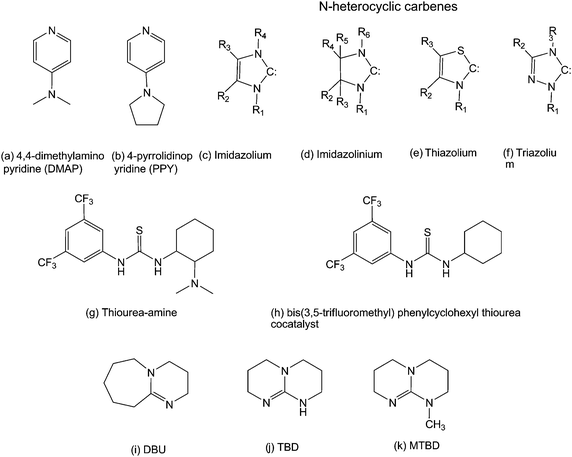 | ||
| Scheme 4 Overview of commonly used organic catalysts for ROP. | ||
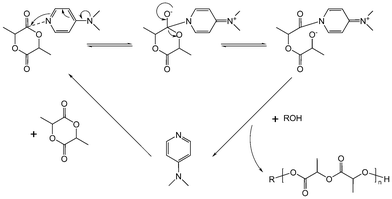 | ||
| Scheme 5 Proposed mechanism of monomer-activated ROP in the presence of pyridine-based catalyst illustrated with a lactide monomer. | ||
Taking advantage of their high nucleophilicity, N-heterocyclic carbenes (NHCs) have emerged as a new class of highly active catalysts. Their important reactivity for transesterification reactions is manifested in their ability to catalyze ROP of lactones and carbonates.26 A wide range of NHCs based on imidazolium, imidazolinium, thiazolium and triazolium carbenes (Scheme 4c–f) were shown to be remarkable catalyst for the ROP of cyclic monomers such as carbonates but also lactides, lactones, as well as silyl ethers. From a mechanistic point of view, ROP mediated by NHCs exhibits many of the features of those catalyzed by DMAP, although it occurs much faster. On the basis of the monomer-activated mechanism, every growing chain has an equal probability to accept the activated monomer, thus allowing all chains to grow at the same rate, which is one of the key characteristic of controlled polymerization reactions. Besides, the observed increase in the rate of polymerization may be explained by the higher nucleophilicity and basicity of NHCs relative to DMAP. Nevertheless, NHCs in some cases proved to mediate the polymerization by an anionic mechanism as recently highlighted by Coulembier et al. The polymerization of β-butyrolactone using 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene (triazole) as catalyst proceeded via anionic mechanism with subsequent crotonate formation when the polymerization was only conducted in toluene. On the contrary, the formation of such undesirable crotonates could be avoided with addition of t-BuOH to the solvent mixture thus allowing control over molecular parameters and end-group fidelity.27,28 Finally, a further advantage of NHCs catalysts is their ability to have both their nucleophilicity and basicity tuned by electronic and steric effects introduced by their substituents.29
Other organic catalysts have been developed that provide simultaneous electrophilic and nucleophilic activations. Amine-substituted ureas and thioureas (Scheme 4g and h) proved to be highly selective for the ROP of cyclic carbonates to give predictable molecular weights, narrow polydispersities along with end-group fidelity. Polymerization rates were lower than those obtained with NHCs, but interestingly, chain extension could be obtained by addition of more monomer. Moreover, after complete monomer conversion and extended reaction times, no transesterification reaction could be detected. This observation was later attributed to the selective activation of cyclic esters over linear polyesters by the thiourea moiety. Mixture of the electrophile-activating thiourea with nucleophile-activating amine enabled a thorough assessment of the catalyst component.30 Whereas the requirement for a highly electron-withdrawing thiourea was confirmed, the ability to increase the basicity of the tertiary amine enabled large increases in activity. For instance, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) proved to polymerize LA without added thiourea, enabling a proposed mechanism based on a chain-end activation/general base catalyzed mechanism (Scheme 6a).31
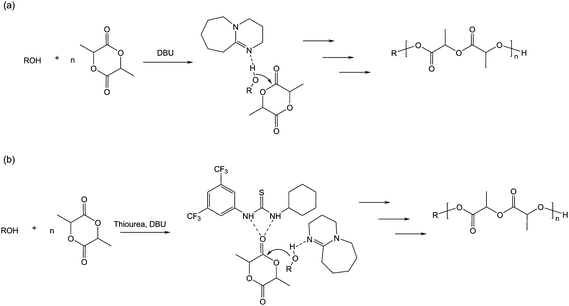 | ||
| Scheme 6 (a) Chain-end/general base activation in the presence of LA and DBU and (b) bifunctional activation of LA in the presence of ROH from an equimolar mixture of thiourea and DBU. | ||
Combination of bis(3,5-trifluoromethyl)phenylcyclohexylthiourea and DBU led to quantitative monomer conversion in much shorter times while maintaining the excellent control over the polyester molecular parameters (Scheme 6b).31 For example, trimethylene carbonate (TMC) could be polymerized by thiourea-amine catalysts with control similar to that found when polymerizing LA.14 More basic tertiary amines were then investigated such as guanidines (1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 7-methyl-1,5,7-triazabicyclo[4.4.0]-dec-5-ene (MTBD), Scheme 4j and k) that also proved to be highly efficient catalysts in ROP.
Organocatalytic ring-opening polymerization (ROP) allows for the synthesis of functional trimethylene carbonate (TMC) derivatives. Functional carbonate monomers have attracted much interest with the myriad of side groups able to be attached to a cyclic carbonate entity.32 Such strategy even offers the possibility to attach protected hydrophilic functional groups, similarly to what is now extensively used with methacrylates.33 Tuning the nature of side groups along the polycarbonate chains allows for the preparation of hydrogels14a,34 and micelles35 presenting, for few, some cell-penetrating properties leading to a new generation of materials.32e,36,37 More particularly for the last point, the synthesis and polymerization of cyclic carbonates containing diacetonide-protected glucose, galactose and mannose to afford carbohydrate-bearing polymers have been reported, using the superbasic DBU catalyst with a thiourea TU co-catalyst, initiated by benzyl alcohol. High conversion of monomer to polymer was achieved within 3 hours at room temperature. It was demonstrated that these sugar-functionalized polycarbonate block copolymers can self-assemble into micelles having surfaces with a high density of sugar molecules. Glucose-, galactose- and mannose-coated micelles are designed for targeting cancer (as many types of cancer cells over-express carbohydrate transporters),38 liver39–41 and dendritic cells,42 respectively. As an example, we used galactose-coated micelles, and prove their ability of targeting liver cancer cells (ASGP-R) through qualitative and quantitative cellular uptake, competition and cytotoxicity studies.43
4. Conclusions
As reported, aliphatic polycarbonates can be obtained viaring-opening polymerization of their respective cyclic monomers with a broad diversity of catalysts, ranging from aluminium alkoxides and tin octoate to metal-free catalysts allowing full control over molecular parameters along with chain-end group fidelity.They also proved to be very promising biodegradable polymers for many types of applications due to the myriad of key-properties they exhibit. In particular, their biodegradability and biocompatibility make them particularly useful for biomedical applications.
Acknowledgements
This work has been supported by the European Commission and Région Wallonne FEDER program (Materia Nova) and OPTI2MAT program of excellence, by the Interuniversity Attraction Pole program of the Belgian Federal Science Policy Office (PAI 6/27) and by FNRS-FRFC. O.C. is Research Associate for the FNRS.References
- S. Vaininopää, P. Rokkanen and R. Törmälä, Prog. Polym. Sci., 1989, 14, 679 CrossRef.
- W. Amass, A. Amass and B. Tighe, Polym. Int., 1998, 47, 89 CrossRef CAS.
- H. Omichi, in Handbook of Polymer Degradation, ed. S. H. Hamid, , M. B. Amin and A. Maadhah, Dekker, N. Y., 1992, p. 335 Search PubMed.
- R. Chandra and R. Rustgi, Prog. Polym. Sci., 1998, 23, 1273 CrossRef CAS.
- (a) H. R. Kricheldorf, R. Dunsing and A. Serra, Makromol. Chem., 1987, 188, 2453 CrossRef CAS; (b) A.-C. Albertsson and M. Sjoeling, J. Macromol. Sci., Part A: Pure Appl. Chem., 1992, 29, 43 Search PubMed; (c) T. Ariga, T. Takata and T. Endo, Macromolecules, 1997, 30, 737 CrossRef CAS; (d) H. Keul, R. Baecher and H. Hoecker, Makromol. Chem., 1986, 187, 2579 CrossRef CAS; (e) J. Matsuo, K. Aoki, F. Sanda and T. Endo, Macromolecules, 1998, 31, 4432 CrossRef CAS; (f) H. R. Kricheldorf, J. Jenssen and I. Kreiser-Sanders, Makromol. Chem., 1991, 192, 2391 CrossRef CAS; (g) K. R. Carter, R. Richter, H. R. Kricheldorf and J. L. Hedrick, Macromolecules, 1997, 30, 6074 CrossRef CAS; (h) P. Dobrzynski and J. Kasperczyk, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3184 CrossRef CAS.
- (a) N. Andronova and A. C. Albertsson, Biomacromolecules, 2006, 7, 1489 CrossRef CAS; (b) H. R. Kricheldorf and S. Rost, Macromolecules, 2005, 38, 8220 CrossRef CAS; (c) J. Feng, F. He and R. Zhuo, Macromolecules, 2002, 35, 7175 CrossRef CAS; (d) C. Kim, S. C. Lee, J. H. Shin and J. S. Yoon, Macromolecules, 2000, 33, 7448 CrossRef CAS; (e) T. F. Al-Azemi, J. P. Harmon and K. S. Bisht, Biomacromolecules, 2000, 1, 493 CrossRef CAS.
- Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma and J. Feijen, Biomaterials, 2006, 27, 1741 CrossRef CAS.
- K. Sehanobish, H. T. Pham and C. P. Bosnyak, in Polymeric Materials Encyclopedia, ed. J. C. Salamone, CRC Press, 1996, vol. 8, p. 5697 Search PubMed.
- G. Rockicki, Prog. Polym. Sci., 2000, 25, 259 CrossRef CAS.
- S. Inoue, CHEMTECH, 1976, 6, 588 CAS.
- M. Penco, R. Donetti, R. Mendichi and P. Ferruti, Macromol. Chem. Phys., 1998, 199, 1737 CrossRef CAS.
- S. Sarel and L. A. Pohoryles, J. Am. Chem. Soc., 1958, 80, 4596 CrossRef CAS.
- S. Sarel, L. A. Pohoryles and R. Ben-Shoshan, J. Org. Chem., 1959, 24, 1873 CrossRef CAS.
- (a) F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 153 CrossRef CAS; (b) R. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114 RSC.
- K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 24, 1736 CrossRef CAS.
- M. Acemoglu, Int. J. Pharm., 2004, 277, 133 CrossRef CAS.
- S. Nakano, Prog. Org. Coat., 1999, 35, 141 CrossRef CAS.
- S. L. Bennett, K. Connolly, E. Gruskin and Y. JiangEP 0837084, United States surgical Corp., USA, invs., 1998.
- (a) X. Chen and R. A. Gross, Macromolecules, 1999, 32, 308 CrossRef CAS; (b) Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 2799 CrossRef CAS; (c) Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 3891 CrossRef CAS; (d) K. S. Bisht, Y. Y. Svirkin, L. A. Henderson, R. A. Gross, D. L. Kaplan and G. Swift, Macromolecules, 1997, 30, 7735 CrossRef CAS; (e) H. R. Kricheldorf, S.-R. Lee and B. Weegen-Schulz, Macromol. Chem. Phys., 1996, 197, 1043 CrossRef CAS; (f) I. K. Varma, A.-C. Albertsson, R. Rajkhowa and R. K. Srivastava, Prog. Polym. Sci., 2005, 30, 949 CrossRef CAS.
- (a) A. P. Dove, Chem. Commun., 2008, 6446 RSC; (b) M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093 CrossRef CAS.
- M. Myers, E. F. Connor, T. Glauser, A. Mock, G. Nyce and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 844 CrossRef CAS.
- F. Nederberg, E. F. Connor, M. Moller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712 CrossRef CAS.
- T. Biela, S. Penczek, S. Slomkowski and O. Vogl, Makromol. Chem., 1983, 184, 811 CrossRef CAS.
- M. Gadzinowski, S. Sosnowski and S. Slomkowski, Macromolecules, 1996, 29, 6404 CrossRef CAS.
- C. Bonduelle, B. Martin-Vaca, F. P. Cossio and D. Bourissou, Chem.–Eur. J., 2008, 14, 5304 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, E. C. Hagberg, G. W. Nyce, R. M. Waymouth and J. L. Hedrick, Polymer, 2006, 47, 4018 CrossRef CAS.
- O. Coulembier, B. G. G. Lohmeijer, A. P. Dove, R. C. Pratt, L. Mespouille, D. A. Culkin, S. J. Benight, P. Dubois, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 5617 CrossRef CAS.
- O. Coulembier, X. Delva, J. L. Hedrick, R. M. Waymouth and P. Dubois, Macromolecules, 2007, 40, 8560 CrossRef CAS.
- G. W. Nyce, T. Glauser, E. F. Connor, A. Mock, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2003, 125, 3046 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. B. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863 CrossRef CAS.
- (a) R. Wu, R. F. Al-Azemi and K. Bisht, Biomacromolecules, 2006, 9, 2921; (b) Y. Zhou, R.-X. Zhuo and Z.-L. Lui, Macromol. Rapid Commun., 2005, 26, 1309 CrossRef CAS; (c) K. D. Weilandt, H. Keul and H. Hocker, Macromol. Chem. Phys., 1996, 197, 3851 CrossRef CAS; (d) B. D. Mullen, C. N. Tang and R. F. Storey, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1978 CrossRef CAS; (e) C. Lu, Q. Shi, X. Chen, T. Lu, Z. Xie, X. Hu, J. Ma and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3204 CrossRef CAS; (f) Z. Xie, X. Hu, X. Chen, J. Sun, Q. Shi and X. Jing, Biomacromolecules, 2008, 9, 376 CrossRef CAS; (g) Z.-L. Liu, Y. Zhou and R. X. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 4001 CrossRef CAS; (h) M. Shi and M. S. Shoichet, J. Biomater. Sci., Polym. Ed., 2008, 19, 1143 CrossRef CAS; (i) Q. Shi, X. Chen, T. Lu and X. Jing, Biomaterials, 2008, 29, 1118 CrossRef CAS; (j) Y. Han, Q. Shi, J. Hu, Q. Du, X. Chen and X. Jing, Macromol. Biosci., 2008, 8, 638 CrossRef CAS; (k) A. P. Goodwin, S. S. Lam and J. M. Frechet, J. Am. Chem. Soc., 2007, 129, 6994 CrossRef CAS.
- (a) R. F. Storey, B. D. Mullen and K. M. Melchert, J. Macromol. Sci., Part A: Pure Appl. Chem., 2001, 38(9), 897 Search PubMed; (b) T. F. Al-Azemi and K. S. Bisht, Macromolecules, 1999, 32, 6536 CrossRef CAS; (c) J. M. Messman and R. F. Storey, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6817 CrossRef CAS; (d) M. Shi, J. H. Wosnick, K. Ho, A. Keating and M. S. Shoichet, Angew. Chem., Int. Ed., 2007, 46, 6126 CrossRef CAS; (e) H. Guan, Z. Xie, Z. Tang, X. Xu, X. Chen and X. Jing, Polymer, 2005, 46, 2817 CrossRef; (f) J. Lu, M. Shi and M. S. Shoichet, Bioconjugate Chem., 2009, 20, 87 CrossRef CAS; (g) Z. Xie, X. Hu, X. Chen, T. Lu, X. Lu and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 110, 2961 CAS.
- F. Nederberg, V. Trang, R. C. Pratt, S. H. Kim, J. Colson, A. Nelson, C. W. Franck, J. L. Hedrick, P. Dubois and L. Mespouille, Soft Matter, 2010, 6, 2006 RSC.
- (a) W. Chen, F. Meng, F. Li, S.-J. Ji and Z. Zhong, Biomacromolecules, 2009, 10, 1727 CrossRef CAS; (b) L. Jubo, Z. Faquan and Ch. Allen, J. Controlled Release, 2005, 103, 481 CrossRef.
- X. Hu, X. Chen, S. Liu, Q. Shi and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1852 CrossRef CAS.
- (a) C. B. Cooley, B. M. Trantow, F. Nederberg, M. K. Kiesewetter, J. L. Hedrick, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2009, 131, 16401 CrossRef CAS; (b) S. H. Kim, J. P. Tan, F. Nederberg, K. Fukushima, J. Colson, A. Nelson, Y.-Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063 CrossRef CAS; (c) L. Li, H. Wang, Z. Y. Ong, K. Xu, P. L. R. S. Ee, S. Zheng, J. L. Hedrick and Y. Y. Yang, Nano Today, 2010, 5, 296 CrossRef CAS; (d) J. P. K. Tan, S. H. Kim, F. Nederberg, K. Fukushima, D. J. Coady, A. Nelson, Y. Y. Yang and J. L. Hedrick, Macromol. Rapid Commun., 2010, 31, 1187 CrossRef CAS.
- Y. C. Wang, X. Q. Liu, T. M. Sun, M. H. Xiong and J. Wang, J. Controlled Release, 2008, 128, 32 CrossRef CAS.
- M. Spiess, Biochemistry, 1990, 29, 10009 CrossRef CAS.
- X. Q. Zhang, X. L. Wang, P. C. Zhang, Z. L. Liu, R. X. Zhuo, H. Q. Mao and K. W. Leong, J. Controlled Release, 2005, 102, 749 CrossRef CAS.
- D. Q. Wu, B. Lu, C. Chang, C. S. Chen, T. Wang, Y. Y. Zhang, S. X. Cheng, X. J. Jiang, X. Z. Zhang and R. X. Zhuo, Biomaterials, 2009, 30, 1363 CrossRef CAS.
- Y. Hattori, S. Kawakami, Y. Lu, K. Nakamura, F. Yamashita and M. Hashida, J. Gene Med., 2006, 8, 824 CrossRef CAS.
- F. Suriano, R. C. Pratt, J. P. K. Tan, N. Wiradharma, A. Nelson, Y. Y. Yang, P. Dubois and J. L. Hedrick, Biomaterials, 2010, 31, 2637 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |