
Discovery of N-substituted-2-oxoindolin benzoylhydrazines as c-MET/SMO modulators in EGFRi-resistant non-small cell lung cancer†
Stefano
Tomassi‡
 ae,
Benito
Natale‡
a,
Michele
Roggia
a,
Luisa
Amato
b,
Caterina
De Rosa
b,
Carminia Maria
Della Corte
b,
Emma
Baglini
c,
Giorgio
Amendola
a,
Anna
Messere
a,
Salvatore
Di Maro
a,
Elisabetta
Barresi
d,
Federico
Da Settimo
d,
Maria Letizia
Trincavelli
d,
Fortunato
Ciardiello
b,
Sabrina
Taliani
*d,
Floriana
Morgillo
*b and
Sandro
Cosconati
*a
ae,
Benito
Natale‡
a,
Michele
Roggia
a,
Luisa
Amato
b,
Caterina
De Rosa
b,
Carminia Maria
Della Corte
b,
Emma
Baglini
c,
Giorgio
Amendola
a,
Anna
Messere
a,
Salvatore
Di Maro
a,
Elisabetta
Barresi
d,
Federico
Da Settimo
d,
Maria Letizia
Trincavelli
d,
Fortunato
Ciardiello
b,
Sabrina
Taliani
*d,
Floriana
Morgillo
*b and
Sandro
Cosconati
*a
aDiSTABiF, University of Campania “Luigi Vanvitelli”, Via Vivaldi 43, 81100, Caserta, Italy. E-mail: sandro.cosconati@unicampania.it
bDepartment of Precision Medicine, University of Campania “Luigi Vanvitelli”, Via Pansini, 5, 80138 Naples, Italy. E-mail: floriana.morgillo@unicampania.it
cCNR IFC, Institute of Clinical Physiology, National Research Council of Italy, CNR Research Area, Via G. Moruzzi 1, Pisa 56124, Italy
dDepartment of Pharmacy, University of Pisa, Via Bonanno 6, 56126, Pisa, Italy. E-mail: sabrina.taliani@unipi.it
eDepartment of Life Science, Health, and Health Professions, LINK Campus University, Via del Casale di San Pio V, 44, 00165, Rome, Italy
First published on 3rd October 2024
Abstract
Non-small cell lung cancer (NSCLC), the leading cause of cancer-related mortality worldwide, poses a formidable challenge due to its heterogeneity and the emergence of resistance to targeted therapies. While initially effective, first- and third-generation EGFR-tyrosine kinase inhibitors (TKIs) often fail to control disease progression, leaving patients with limited treatment options. To address this unmet medical need, we explored the therapeutic potential of multitargeting agents that simultaneously inhibit two key signalling pathways, the mesenchymal-epithelial transition factor (c-MET) and the G protein-coupled receptor Smoothened (SMO), frequently dysregulated in NSCLC. By employing a combination of in silico drug repurposing and structure-based structure–activity relationship (SAR) studies, we identified and developed novel c-MET/SMO-targeting agents with antiproliferative activity against first- as well as third-generation EGFR-TKI-resistant NSCLC cells suggesting a synergistic effect arising from the simultaneous inhibition of c-MET and SMO.
Introduction
Non-small cell lung cancer (NSCLC) is a heterogeneous malignancy including diverse cellular subtypes and accounts for approximately 85% of all lung cancers.1 Its pharmacological treatment involves chemo-, radio-, targeted-, and immuno-therapy, or their combinations, and depends on clinical and prognostic factors, including the presence of genetic alterations behaving as oncogenic drivers.2 Despite the overall initial response to the available therapies, the occurrence of low efficacy, drug resistance, spread of metastasis, and severe side effects represents a major pitfall for patients suffering from NSCLC. Moreover, timely medical intervention is often hampered by metastatic dissemination and the degree of tumour cell transformation at the time of the diagnosis.3,4 Therefore, though recent advances in targeted- and immuno-therapy have significantly improved the unfavourable reputation of this malignancy, NSCLC remains a therapeutic challenge, and the search for novel and efficient therapeutical alternatives persists.5In this concern, one paradigm achieving recognition is that the use of a single-targeted approach may not be sufficient to address all the underlying molecular and genetic alterations that drive the growth and spread of NSCLC cells and cancer in general. In contrast, targeting multiple pathways by a single chemical agent may represent a more effective and durable strategy.6 Multitargeting may indeed provide various advantages in cancer:6 overcoming drug resistance, as cancer cells are more unlikely to evade treatments acting on different pathways simultaneously; reducing toxicity since it may permit the use of reduced doses and avoid interactions between individual chemical entities; finally, a personalized and smooth dosing regimen with respect to combination therapy.7
Currently, several multitarget drugs have emerged or were evaluated in clinical trials for NSCLC, with some arising as a result of drug discovery programs and others whose mechanism of action was uncovered by retrospective analysis.2 This scenario is well-portrayed by several examples: amivantamab-vmjw (JNJ-61186372), an anti-EGFR/c-MET bispecific antibody, approved for locally advanced or metastatic NSCLC with EGFR exon20 insertion mutations;8 a series of PI3K/Akt/mTOR inhibitors (pictilisib, PX-866, buparlisib, pilaralisib) clinically evaluated in association with chemotherapy;9 crizotinib, an ALK/c-MET/ROS1 inhibitor approved as first-line treatment for ALK rearrangement-positive patients;10 cabozantinib, a multikinase inhibitor of c-MET, VEGFR2, AXL, c-KIT, and RET, currently under clinical investigation for metastatic NSCLC.11
Despite these successful examples, the rational design of multitarget agents is not effortless and requires key prerequisites: i) a deep understanding of the genetic and molecular determinants supporting the pathological state; ii) the identification of molecular targets that once simultaneously hit can elicit synergism; iii) the possibility of merging single-target pharmacophores into a common structure, preferably preventing promiscuity on biological off-targets.6
In this context, targeting biological pathways that sustain recurrent and aggressive cancer phenotypes, such as the epithelial-to-mesenchymal transition (EMT), may be beneficial in NSCLC. The EMT determines the loss of epithelial hallmarks, including the E-cadherin expression, and the reduction of the cell-to-cell/matrix adhesion and polarity, and lends remodelled cells that are motile and with stem-like traits.12 This physiological process is often hijacked in malignant cells to fuel the abnormal proliferation, survival, and acquisition of a metastatic character.13 Moreover, the mesenchymal signature is characteristic of advanced NSCLC with a poor prognosis and with acquired tolerance to cytotoxic and targeted agents, including EGFR tyrosine kinase inhibitors (TKIs).14–17
Among the cellular mediators sustaining the EMT process in cancer, we recently focused on two unrelated transducing systems: the mesenchymal-epithelial transition factor (c-MET) and hedgehog (Hh) pathways.18–20 The former is a tyrosine kinase receptor that upon binding to its ligand, the hepatocyte growth factor (HGF), activates multiple cytosolic transducing cascades (RAS/MAPK, PI3K/AKT, JAK/STAT3, Wnt/β-catenin, etc.) and by this fosters cell proliferation, survival, migration, and angiogenesis.21,22 The involvement of c-MET in NSCLC is not limited to its role in EMT, indeed the exon14 skipping mutation aberrantly activates the receptor and is deemed as an oncogenic driver for tumour growth,23,24 while c-MET amplification and/or overexpression are prevailing acquired resistance mechanisms to first- and second-line EGFR-TKI.25,26 Contrarily, the hedgehog (Hh) signalling pathway consists of three receptor ligands, the Indian, sonic, and desert hedgehog, and the transmembrane receptor patched (PTCH), with suppression functions on the G protein-coupled receptor Smoothened (SMO). In the canonical Hh signal transduction, ligand binding to PTCH evokes SMO activation that in turn triggers the expression of GLI1 (or glioma-associated oncogene homolog 1) transcription factor-dependent genes. The Hh pathway exerts functions in human embryonic development and tissue healing; however, it is also involved in the renewal and survival of cancer stem cells.27 In NSCLC, upregulation of Hh signalling correlates with a TGFβ1-dependent EMT signature,28 while genomic alterations of the gene encoding for SMO have been found in about 12% of lung adenocarcinoma.29
Previous research by some of us reported the functional interplay between c-MET hyperactivation and SMO gene aberrations in an EGFR-TKI-resistant human NSCLC model.30In vitro and in vivo experiments demonstrated that their concurrent inhibition decreased cancer cell growth and invasiveness, restored the tumour susceptibility to anti-EGFR TKIs, and reverted the EMT process, thus providing the rationale for a multitargeting approach. Along this line, we challenged in an in silico ligand repurposing campaign to identify dual-targeting agents as antiproliferative effectors on EGFR-TKI-resistant lung cancer cells.31 Using a docking-based virtual screening (VS) protocol, we uncovered a set of known c-MET inhibitors with binding capability to and an antagonistic effect on the SMO receptor, thus showing for the first time that these two phylogenetically distinct targets (a class IV TK and a Frizzled class F GPCR, respectively) can recognize a common pharmacophore. Among these, the two most potent compounds were eventually recognized as foretinib (XL880, Exelixis, GSK1363089, GlaxoSmithKline, 1, Fig. 1) and glesatinib (MGCD265, Mirati Therapeutics, 2, Fig. 1), two TKIs also targeting AXL protein, with the latter recently completing a clinical trial on NSCLC in combination with nivolumab.32,33 These showed enhanced cell growth inhibition and induction of apoptosis with respect to single-targeting agents, alone or in combination, in NSCLC cells with a resistant phenotype to first-generation EGFR-TKIs; importantly, their administration restored the tumour sensitivity to existing EGFR-targeted drugs in a mouse xenograft model.31
 | ||
| Fig. 1 Structure of foretinib (1), glesatinib (2), and compound 3. | ||
The aforementioned compounds have been categorized as single-targeted TKIs, but within the set of c-MET/SMO binders disclosed by us,31 compound 3 in Fig. 1 structurally resembled a multi-targeted TKI (MT-TKI). More precisely, 3 closely recalls the structure of sunitinib, a MT-TKI that hits PDGFRs, VEGFRs, c-KIT, and RET, among others, and is marketed for the treatment of renal cell carcinoma and imatinib-resistant gastrointestinal stromal tumours. As sunitinib, compound 3 features an oxindole module serving as a structural platform for establishing hydrogen bonds with the hinge region of kinase proteins.34
To date, the US FDA has greenlit over 80 small-molecule protein kinase inhibitors,35 with over two dozen falling under the category of MT-TKIs. Given that the specificity of numerous protein kinase inhibitors mostly remains unexplored, many other approved drugs will likely be reclassified as MT kinase inhibitors in the future, with their efficacy correlated to the updated pharmacodynamic profile. Indeed, concurrently, inhibiting several protein kinases holds both potential benefits and drawbacks.34 Intrigued by the potential availability of a MT-TKI endowed with SMO affinity and antagonism, herein we used the structure of compound 3 as a seed to explore novel mechanisms of action on cancer cells by taking advantage of its exquisite pharmacological properties. Here, our strategy consisted of coupling the intrinsic advantages of MT-TKIs in the cure of tumours with an add-on activity against a phylogenetically distant target previously demonstrated to be relevant against drug recalcitrant NSCLC.
Thus, a series of derivatives with point structural modifications were designed, synthesized, and tested for their inhibiting properties against c-MET TK, as the main target. As the preservation of the multi-targeted profile was relevant too, the most potent compounds were later investigated for their affinity for SMO in competition binding experiments using NSCLC cells as well as against a set of 19 relevant TKs. Finally, the compounds retaining the wanted multi-targeting properties were tested for their antiproliferative and functional activities in cell-based experiments on gefitinib-resistant and osimertinib-resistant NSCLC models.
Results and discussion
Design and synthesis
To explore the initial idea that compound 3 might indeed feature MT-TKI properties, this latter was tested for its inhibition against a panel of 20 TKs, representative of the TKinome, including our main target c-MET. The results of this inspection are reported in Table 1.| a % of inhibition of 3 at 20 μM. |
|---|

|
This analysis substantiated our initial assumption on the MT-TKI properties of 3 as of the 20 tested kinases only three of them (namely PIK2, SRC, and TYRO3) were inhibited by less than 50% at 20 μM. Interestingly, the remaining 17 tested kinases were inhibited by more than 70%. To explore possible modifications of our lead 3, molecular modelling studies were undertaken. Two X-ray crystal structures were selected to obtain a viable model of compound 3 in c-MET, considering the conserved Asp-Phe-Gly (DFG) motif alternatively adopting the “in” and “out” conformations (PDB codes: 7B40 (ref. 36) and 3RHK,37 respectively). Analysis of the docking results achieved on these protein conformations revealed that in both cases the most frequently calculated binding pose of 3 calculates an H-bond interaction between the ligand hydrazide group and the backbone NH of M1160 in the hinge region of c-MET. However, the other portions of the lead compound seem to explore different regions depending on the protein conformation considered. Specifically, for the “DFG-in” protein conformation, the m-Br-o-OH-benzoyl moiety is embedded in a narrow and highly hydrophobic cleft of the receptor (made up by M1160, F1089, V1092, L1140, and L1157). The central 2-oxoindoline core is positioned so that its carbonyl group points towards the same hydrophobic cleft with the other portion of the core being partially solvent-exposed. The pendant p-tolylaminocarbonyl moiety occupies a π-rich zone where F1168, H1174, and H1162 are located (Fig. S1A in the ESI†). Conversely, in the “DFG-out” folding, the m-Br-o-OH-benzoyl moiety points outside the protein, with the 2-oxoindoline core that occupies the hydrophobic cleft defined by M1160, F1089, V1092, L1140, and L1157, and the pendant p-tolylaminocarbonyl group pointing towards a rather polar protein region lined by R1227, F1223, and D1164 (Fig. S2B in the ESI†). This analysis did not give a clear indication of the viability of the two alternative binding poses calculated using the docking software. Therefore, the stability of the afore-described 3/c-MET complexes was probed by running a 500 ns long molecular dynamics (MD) simulation. A preliminary analysis of the MD trajectories revealed important differences in terms of ligand RMSD (L-RMSD) fluctuations. In particular, compound 3 in the “DFG-out” conformation was more unstable with respect to the “DFG-in” one (see Fig. S2 in the ESI†). This evidence is confirmed by a sudden relocation of the ligand at around 350 ns, which is responsible for the loss of the important H-bond interaction with M1160. Indeed, the formation of H-bond interactions with the kinase hinge region is generally conducive to enzyme inhibition by the majority of kinase inhibitors.38 In contrast, 3 in the “DFG-in” folding showed a more robust and consistent H-bond interaction with M1160 for the vast majority of the simulation time (around 70%), while also being in contact with the hinge region with a second H-bond involving K1161 (see Fig. S3A and S3B in the ESI†). Overall, this analysis allows us to postulate that the ligand itself behaves as a type I TKI (see Fig. 2). Considering the achieved theoretical results, it was possible to envisage modifications of compound 3 that could allow a tailored SAR profile of the c-MET inhibition to be drawn. It is important to note that analysis of the binding mode of 3 in the protein's “DFG-in” conformation showed two intramolecular H-bond interactions, both involving the hydrazido-NH: one is made with the carbonyl group of the central core, and the other with the hydroxyl of the m-Br-o-OH-benzoyl moiety. Specifically, these two interactions contribute to the correct orientation of the m-Br-o-OH-benzoyl moiety, thanks to the positioning of the Br atom pointing towards the hydrophobic cleft (see Fig. 2). For these considerations, while the m-Br-o-OH-benzoyl moiety seems to have sufficient steric and electrostatic complementarity with the enzyme counterpart, the 2-oxindoline core and the p-tolylaminocarbonyl regions could be further evaluated for point structural modifications.
 | ||
| Fig. 2 Predicted binding mode of 3 in complex with the “DFG-in” conformation of c-MET, as representative of the 500 ns long MD simulation. The ligand and the kinase (PDB code: 7B40) are in orange sticks and blue sticks and ribbons, respectively. H-bonds are represented as dashed green lines. | ||
Thus, it was postulated that the insertion of electron-withdrawing groups (such as halogens) on the 2-oxoindoline core could enhance π–π interactions with the nearby Y1159 (compounds 4–13 in Fig. 3). Similarly, phenylaminocarbonyl moieties bearing electron-withdrawing groups in place of the p-tolylaminocarbonyl portion might stabilize charge-transfer interactions with neighbouring residues such as F1168, H1174, and H1162 (compounds 14–19 in Fig. 3). Enhancement of the interactions with these latter residues could be also achieved through the extension of the aromaticity of this ligand region (compounds 20–24 in Fig. 3). Finally, it could be useful to evaluate combinations of these point structural modifications on the overall structure of 3 (compounds 25–29 in Fig. 3), envisaging that these might not be mutually exclusive.
 | ||
| Fig. 3 Substitution patterns of compounds 4–29. | ||
For the chemical synthesis of 4–29, we proceeded according to a general synthetic route (Scheme 1A and B) that includes the N-alkylation of the isatin cores with suitable reagents (30–36, 64, 65, and commercially available reagents), followed by condensation of the obtained intermediates (37–57 and 66–70) with the commercially available 5-bromo-2-hydroxybenzohydrazide. The 2-chloro-N-phenylacetamides (30–36) endowed with the desired substitution pattern were obtained by the reaction of the corresponding anilines with chloroacetyl chloride (Scheme 1A). Isatins with methylene-bicyclic pendants (66–70) were assembled by the reaction with commercially available reagents (2-(bromomethyl)naphthalene, 2-(chloromethyl)benzimidazole, and 2-(bromomethyl)-5-chloro-1-benzothiophene for 68, 69, and 70, respectively) or with synthons prepared on purpose (for 66 and 67). In this concern, suitable aldehydes (58, 59) were cyclized to the corresponding heterocyclic intermediates (60, 61) in the presence of ethyl bromoacetate or methylthioglycolate (for 60 and 61, respectively), and benzylic reactive sites were generated through an ester-to-alcohol reduction by LiAlH4 (62, 63) and final bromination with PBr3 (64, 65) (Scheme 1B).
 | ||
Scheme 1 Synthetic procedures for compounds 4–19 and 25–29 (A) and 20–24 (B). Reagents and conditions: a) chloroacetyl chloride, NaHCO3, dry THF, room temperature, 16 h; b) isatins, K2CO3, KI, dry DMF, room temperature, 16 h; c) 5-bromo-2-hydroxybenzohydrazide, abs. EtOH/AcOH (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 90 °C, 16 h or μW, 110 °C, 20 min; d) BrCH2CO2Et, Cs2CO3, CH3CN/DMF, reflux, 16 h; e) HSCH2CO2Me, K2CO3, dry DMF, 60 °C, 16 h; f) LiAlH4, dry THF, room temperature, 2 h; g) PBr3, dry THF, room temperature, 3 h. :1), 90 °C, 16 h or μW, 110 °C, 20 min; d) BrCH2CO2Et, Cs2CO3, CH3CN/DMF, reflux, 16 h; e) HSCH2CO2Me, K2CO3, dry DMF, 60 °C, 16 h; f) LiAlH4, dry THF, room temperature, 2 h; g) PBr3, dry THF, room temperature, 3 h. | ||
Biochemical evaluation on c-MET and SMO targets
The parent compound 3 showed an unbalance between the inhibitory capability against the c-MET kinase and the binding affinity for the SMO receptor (IC50 = 8.1 μM and 0.060 μM, respectively); therefore, our first step was to screen compounds 4–29 on c-MET. In this respect, we proceeded with fixed-dose activity evaluation on the c-MET kinase using 1 μM and 20 μM as test concentrations (Table 1) and with a protocol reported in the Experimental section. Among the compounds of the first series, with the sole exception of 12, 19, 20, 23 and 24, all the others were demonstrated to induce inhibition of the c-MET kinase activity of 50% or higher, at 20 μM.In particular, the aromatic substitution of the 2-oxoindoline core with halogen atoms (Cl and Br) seemed to be quite tolerated, while the increase of the atomic radius, as in the case of 12, seemed to be detrimental and it was not further investigated. Parallel to this, the double aromatic substitution (13) was also beneficial for the activity, eliciting 72% inhibitory activity against c-MET at 20 μM. In contrast at the same concentration, the 2-oxoindoline N-substitution seemed to have a mixed impact on the inhibitory activity with the p-Cl group leading to the highest potency (see compounds 14–16, 18), whereas the m-double substitution with the CF3 group (17) evoked an almost complete abrogation of the kinase activity at 20 μM. Among the plausible structural modifications on the 2-oxoindoline N-substituent, we planned to cyclize the acetamido moiety into a bicyclic structure as a means to rigidify this molecular portion and extend its aromaticity. In this concern, the 2-benzofuran (20) and the 2-benzothiophene (21) moieties prompted a more pronounced effect with half residual activity of c-MET at 20 μM if compared with the 2-naphthyl (22) and 2-benzimidazole (23) ones; interestingly, the introduction of an electro-withdrawing group at position 5 of the 2-benzothiophene ring of 21 yielded compound 24 with a completely abolished activity. Envisaging plausible cooperativity between the already explored modifications, we designed a second series of compounds according to the fixed-dose results of the first round of modifications. In this respect, we merged the 2-oxoindoline core substitutions with Cl and Br at C4/C6/C7 (4, 6–7) and C4/C7 (8 and 11), respectively, together with the p-NO2 (15) and m-CF3 (17) double substitution on the N-pendant, as these induced the highest c-MET inhibition at 20 μM and at the same time complied with the computational model we devised. In the same experimental setup, this approach did not produce the desired additive effect (26–29); however, compound 25 exhibited c-MET inhibition at both the fixed concentrations used in the assay. The fixed-dose assay setup was used to narrow down the subset of compounds eligible for the determination of the dose–response curve on both c-MET and SMO proteins. Compounds 6, 10, and 25 evoked consistent inhibition of the kinase activity at both doses and were selected for further biochemical characterization. In the IC50 and Ki determinations, we used the pan-kinase modulator staurosporine and the antagonist vismodegib as single-target references for c-MET and SMO inhibition, respectively. The SMO affinity was measured by carrying out competition binding experiments on HEK293T cells expressing SMO and using [3H]-cyclopamine or ((2′R,3S,3′R,3′aS,6′S,6aS,6bS,7′aR,11aS,11bR)-1,2,3,3′a,4,4′,5′,6,6′,6a,6b,7,7′,7′a,8,11,-11a,11b-octadecahydro-3′,6′,10,11b-tetramethyl-spiro[9H-benzo[a]fluorene-9,2′(3′H)-furo[3,2-b]pyridin]-3-ol, as a radioligand, Table 2).
| Cpd |
|
c-MET % inhibition | ||
|---|---|---|---|---|
| X | R | @1 μM | @20 μM | |
| 4 | 4-Cl |

|
18.8 | 61.1 |
| 5 | 5-Cl | 11.5 | 72.9 | |
| 6 | 6-Cl | 60.4 | 78.2 | |
| 7 | 7-Cl | 14.7 | 74.5 | |
| 8 | 4-Br | 7.7 | 55.5 | |
| 9 | 5-Br | 9.3 | 50.2 | |
| 10 | 6-Br | 52.2 | 71.8 | |
| 11 | 7-Br | 19.1 | 66.1 | |
| 12 | 7-I | −1.9 | 17.5 | |
| 13 | 4-F; 5-Me | 35.5 | 72.2 | |
| 14 | H |
|
34.5 | 73.4 |
| 15 |

|
36.7 | 42.5 | |
| 16 |

|
40.8 | 49.6 | |
| 17 |
|
11 | 82.9 | |
| 18 |

|
23.9 | 67.4 | |
| 19 |

|
2.8 | 31.4 | |
| 20 |

|
25.1 | 52.2 | |
| 21 |

|
31.3 | 55.1 | |
| 22 |

|
−2.2 | 23.8 | |
| 23 |

|
3.4 | 39.2 | |
| 24 |

|
−3.4 | 17.3 | |
| 25 | 4-Br |

|
46.4 | 70.6 |
| 26 | 7-Br | −5.4 | 15.3 | |
| 27 | 4-Cl |
|
3.2 | 12.4 |
| 28 | 6-Cl | 7.8 | 18.3 | |
| 29 | 7-Cl | 5.6 | 19.4 | |

As reported in Table 3, derivatives 6, 10, and 25 showed IC50 values for c-MET in the low-micromolar range (IC50 = 1.3–1.8 μM), resulting in being approximately 8 times more potent than the corresponding parent molecule 3 (IC50 = 8.1 μM). Intriguingly, an even more pronounced increase of activity for 6, 10, and 25 on SMO was observed, with Ki values in the low-nanomolar span, whereas the ability of 3 in the same setup corresponded to a Ki value of 0.185 μM. Compounds 6 and 10 with a Cl or a Br atom at C6 of the 2-oxoindoline core structure exhibited Ki values ranging from 0.028 to 0.011 μM, while the presence of a Br atom at the core C4 position, together with the p-NO2 substitution on the aromatic N-pendant region, (25) yielded a 20-fold increase in the binding affinity (Ki 0.009 μM). Most importantly, the newly identified compounds preserved the MT-TK inhibition potential as reported in Table 4.



|
Antiproliferative activity in EGFR-TKI resistant human NSCLC
Compounds 6, 10, and 25 were identified as the most effective c-MET inhibitors with the highest affinity for SMO. These compounds, together with the parent lead 3, were tested on first-generation EGFR-TKI gefitinib-resistant HCC827 (HCC827-GR) NSCLC cells, which have been previously described to possess amplification of SMO and overexpression of c-MET and displayed a typical mesenchymal behavior.30 In addition, given the occurrence of acquired resistance in EGFR patients treated with the third-generation EGFR-TKI osimertinib (N-(2-{[2-(dimethylamino)ethyl]-(methyl)amino}-4-methoxy-5-{[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]amino}phenyl)acrylamide, AZD9291),41 we have generated an osimertinib-resistant PC9 NSCLC cell line (with both exon 19 deletion and T790M acquired mutation, PC9-OR, Fig. S5 in the ESI†) to test whether the c-MET/SMO targeting strategy could be applied to EGFR-mutant NSCLC models with acquired resistance to third-generation EGFR inhibitors.We first examined the expression of c-MET and SMO in resistant cell lines compared to their parental counterparts. Strong overexpression of SMO was found in both HCC827-GR and PC9-OR cell lines compared to their parental cell lines, as shown in Fig. 4.
 | ||
| Fig. 4 Western blotting of phosphorylated and total AKT, c-MET, SMO, vimentin, and GLI1 parental and resistant PC9 and HCC827. Tubulin was used to ensure equal loading. | ||
In addition, we found an increase of the mesenchymal marker vimentin42 as well as the proliferation marker p-AKT,43 compared to the parental cell lines. Based on these findings, an MTS assay was performed to assess the antiproliferative effects of compounds 3, 6, 10, and 25 at increasing concentrations (0.01–10 μM) on both HCC827-GR and PC9-OR cells. Compounds 3, 6, 10, and 25 demonstrated dose-dependent inhibition of cancer cell growth and all exhibited IC50 values in the low μM concentrations as depicted in Fig. 5A and B, with an improvement of the newly synthesized compounds (6, 10, and 25) in terms of IC50 values with respect to the parent lead 3.
 | ||
| Fig. 5 Biological activity of experimental compounds on the HCC827-GR and PC9-OR NSCLC cell lines. A) Cell toxicity assay in parental HCC827-GR and PC9-OR cells exposed to increasing concentrations of compounds 3, 6, 10, and 25 for 72 h. B) IC50 values of compounds 3, 6, 10, and 25 on PC9-OR and HCC827-GR cells. Data are expressed as a mean of three independent experiments ± SD. At least three independent experiments were performed. Statistical significance **p < 0.01, ***p < 0.001 and ****p < 0.0001. | ||
Importantly, PC9-OR cells showed the lowest IC50 values compared to HCC827-GR, demonstrating good sensitivity to compounds despite the acquired mutations in the EGFR gene. Exposing the HCC927-GR and PC9-OR cell lines to the combination of the four compounds with gefitinib or osimertinib, respectively, did not result in cooperative antiproliferation (see Fig. S6 in the ESI†). To confirm the mechanism of action in the studied cellular environment, resistant cell lines were treated for 72 hours with each compound at a concentration corresponding to their IC50 values, and the in vitro functional effect of inhibition of c-MET and SMO was assessed at the protein level by western blot analysis (Fig. 6). The tested compounds induced a reduction of phosphorylated c-MET (p-c-MET) and GLI1 in both HCC827-GR and PC9-OR cells. In addition, 6 and 25 were effective in reducing the expression of vimentin, a marker of EMT. Taken together, these results suggest inhibitory activity and antiproliferative capacity in vitro in two models of resistant NSCLC cell lines harbouring different mutations in the EGFR gene via the concurrent inhibition of c-MET and SMO-regulated pathways.
 | ||
| Fig. 6 Representative western blotting of whole cell lysates showing phosphorylated and total forms of AKT, c-MET, SMO, vimentin, and GLI1 in resistant NSCLC cell lines treated with IC50 values of tested compounds. α-Tubulin was used to ensure equal loading. | ||
Assessment of compound-induced cell death in TKI-resistant NSCLC cell lines
We wanted to investigate further the mechanisms of cell death induced by the tested compounds in TKI-resistant cell lines based on the data obtained from the anti-proliferative assay. Specifically, the ligands were tested for their ability to induce apoptosis on PC9-OR and HCC827-GR cell lines. To determine the percentage of early and late apoptotic cells in resistant NSCLC cell lines, flow cytometry was performed using the annexin V-Alexa 488 conjugate and PI according to the manufacturer's protocol. Early apoptotic cells exhibit annexin V-Alexa 488+/PI- staining patterns, whereas late apoptotic cells exhibit annexin V-Alexa 488+/PI+ staining patterns due to the loss of plasma membrane integrity.44 After 24 hours of exposure to tested compounds at a 20 μM concentration, a significant increase of early and late apoptotic cells was identified in both resistant-NSCLC cell lines (Fig. 7A and B) with some differences.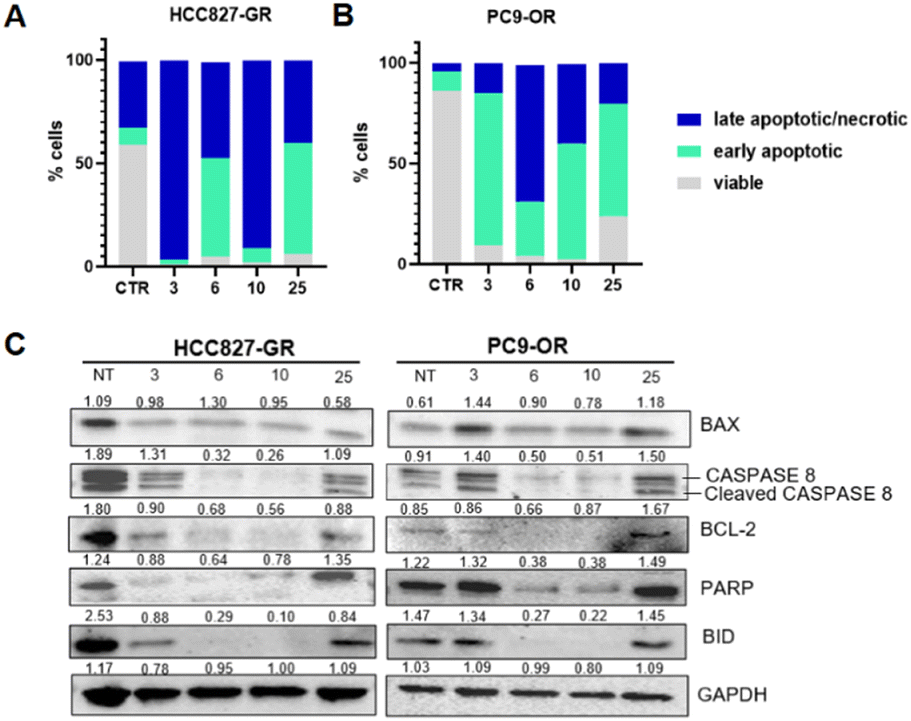 | ||
| Fig. 7 Stacked column graph representative of the percentage of live, early apoptotic, and late apoptotic/necrotic cells in the HCC827-GR (A) and PC9-OR (B) cell lines after 24 h treatment with 20 μM of the four compounds. (C) Western blotting of whole cell lysates showing levels of BAX, caspase-8, BCL-2, PARP, and BID in TKI-resistant NSCLC cell lines. Cells were treated with compounds (3, 6, 10, and 25) at 20 μM for 24 h. GAPDH was used to ensure equal loading. | ||
In HCC827-GR, compounds 3 and 10 showed a higher percentage of late apoptotic/necrotic cells after 24 hours (96.2% and 90.8%, respectively). In PC9-OR, the same compounds showed a lower percentage of late apoptotic/necrotic cells and a higher percentage of early apoptotic cells (75.5% for compound 3 and 57.5% for compound 10). Then, we evaluated alterations in the expression of proteins involved in apoptosis by western blot analysis in the compound-treated cells (Fig. 7C). The expression of Bax, a key component of cell-induced apoptosis through mitochondrial stress, was increased in PC9-OR treated with compounds 3 and 25. An increase in the expression of cleaved caspase-8 (43 kDa) was also observed in the same treated cells. Conversely, an examination of the effects of compounds 6 and 10 in PC9-OR reveals a reduction in the anti-apoptotic factor BCL-2, which also exhibited a lower expression in all HCC827-GR-treated cells. In both cell lines, compounds 6 and 10 determine a reduction in PARP full-length along with a reduction in BID full-length protein expression. Overall, these results indicate that the four tested compounds induce a shift in the balance of pro- and anti-apoptotic protein expression, thus resulting in the activation of apoptosis in resistant-NSCLC cell lines.
Conclusions
In the present study, we investigated the structure–activity relationships (SARs) of a series of 2-oxoindoline derivatives aimed at achieving c-MET inhibitory properties of a previously developed compound (3) while maintaining its SMO-targeting activity. Through a systematic approach involving molecular modelling, synthetic methodologies, and in vitro assays, we identified structural modifications that preserve the c-MET and SMO inhibition profiles of this class of compounds. Molecular modelling studies revealed that the most favourable binding pose of compound 3 in the c-MET active site involves the 2-oxoindoline core interacting with an enzyme hydrophobic cleft and the p-tolylaminocarbonyl group engaging in charge transfer interactions with nearby residues. This analysis guided the selection of structural modifications aimed at enhancing these interactions and improving c-MET inhibition. Synthesis of a diverse library of derivatives with point structural modifications revealed that the introduction of halogen atoms (Cl or Br) at C4 and C6-positions of the 2-oxoindoline core, together with a p-NO2 substitution on the aromatic N-pendant, led to improvements in c-MET inhibition. These modifications also significantly increased SMO-targeting activity, with Ki values in the low nanomolar range. Compounds 3, 6, 10, and 25 emerged as the most promising candidates with c-MET and SMO inhibition profiles. In vitro experiments on first- and third-generation EGFR-TKIs resistant cell lines, GR-resistant HCC827 and OR-resistant PC9 NSCLC, demonstrated that these ligands exhibited antiproliferative activity with IC50 values in the low μM range. These compounds demonstrated the ability to reduce p-c-MET and GLI1 protein expression, as well as vimentin and p-AKT levels, suggesting a double modulation of c-MET and SMO-amplified pathways in resistant NSCLC cells. The results on PC9-OR cell lines, representing a relevant model of EGFR-mutant NSCLC with acquired resistance to third-generation EGFR-TKIs, are particularly encouraging. The lack of responsiveness to therapeutics such as osimertinib represents a significant challenge in NSCLC treatment, often leaving patients with limited therapeutic options. While the compounds evaluated in this study share a common mechanism of action, namely an MTKI effect accompanied by SMO negative modulation, the observed variations in their effects on the same cell line underscore the complexity of cellular responses. These differences likely arise from nuanced variations in compound–target interactions, differential activation of downstream apoptotic pathways, and inherent heterogeneity within the cell lines. Notably, the 24 hour time point chosen for analysis represents a single snapshot of a dynamic process. This observed variability highlights the importance of thoroughly investigating the individual compounds within a class sharing a common mechanism of action, as subtle differences can translate into significant variations in therapeutic efficacy against TKI-resistant NSCLC. In this context, the compounds identified in this study offer a promising avenue to address this unmet clinical need. Further investigations into the development of inhibitors of c-MET and SMO are warranted to fully unleash their potential in advancing NSCLC treatment strategies.Experimental section
Computational studies
The two crystal structures were prepared with Protein Preparation Wizard within Schrodinger's Maestro molecular modelling suite.45 For both crystals, only chain A was selected, and chain B was deleted. Bond orders were assigned, and all the hydrogen atoms were added. Using the Epik tool within the Maestro suite, the ionization and tautomeric states of the amino acidic side chains were predicted at physiological pH. Optimization of the hydrogen bonds, followed by minimization of the structure, was then performed. Finally, all the water molecules were deleted.The selected DFG-in c-MET crystal structure (PDB code: 7B40) was missing two loops: a smaller one, comprising residues 1148 to 1150 in the N-lobe region, and part of the activation loop, comprising residues 1236–1245. All these were reconstructed using AlphaFold46 (UniProt code: P08581) and subjected to a minimization round applying constraints on the remaining residues (2500 steps of steepest descent followed by 5000 steps employing a Polak–Ribiere conjugate gradient). The 3D structure of compound 3 was generated with Maestro's 2D Sketcher.
The AutoDock-GPU software package47 was used to perform molecular docking. Using Autogrid4, grid points of 67 × 67 × 67 with a 0.375 Å grid spacing were calculated around the respective co-crystal binding cavity for each protein, in particular including the M1160 residue. 200 separate docking runs were performed with the Lamarckian genetic algorithm search method. Unless otherwise specified, default docking parameters were applied. Docking conformations were clustered based on their RMSD with a tolerance of 2.0 Å, and eventually ranked based on their most populated cluster.
The ligand–protein complexes obtained from the docking experiments were used to construct a molecular dynamics system that was solvated in water within an orthorhombic box with a buffer distance of 10 Å. Additionally, the overall charge of the system was automatically neutralized using Maestro's System Builder utility. The salt concentration was set to 0.15 M NaCl. The chosen force field for constructing the ligand–receptor–membrane system was OPLS3.48 The Desmond module within the Schrodinger suite was used for 500 ns MD simulations. NPT was chosen as the ensemble class constant number of particles (constant pressure of 1.01325 bar and constant temperature of 300 K). In detail, the temperature was controlled using the Nosé–Hoover thermostat49 with a 1.0 ps relaxation time. The pressure was controlled using the Martyna–Tobias–Klein barostat47 with an isotropic coupling style and a 2.0 ps relaxation time. The cutoff distance for short-range nonbonded interactions was 9 Å. To minimize the computation time, nonbonded forces were calculated using a RESPA integrator where the short-range forces were updated every two steps, and the long-range forces were updated every six steps. The Desmond Simulation Interaction Diagram tool was used to analyze the receptor–ligand interactions during the MD trajectory.
Chemistry
General procedure for the synthesis of N′-(1-substituted-2-oxoindolin-3-ylidene)(5-bromo-2-hydroxybenzo)hydrazides (4–29)
A suspension of the appropriate N-alkylated isatin (37–57, 66–70) (0.137 mmol) and 5-bromo-2-hydroxybenzohydrazide (32 mg, 0.137 mmol) in a mixture of ethanol and acetic acid (1.5 mL, 9:1) was irradiated in a microwave reactor at 110 °C for 20 minutes. The obtained yellow suspension was filtered and the solid was washed with cold ethanol three times and then with dichloromethane, EtOAc, diethyl ether, and hexane to give the desired products 4–29.
(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-4-chloro-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (4)
Yellow powder. Yield: 74%. HRMS (ESI-MS): calculated: 539.01272 for C24H1779BrClN4O4 [M–H]−, found: 539.01306. HRMS (ESI-MS): calculated: 541.01067 for C24H1781BrClN4O4 [M–H]−, found: 541.01093. 1H NMR (400 MHz, DMSO-d6) δ 14.52 (bs, 1H), 12.17 (bs, 1H), 10.21 (bs, 1H), 8.30–7.80 (m, 1H), 7.77–6.07 (m, 9H), 4.65 (s, 2H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.25, 161.36, 159.49, 155.83, 144.44, 136.53, 135.74, 133.25, 132.63, 131.91, 129.03, 127.80, 127.73, 124.24, 119.41, 119.21, 116.45, 110.66, 109.41, 108.53, 42.92, 20.28.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-5-chloro-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (5)
Yellow solid. Yield: 84%. HRMS (ESI-MS): calculated: 539.01272 for C24H1779BrClN4O4 [M–H]−, found: 539.01318. HRMS (ESI-MS): calculated: 541.01067 for C24H1781BrClN4O4 [M–H]−, found: 541.01105. 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 12.40 (s, 1H), 10.81 (s, 1H), 8.16–7.78 (m, 1H), 7.92–6.54 (m, 9H), 4.74 (s, 2H), 2.24 (s, 3H). 1H NMR (400 MHz, DMF-d7) δ 10.83 (bs, 1H), 8.18 (m, 1H), 7.70 (s, 1H), 7.65–7.61 (m, 3H), 7.50 (d, J = 8.2 Hz, 1H), 7.33–7.28 (m, 2H), 7.13 (d, J = 8.1 Hz, 2H), 6.97 (s, 1H), 6.70 (s, 1H), 4.84 (s, 2H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.98, 167.68, 164.57, 159.76, 142.11, 137.12, 136.12, 133.32, 132.56, 130.86, 129.16, 127.29, 121.37, 120.17, 119.30, 114.57, 111.77, 110.69, 103.99, 102.33, 42.93, 20.47.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-6-chloro-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (6)
Yellow solid. Yield: 75%. HRMS (ESI-MS): calculated: 539.01272 for C24H1779BrClN4O4 [M–H]−, found: 539.01343. HRMS (ESI-MS): calculated: 541.01067 for C24H1781BrClN4O4 [M–H]−, found: 541.01105. 1H NMR (400 MHz, DMSO-d6) δ 14.36 (bs, 1H), 12.12 (bs, 1H), 10.24 (bs, 1H), 8.07 (d, J = 2.4 Hz, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.62 (dd, J = 8.0 Hz, J = 2.6 Hz, 1H), 7.46–7.45 (m, 2H), 7.42 (d, J = 1.6 Hz, 1H), 7.22 (dd, J = 8.1 Hz, J = 2.6 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 8.1 Hz, 1H), 4.67 (s, 2H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.50, 161.43, 160.07, 155.94, 144.65, 136.82, 135.96, 135.83, 135.56, 133.39, 132.65, 129.24, 122.95, 122.02, 119.33, 119.28, 118.52, 110.85, 110.64, 43.02, 20.44.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-7-chloro-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (7)
Yellow solid. Yield: 68%. HRMS (ESI-MS): calculated: 539.01272 for C24H1779BrClN4O4 [M–H]−, found: 539.01300. HRMS (ESI-MS): calculated: 541.01067 for C24H1781BrClN4O4 [M–H]−, found: 541.01074. 1H NMR (400 MHz, DMSO-d6) δ 14.33 (bs, 1H), 12.15 (bs, 1H), 10.10 (bs, 1H), 8.23–7.94 (m, 1H), 7.81–7.54 (m, 2H), 7.52–7.31 (m, 3H), 7.28–6.87 (m, 4H), 4.87 (s, 2H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.65, 161.99, 160.69, 156.49, 138.92, 137.37, 136.38, 135.71, 133.88, 133.40, 133.21, 129.66, 124.92, 123.36, 120.11, 120.05, 119.84, 119.63, 115.91, 111.34, 44.76, 20.90.(Z)-2-(4-Bromo-3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (8)
Yellow solid. Yield: 78%. HRMS (ESI-MS): calculated: 584.96016 for C24H1779Br81BrN4O4 [M–H]−, found: 584.96088. HRMS (ESI-MS): calculated: 582.96221 for C24H1779Br79BrN4O4 [M–H]−, found: 582.96183. HRMS (ESI-MS): calculated: 586.95811 for C24H1781Br81BrN4O4 [M–H]−, found: 586.95752. 1H NMR (400 MHz, DMSO-d6) δ 14.53 (s, 1H), 12.13 (s, 1H), 10.19 (s, 1H), 8.31–7.87 (m, 1H), 7.77–6.81 (m, 9H), 4.65 (s, 2H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.51, 161.41, 159.67, 156.01, 144.84, 136.87, 135.95, 135.72, 133.51, 132.76, 132.22, 129.29, 127.64, 119.74, 119.47, 119.42, 118.21, 115.71, 110.88, 109.20, 42.98, 20.53.(Z)-2-(5-Bromo-3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (9)
Yellow solid. Yield: 68%. HRMS (ESI-MS): calculated: 584.96016 for C24H1779Br81BrN4O4 [M–H]−, found: 584.96115. HRMS (ESI-MS): calculated: 582.96221 for C24H1779Br79BrN4O4 [M–H]−, found: 582.96178. HRMS (ESI-MS): calculated: 586.95811 for C24H1781Br81BrN4O4 [M–H]−, found: 586.95915. 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 12.42 (s, 1H), 10.50 (s, 1H), 8.20–8.01 (m, 1H), 7.91 (m, 1H) 7.55–6.54 (m, 8H), 4.68 (s, 2H), 2.20 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.5, 168.81, 163.71, 161.70, 147.44, 143.30, 136.72, 134.08, 133.51, 130.03, 130.45, 128.90, 124.46, 122.70, 119.67, 115.32, 113.50, 110.90, 106.25, 103.18, 43.50, 21.23.(Z)-2-(6-Bromo-3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (10)
Yellow solid. Yield: 63%. HRMS (ESI-MS): calculated: 584.96016 for C24H1779Br81BrN4O4 [M–H]−, found: 584.95970. HRMS (ESI-MS): calculated: 582.96221 for C24H1779Br79BrN4O4 [M–H]−, found: 582.96304. HRMS (ESI-MS): calculated: 586.95811 for C24H1781Br81BrN4O4 [M–H]−, found: 586.95910. 1H NMR (400 MHz, DMSO-d6) δ 14.35 (bs, 1H), 12.14 (bs, 1H), 10.18 (bs, 1H), 8.10–7.95 (m, 1H), 7.84–7.27 (m, 6H), 7.22–6.88 (m, 3H), 4.67 (s, 2H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.53, 161.46, 159.96, 155.96, 144.64, 136.83, 135.97, 135.65, 133.39, 132.65, 129.25, 125.85, 124.39, 122.20, 119.34, 119.28, 118.88, 113.34, 110.84, 43.01, 20.44.(Z)-2-(7-Bromo-3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (11)
Yellow solid. Yield: 70%. HRMS (ESI-MS): calculated: 584.96016 for C24H1779Br81BrN4O4 [M–H]−, found: 584.96110. HRMS (ESI-MS): calculated: 582.96221 for C24H1779Br79BrN4O4 [M–H]−, found: 582.96298. HRMS (ESI-MS): calculated: 586.95811 for C24H1781Br81BrN4O4 [M–H]−, found: 586.95884. 1H NMR (400 MHz, DMSO-d6) δ 14.36 (s, 1H), 12.25 (s, 1H), 10.15 (s, 1H), 8.07 (s, 1H), 7.71 (d, J = 7.4 Hz, 1H), 7.67–7.55 (m, 2H), 7.42 (d, J = 8.0 Hz, 2H), 7.20–7.05 (m, 3H), 6.98 (d, J = 8.8 Hz, 1H), 4.90 (s, 2H), 2.24 (s, 3H). 13C NMR (176 MHz, DMSO-d6) δ 165.11, 161.54, 160.45, 156.03, 139.90, 136.91, 136.26, 135.94, 135.23, 133.41, 132.74, 129.17, 124.78, 123.23, 120.01, 119.70, 119.39, 119.19, 110.87, 102.86, 44.11, 20.44.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-7-iodo-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (12)
Yellow solid. Yield: 70%. HRMS (ESI-MS): calculated: 630.94834 for C24H1779BrIN4O4 [M–H]−, found: 630.94897. HRMS (ESI-MS): calculated: 632.94629 for C24H1781BrIN4O4 [M–H]−, found: 632.94702. 1H NMR (400 MHz, DMSO-d6) δ 14.36 (s, 1H), 12.14 (s, 1H), 10.12 (s, 1H), 8.07 (s, 1H), 7.96–7.77 (m, 1H), 7.71 (d, J = 5.2 Hz, 1H), 7.61 (d, J = 5.2 Hz, 1H), 7.43 (d, J = 5.0 Hz, 2H), 7.12 (d, J = 5.0 Hz, 2H), 7.04–6.86 (m, 2H), 4.94 (s, 2H), 2.25 (s, 3H). 13C NMR (176 MHz, DMSO-d6) δ 165.02, 161.51, 160.78, 155.99, 143.25, 143.00, 136.87, 135.98, 135.33, 133.41, 132.73, 129.15, 125.03, 122.83, 120.40, 119.77, 119.51, 119.37, 119.23, 110.87, 43.72, 20.45.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-4-fluoro-5-methyl-2-oxoindolin-1-yl)-N-(p-tolyl)acetamide (13)
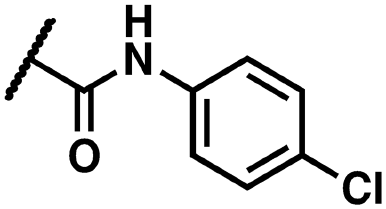
Yellow solid. Yield: 78%. HRMS (ESI-MS): calculated: 537.05792 for C25H1979BrFN4O4 [M–H]−, found: 537.05847. HRMS (ESI-MS): calculated: 539.05587 for C25H1981BrFN4O4 [M–H]−, found: 539.05652. 1H NMR (400 MHz, DMSO-d6) δ 14.29 (s, 1H), 12.03 (s, 1H), 10.17 (s, 1H), 8.20–7.96 (m, 1H), 7.70–7.36 (m, 4H), 7.05 (d, J = 59.0 Hz, 4H), 4.61 (s, 2H), 2.43–1.93 (m, 6H). 13C NMR (176 MHz, DMSO-d6) δ 164.55, 163.32, 161.92, 161.46, 160.26, 156.01, 143.34, 143.27, 136.71, 135.98, 135.93, 135.81, 133.40, 132.69, 129.26, 123.75, 123.71, 119.34, 118.73, 118.62, 115.61, 115.59, 110.79, 98.74, 98.58, 43.09, 20.47.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(4-chlorophenyl)acetamide (14)
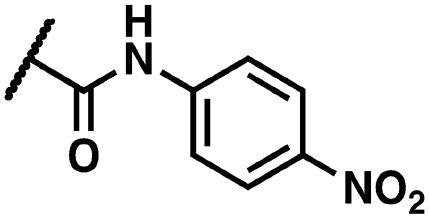
Yellow solid. Yield: 43%. HRMS (ESI-MS): calculated: 524.99707 for C23H1579BrClN4O4 [M–H]−, found: 524.99634. HRMS (ESI-MS): calculated: 526.99502 for C23H1581BrClN4O4 [M–H]−, found: 526.99601. 1H NMR (400 MHz, DMSO-d6) δ 14.41 (s, 1H), 12.15 (s, 1H), 10.45 (s, 1H), 8.07 (s, 1H), 7.68 (d, J = 7.5 Hz, 1H), 7.62–7.57 (m, 3H), 7.45 (t, J = 7.8 Hz, 1H), 7.37 (d, J = 8.5 Hz, 2H), 7.22–7.12 (m, 2H), 6.97 (dd, J = 8.7, 1.5 Hz, 1H), 4.66 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 165.11, 160.01, 151.47, 143.27, 139.20, 137.42, 136.71, 136.46, 133.36, 131.51, 128.79, 128.04, 127.29, 124.92, 123.19, 120.91, 120.70, 119.70, 119.40, 109.99, 42.89.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(4-nitrophenyl)acetamide (15)
Yellow solid. Purified on silica gel (eluent hexane/DCM/EtOAc 4:4:2). Yield: 30%. HRMS (ESI-MS): calculated: 536.02112 for C23H1579BrN5O6 [M–H]−, found: 536.02124. HRMS (ESI-MS): calculated: 538.01907 for C23H1581BrN5O6 [M–H]−, found: 538.01947. 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 12.19 (s, 1H), 10.96 (s, 1H), 8.24 (dd, J = 9.4, 3.7 Hz, 2H), 8.07 (s, 1H), 7.92–7.77 (m, 2H), 7.69 (d, J = 7.4 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.0 Hz, 2H), 6.97 (dd, J = 9.1, 3.5 Hz, 1H), 4.74 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 166.03, 161.53, 160.04, 156.03, 144.56, 143.21, 142.58, 136.75, 136.38, 133.39, 131.56, 125.11, 123.27, 120.76, 119.66, 119.37, 119.10, 110.78, 110.05, 43.09.
(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(3-nitrophenyl)acetamide (16)
Yellow solid. Purified on silica gel (eluent hexane/DCM/Ethyl acetate 4:4:2). Yield: 42%. HRMS (ESI-MS): calculated: 536.02112 for C23H1579BrN5O6 [M–H]−, found: 536.02124. HRMS (ESI-MS): calculated: 538.01907 for C23H1581BrN5O6 [M–H]−, found: 538.01947. 1H NMR (400 MHz, DMSO-d6) δ 14.40 (s, 1H), 12.21 (s, 1H), 10.83 (s, 1H), 8.57 (s, 1H), 8.08 (s, 1H), 7.99–7.86 (m, 2H), 7.69 (d, J = 7.6 Hz, 1H), 7.67–7.56 (m, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.23–7.13 (m, 2H), 6.98 (d, J = 8.7 Hz, 1H), 4.72 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 166.29, 161.94, 160.54, 156.43, 148.45, 143.68, 140.00, 137.21, 136.91, 133.86, 132.04, 130.87, 125.77, 123.73, 121.22, 120.15, 119.83, 119.81, 118.77, 113.94, 111.28, 110.51, 43.46.
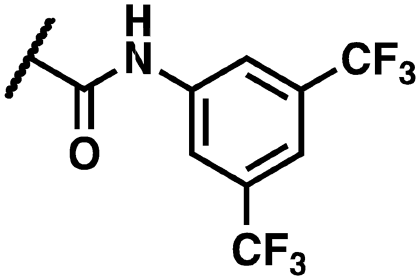
(Z)-N-(3,5-Bis(trifluoromethyl)phenyl)-2-(3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)acetamide (17)
Yellow solid. Yield: 67%. HRMS (ESI-MS): calculated: 627.01081 for C25H1479BrF6N4O4 [M–H]−, found: 627.01074. HRMS (ESI-MS): calculated: 629.00877 for C25H1481BrF6N4O4 [M–H]−, found: 629.00891. HRMS (ESI-MS): calculated: 741.00368 for C27H1579BrF9N4O6 [M + CF3CO2]−, found: 741.00494. HRMS (ESI-MS): calculated: 743.00163 for C27H1581BrF9N4O6 [M + CF3CO2]−, found: 743.00317. 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 12.18 (s, 1H), 10.99 (s, 1H), 8.24 (s, 2H), 8.08 (s, 1H), 7.82 (s, 1H), 7.69 (d, J = 5.5 Hz, 1H), 7.62 (d, J = 6.0 Hz, 1H), 7.52–7.39 (m, 1H), 7.19 (d, J = 8.2 Hz, 2H), 6.98 (d, J = 8.7 Hz, 1H), 4.73 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 166.25, 161.47, 160.05, 155.97, 143.10, 140.28, 136.74, 136.34, 133.38, 131.56, 131.17, 130.98, 130.79, 130.61, 125.45, 123.90, 123.30, 122.35, 120.80, 120.74, 119.67, 119.34, 119.06, 119.05, 116.71, 116.69, 116.67, 110.80, 110.08, 43.00.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(2-cyanophenyl)acetamide (18)
Yellow solid. Yield: 72%. HRMS (ESI-MS): calculated: 516.03129 for C24H1579BrN5O4 [M–H]−, found: 516.03131. HRMS (ESI-MS): calculated: 518.02924 for C24H1581BrN5O4 [M–H]−, found: 518.02942. 1H NMR (400 MHz, DMSO-d6) δ 14.41 (s, 1H), 12.25 (s, 1H), 10.58 (s, 1H), 8.08 (d, J = 2.7 Hz, 1H), 7.84 (dd, J = 7.8, 1.6 Hz, 1H), 7.74–7.66 (m, 2H), 7.66–7.57 (m, 2H), 7.46 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.24–7.14 (m, 2H), 6.99 (d, J = 8.7 Hz, 1H), 4.74 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 165.86, 161.47, 159.97, 155.98, 151.48, 143.08, 139.47, 139.20, 136.76, 133.99, 133.41, 131.52, 128.05, 125.56, 124.92, 123.29, 120.70, 119.35, 116.73, 110.82, 110.11, 107.31, 67.03, 42.55.(Z)-2-(3-(2-(5-Bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(4-bromo-3-chlorophenyl)acetamide (19)
Yellow solid. Purified on silica gel (eluent hexane/DCM/EtOAc 4:4:3). Yield: 64%. HRMS (ESI-MS): calculated: 604.90554 for C23H1479Br81BrClN4O4 [M–H]−, found: 604.90607. HRMS (ESI-MS): calculated: 602.90758 for C23H1479Br79BrClN4O4 [M–H]−, found: 602.90820. HRMS (ESI-MS): calculated: 606.90349 for C23H1481Br81BrClN4O4 [M–H]−, found: 606.90369. 1H NMR (400 MHz, DMSO-d6) δ 14.36 (bs, 1H), 12.43 (bs, 1H), 10.60 (bs, 1H), 7.98–7.93 (m, 2H), 7.71–7.63 (m, 1H), 7.59 (dd, J = 8.8, 2.8 Hz, 1H), 7.49 (dd, J = 8.8, 2.5 Hz, 2H), 7.43–7.35 (m, 1H), 7.15 (dd, J = 7.9, 3.4 Hz, 1H), 6.85 (d, J = 9.2 Hz, 2H), 4.65 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 166.35, 166.03, 160.38, 158.39, 143.48, 139.36, 137.16, 136.85, 136.09, 134.47, 133.64, 132.01, 130.45, 123.75, 121.31, 121.03, 120.12, 120.09, 120.02, 117.72, 115.55, 110.28, 43.19.
(Z)-N′-(1-(Benzofuran-2-ylmethyl)-2-oxoindolin-3-ylidene)-5-bromo-2-hydroxybenzohydrazide (20)
Yellow solid. Purified on silica gel (eluent hexane/EtOAc 7:3). Yield: 44%. HRMS (ESI-MS): calculated: 488.02514 for C24H1579BrN3O4 [M–H]−, found: 488.02563. HRMS (ESI-MS): calculated: 490.02310 for C24H1581BrN3O4 [M–H]−, found: 490.02374. 1H NMR (400 MHz, DMSO-d6) δ 14.45 (s, 1H), 12.31 (s, 1H), 8.08 (s, 1H), 7.74–7.56 (m, 3H), 7.52 (d, J = 8.0 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.35–7.13 (m, 4H), 7.09–6.93 (m, 2H), 5.20 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 170.35, 161.44, 159.49, 155.97, 154.36, 151.78, 142.46, 136.77, 136.29, 133.40, 131.49, 127.71, 124.51, 123.33, 123.07, 121.17, 120.81, 119.78, 119.35, 119.33, 111.16, 110.82, 110.27, 105.70, 36.52.
(Z)-N′-(1-(Benzo[b]thiophen-2-ylmethyl)-2-oxoindolin-3-ylidene)-5-bromo-2-hydroxybenzohydrazide (21)
Yellow solid. Purified on silica gel (eluent hexane/EtOAc 8:2). Yield: 46%. HRMS (ESI-MS): calculated: 504.00230 for C24H1579BrN3O3S [M–H]−, found: 504.00296. HRMS (ESI-MS): calculated: 506.00025 for C24H1581BrN3O3S [M–H]−, found: 506.00046. 1H NMR (400 MHz, DMSO-d6) δ 14.44 (s, 1H), 12.32 (s, 1H), 8.08 (d, J = 2.7 Hz, 1H), 7.88 (d, J = 7.7 Hz, 1H), 7.85–7.75 (m, 1H), 7.70–7.59 (m, 2H), 7.55 (s, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.40–7.26 (m, 3H), 7.17 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 8.7 Hz, 1H), 5.30 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 161.44, 159.43, 155.98, 142.23, 139.12, 139.11, 139.00, 136.78, 136.23, 133.40, 131.48, 124.61, 124.58, 123.69, 123.58, 123.38, 122.54, 120.86, 119.82, 119.32, 119.30, 110.82, 110.24, 38.49.
(Z)-5-Bromo-2-hydroxy-N′-(1-(naphthalen-2-ylmethyl)-2-oxoindolin-3-ylidene)benzohydrazide (22)
Yellow solid. Yield: 55%. HRMS (ESI-MS): calculated: 498.04588 for C26H1779BrN3O3 [M–H]−, found: 498.04663. HRMS (ESI-MS): calculated: 500.04383 for C26H1781BrN3O3 [M–H]−, found: 500.04449. 1H NMR (400 MHz, DMSO-d6) δ 14.44 (s, 1H), 12.15 (s, 1H), 8.09 (s, 1H), 7.88 (s, 4H), 7.74–7.23 (m, 6H), 7.23–6.87 (m, 3H), 5.16 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 161.94, 160.41, 156.45, 143.29, 137.20, 137.16, 133.87, 133.86, 133.32, 132.83, 131.90, 128.99, 128.13, 128.05, 126.91, 126.59, 126.38, 125.88, 123.64, 121.27, 120.41, 119.87, 119.80, 111.27, 110.74, 43.33.(Z)-N′-(1-((1H-Benzo[d]imidazol-2-yl)methyl)-2-oxoindolin-3-ylidene)-5-bromo-2-hydroxybenzohydrazide (23)
Yellow solid. Purified on silica gel (eluent hexane/DCM/EtOAc 4:4:3). Yield: 22%. HRMS (ESI-MS): calculated: 488.03638 for C23H1579BrN5O3 [M–H]−, found: 488.03650. HRMS (ESI-MS): calculated: 490.03433 for C23H1581BrN5O3 [M–H]−, found: 490.03476. 1H NMR (400 MHz, DMSO-d6) δ 14.47 (s, 1H), 12.50 (s, 1H), 12.26 (s, 1H), 8.09 (d, J = 2.6 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.62 (dd, J = 8.8, 2.6 Hz, 1H), 7.56 (s, 1H), 7.37 (t, J = 7.9 Hz, 2H), 7.15 (d, J = 7.1 Hz, 3H), 6.98 (d, J = 8.3 Hz, 2H), 5.25 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 161.56, 159.79, 156.12, 149.11, 143.10, 142.78, 136.73, 134.32, 133.38, 131.47, 123.24, 122.24, 121.42, 120.75, 120.01, 119.38, 118.61, 111.27, 110.71, 109.92, 37.76.
(Z)-5-Bromo-N-(1-((5-chlorobenzo[b]thiophen-3-yl)methyl)-2-oxoindolin-3-ylidene)-2-hydroxybenzohydrazide (24)
Yellow solid. Purified on silica gel (eluent hexane/DCM/EtOAc 60:35:5). Yield: 86%. HRMS (ESI-MS): calculated: 537.96333 for C24H1479BrClN3O3S [M–H]−, found: 537.96409. HRMS (ESI-MS): calculated: 539.96128 for C24H1481BrClN3O3S [M–H]−, found: 539.96295. 1H NMR (400 MHz, DMSO-d6) δ 14.46 (s, 1H), 12.26 (s, 1H), 8.15–8.06 (m, 2H), 8.04 (dd, J = 8.6, 2.5 Hz, 1H), 7.88 (d, J = 2.4 Hz, 1H), 7.70–7.57 (m, 2H), 7.47–7.34 (m, 2H), 7.19–7.09 (m, 2H), 7.01 (dd, J = 8.7, 2.5 Hz, 1H), 6.91–6.82 (m, 2H), 6.63 (s, 1H), 5.23 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 162.02, 160.28, 156.53, 151.93, 143.13, 139.67, 139.13, 139.01, 137.22, 137.04, 133.87, 131.86, 130.05, 128.52, 128.27, 125.38, 125.26, 123.73, 121.84, 120.49, 119.84, 111.25, 110.81, 67.49.
(Z)-2-(4-Bromo-3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(4-nitrophenyl)acetamide (25)
Yellow solid. Yield: 61%. HRMS (ESI-MS): calculated: 615.92959 for C23H1479Br81BrN5O6 [M–H]−, found: 615.93011. HRMS (ESI-MS): calculated: 613.93163 for C23H1679Br79BrN5O6 [M–H]−, found: 613.93213. HRMS (ESI-MS): calculated: 617.92754 for C23H1681Br81Br N5O6 [M–H]−, found: 617.92719. 1H NMR (400 MHz, DMSO-d6) δ 14.55 (s, 1H), 12.18 (s, 1H), 10.94 (s, 1H), 8.24 (d, J = 8.1 Hz, 2H), 8.07 (s, 1H), 7.82 (d, J = 8.1 Hz, 2H), 7.61 (dd, J = 7.6, 4.1 Hz, 1H), 7.39–7.33 (m, 2H), 7.24 (d, J = 6.8 Hz, 1H), 6.97 (d, J = 8.8 Hz, 1H), 4.76 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.82, 159.58, 156.08, 144.60, 144.48, 142.59, 136.78, 136.74, 135.38, 133.41, 132.12, 127.68, 125.09, 119.49, 119.38, 119.13, 118.16, 115.64, 110.73, 109.19, 43.15.(Z)-2-(7-Bromo-3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-2-oxoindolin-1-yl)-N-(4-nitrophenyl)acetamide (26)
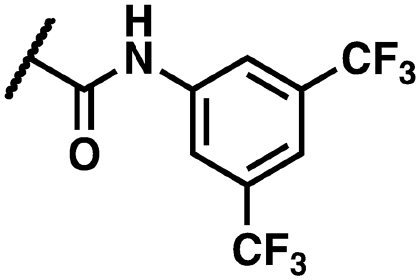
Yellow solid. Yield: 60%. HRMS (ESI-MS): calculated: 615.92959 for C23H1479Br81BrN5O6 [M–H]−, found: 615.92975. HRMS (ESI-MS): calculated: 613.93163 for C23H1679Br79BrN5O6 [M–H]−, found: 613.93066. HRMS (ESI-MS): calculated: 617.92754 for C23H1681Br81Br N5O6 [M–H]−, found: 617.92822. 1H NMR (400 MHz, DMSO-d6) δ 14.36 (s, 1H), 12.28 (s, 1H), 10.96 (s, 1H), 8.24 (d, J = 8.1 Hz, 2H), 8.08 (d, J = 3.6 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 7.2 Hz, 1H), 7.65–7.60 (m, 2H), 7.14 (t, J = 8.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 5.01 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.44, 161.54, 160.34, 156.02, 144.60, 142.60, 139.68, 136.94, 136.26, 134.96, 133.41, 125.07, 124.91, 123.14, 120.08, 119.39, 119.23, 119.17, 110.89, 102.85, 44.35.(Z)-N-(3,5-Bis(trifluoromethyl)phenyl)-2-(3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-4-chloro-2-oxoindolin-1-yl)acetamide (27)
Yellow solid. Yield: 68%. HRMS (ESI-MS): calculated: 660.97184 for C25H1379BrClF6N4O4 [M–H]−, found: 660.97241. HRMS (ESI-MS): calculated: 662.96979 for C25H1381BrClF6N4O4 [M–H]−, found: 662.97052. 1H NMR (400 MHz, DMSO-d6) δ 14.49 (s, 1H), 12.15 (s, 1H), 10.95 (s, 1H), 8.36–8.12 (m, 2H), 8.05 (s, 1H), 7.80 (s, 1H), 7.71–7.52 (m, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.31–7.07 (m, 2H), 6.96 (d, J = 8.8 Hz, 1H), 4.73 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 166.08, 161.46, 159.60, 156.04, 144.34, 140.23, 136.83, 135.20, 133.44, 132.16, 131.19, 131.01, 130.82, 130.63, 127.87, 125.46, 124.55, 123.91, 122.36, 120.81, 119.44, 119.38, 119.16, 119.14, 119.12, 119.10, 116.81, 116.79, 116.77, 116.75, 116.57, 110.82, 108.82, 43.16.(Z)-N-(3,5-Bis(trifluoromethyl)phenyl)-2-(3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-6-chloro-2-oxoindolin-1-yl)acetamide (28)
Yellow solid. Purified on silica gel (eluent hexane/DCM/EtOAc 5:4:1). Yield: 55%. HRMS (ESI-MS): calculated: 660.97184 for C25H1379BrClF6N4O4 [M–H]−, found: 660.97247. HRMS (ESI-MS): calculated: 662.96979 for C25H1381BrClF6N4O4 [M–H]−, found: 662.97040. HRMS (ESI-MS): calculated: 774.96470 for C27H1479BrClF9N4O6 [M + CF3CO2]−, found: 774.96558. HRMS (ESI-MS): calculated: 776.96266 for C27H1481BrClF9N4O6 [M + CF3CO2]−, found: 776.96381. 1H NMR (400 MHz, DMSO-d6) δ 14.36 (s, 1H), 12.27 (s, 1H), 10.97 (s, 1H), 8.22–8.20 (m, 2H), 8.08 (d, J = 4.1 Hz, 1H), 7.82 (s, 1H), 7.70 (d, J = 7.2 Hz, 1H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.47 (d, J = 4.1 Hz, 1H), 7.21 (7, J = 8.1 Hz, 1H), 7.00 (d, J = 8.1 Hz, 1H), 4.97 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.81, 161.54, 160.15, 156.04, 140.29, 138.19, 136.96, 134.96, 133.43, 132.96, 131.43, 131.09, 130.77, 130.45, 124.65, 124.48, 122.87, 121.77, 119.68, 119.40, 119.15, 119.13, 119.12, 119.11, 119.10, 119.09, 119.07, 116.78, 116.76, 116.74, 116.72, 115.44, 110.90, 44.50.
(Z)-N-(3,5-Bis(trifluoromethyl)phenyl)-2-(3-(2-(5-bromo-2-hydroxybenzoyl)hydrazineylidene)-7-chloro-2-oxoindolin-1-yl)acetamide (29)
Yellow solid. Yield: 57%. HRMS (ESI-MS): calculated: 660.97184 for C25H1379BrClF6N4O4 [M–H]−, found: 660.97266. HRMS (ESI-MS): calculated: 662.96979 for C25H1381BrClF6N4O4 [M–H]−, found: 662.97058. 1H NMR (400 MHz, DMSO-d6) δ 14.41 (s, 1H), 10.98 (s, 1H), 8.22–8.20 (m, 2H), 8.07 (d, J = 4.4 Hz, 1H), 7.81 (s, 1H), 7.70 (d, J = 7.2 Hz, 1H), 7.61 (dd, J = 8.8, 2.4 Hz, 1H), 7.45 (d, J = 4.4 Hz, 1H), 7.21 (7, J = 8.0 Hz, 1H), 6.99 (d, J = 8.8 Hz, 1H), 4.92 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 166.83, 161.59, 160.16, 156.08, 140.49, 140.31, 138.19, 136.97, 134.98, 133.44, 132.97, 131.23, 131.05, 130.86, 130.67, 125.47, 124.66, 123.92, 122.88, 122.37, 120.82, 119.70, 119.42, 119.16, 119.14, 119.12, 119.10, 116.79, 116.77, 116.75, 116.73, 115.45, 110.91, 44.50.General procedure for the synthesis of 2-chloro-N-phenylacetamides (30–36)
The appropriate substituted aniline (6 mmol) and NaHCO3 (552 mg, 6.6 mmol) were suspended in THF (7 mL) and the thus-obtained mixture was stirred at 0 °C for ten minutes. Acetyl chloride (0.53 mL 6.6 mmol) was added dropwise, and the reaction was allowed to warm up at room temperature. After one hour, the reaction was complete and the mixture was extracted in EtOAc. The organic layer was washed with a saturated solution of NaHCO3 twice and the combined aqueous phases were counter extracted with one aliquot of EtOAc. The combined organic phases were dried over anhydrous sodium sulfate, filtered, and evaporated to dryness to obtain the desired compounds 30–36.2-Chloro-N-(p-tolyl)acetamide (30)
White crystals, Yield: 95%. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (1H, s); 7.50 (2H, d, J = 7.4 Hz), 7.12 (2H, d, J = 7.4 Hz), 4.22 (2H, s), 2.26 (3H, s). 13C NMR (DMSO-d6) was in accordance with the reported literature.502-Chloro-N-(4-chlorophenyl)acetamide (31)
Off-white solid. Yield: 85%. 1H NMR (CDCl3) was in accordance with the reported literature.512-Chloro-N-(4-nitrophenyl)acetamide (32)
Yellow solid. Yield: 94%. 1H NMR (400 MHz, DMSO-d6): δ 10.98 (1H, s); 8.22 (2H, d, J = 9.0 Hz); 7.84 (2H, d, J = 9.0 Hz); 4.33 (2H, s) was in accordance with the reported literature.502-Chloro-N-(3-nitrophenyl)acetamide (33)
Pale orange solid. Yield: 96%. 1H NMR (DMSO-d6) was in accordance with the reported literature.52N-(3,5-Bis(trifluoromethyl)phenyl)-2-chloroacetamide (34)
Pale yellow liquid. Yield: 92%. 1H NMR (CDCl3) was in accordance with the reported literature.532-Chloro-N-(2-cyanophenyl)acetamide (35)
Off-white solid. Yield: 97%. 1H NMR (CDCl3) was in accordance with the reported literature.54N-(4-Bromo-3-chlorophenyl)-2-chloroacetamide (36)
Off-white solid. Yield: 99%. 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H), 7.96 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.42 (dd, J = 8.4, 2.4 Hz, 1H), 4.27 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.17, 139.12, 134.00, 133.14, 120.49, 119.58, 115.10, 43.47.General procedure for the synthesis of N-alkylated isatins (37–57)
The appropriate substituted isatin (0.5 mmol) and potassium carbonate (69 mg, 0.5 mmol) were suspended in anhydrous DMF (1.5 mL) and stirred at room temperature for 5 minutes. Potassium iodide (83 mg, 0.5 mmol) and the appropriate substituted-acetamide (30–36) (0.55 mmol) were then added and the resulting mixture was stirred at room temperature overnight. Ten-fold volume of water (with respect to DMF) was added and the thus-obtained suspension was allowed to stir for 10 minutes before being filtered. The solid was washed three times with water and the resulting solid was dried in a climate chamber at 60 °C to afford the desired compounds 37–57 in appropriate purity grade.2-(4-Chloro-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (37)
Orange solid. Yield: 91%. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 7.64 (t, J = 8.1 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.27–6.97 (m, 4H), 4.56 (s, 2H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 179.84, 164.47, 157.81, 152.06, 138.92, 135.74, 132.92, 131.11, 129.22, 124.49, 119.73, 114.63, 109.82, 43.34, 20.51.2-(5-Chloro-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (38)
Orange solid. Yield: 74%. 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 7.91–7.59 (m, 2H), 7.54–7.32 (m, 2H), 7.26–6.99 (m, 3H), 4.56 (s, 2H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.91, 164.50, 158.22, 149.35, 137.26, 135.76, 132.90, 129.21, 127.75, 124.03, 119.69, 118.88, 112.85, 43.25, 20.47.2-(6-Chloro-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (39)
Orange solid. Yield: 55%. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 7.76–7.36 (m, 4H), 7.18 (dt, J = 37.6, 12.6 Hz, 3H), 4.58 (s, 2H), 2.25 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 181.45, 164.29, 158.37, 152.06, 142.56, 135.68, 132.68, 129.03, 125.82, 123.26, 119.49, 116.22, 111.55, 43.20, 20.29.2-(7-Chloro-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (40)
Orange solid. Yield: 93%. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.02–7.55 (m, 2H), 7.55–7.28 (m, 2H), 7.28–6.80 (m, 3H), 4.78 (s, 2H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.20, 165.72, 159.36, 146.04, 139.96, 136.14, 133.34, 129.61, 125.38, 124.07, 121.23, 120.33, 116.65, 45.19, 20.89.2-(4-Bromo-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (41)
Orange solid. Yield: 99%. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 7.54 (t, J = 7.9 Hz, 1H), 7.41 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 1H), 7.21–7.06 (m, 3H), 4.55 (s, 2H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 180.83, 164.92, 158.21, 152.91, 139.27, 136.17, 133.36, 129.66, 128.04, 120.19, 119.89, 116.69, 110.65, 43.67, 20.93.2-(5-Bromo-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (42)
Orange solid. Yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.78 (s, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.13 (dd, J = 13.8, 8.3 Hz, 3H), 4.56 (s, 2H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.15, 164.88, 158.47, 150.11, 140.46, 136.18, 133.28, 129.63, 127.17, 120.06, 119.68, 115.68, 113.69, 49.24, 20.90.2-(6-Bromo-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (43)
Orange solid. Yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 7.77–7.50 (m, 2H), 7.50–7.26 (m, 3H), 7.24–6.97 (m, 2H), 4.58 (s, 2H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.31, 164.98, 158.94, 152.39, 136.31, 133.24, 132.43, 129.71, 126.80, 126.39, 119.94, 117.13, 114.90, 43.70, 20.94.2-(7-Bromo-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (44)
Reddish solid. Yield: 96%. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.08–7.76 (m, 1H), 7.67 (m, 1H), 7.61–7.34 (m, 2H), 7.33–7.02 (m, 3H), 4.81 (s, 2H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.27, 165.68, 159.59, 147.50, 143.32, 136.16, 133.36, 129.61, 125.71, 124.53, 121.57, 120.39, 104.07, 44.92, 20.90.2-(7-Iodo-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (45)
Dark orange solid. Yield: 91%. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.18–7.96 (m, 1H), 7.76–7.57 (m, 1H), 7.55–7.30 (m, 2H), 7.29–7.02 (m, 2H), 7.01–6.86 (m, 1H), 4.84 (s, 2H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 182.55, 165.57, 159.82, 150.70, 150.24, 136.19, 133.33, 129.56, 125.88, 124.84, 121.12, 120.45, 119.44, 44.46, 20.95.2-(4-Fluoro-5-methyl-2,3-dioxoindolin-1-yl)-N-(p-tolyl)acetamide (46)
Orange solid. Yield: 93%. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 7.90–7.55 (m, 1H), 7.54–7.29 (m, 2H), 7.29–6.81 (m, 3H), 4.53 (s, 2H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.31, 167.79, 165.26, 164.52, 158.75, 151.89, 151.76, 135.87, 132.80, 129.24, 128.33, 128.25, 119.69, 119.54, 119.34, 113.99, 113.96, 99.85, 99.56, 43.32, 20.46, 13.40, 13.37.N-(4-Chlorophenyl)-2-(2,3-dioxoindolin-1-yl)acetamide (47)
Orange solid. Yield: 99%. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.67 (dt, J = 4.0, 8.0 Hz, 1H), 7.62–7.58 (m, 3H), 7.37 (d, J = 8.8 Hz, 2H), 7.18–7.15 (m, 2H), 4.59 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.94, 165.07, 158.45, 156.82, 150.80, 138.37, 137.33, 128.74, 124.51, 123.53, 121.10, 117.51, 111.03, 40.66.2-(2,3-Dioxoindolin-1-yl)-N-(4-nitrophenyl)acetamide (48)
Orange solid. Yield: 93%. 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 8.24 (d, J = 8.9 Hz, 2H), 7.82 (d, J = 8.9 Hz, 2H), 7.72–7.57 (m, 2H), 7.24–7.10 (m, 2H), 4.66 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.88, 165.99, 158.50, 150.77, 144.49, 142.68, 138.47, 125.07, 124.61, 123.65, 119.29, 117.50, 111.11, 43.35.2-(2,3-Dioxoindolin-1-yl)-N-(3-nitrophenyl)acetamide (49)
Orange solid. Yield: 96%. 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 8.56 (s, 1H), 8.16–7.82 (m, 2H), 7.82–7.45 (m, 3H), 7.18 (d, J = 6.9 Hz, 2H), 4.63 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.86, 165.78, 158.49, 150.72, 147.97, 139.44, 138.42, 130.37, 125.51, 124.57, 123.62, 118.39, 117.54, 113.67, 111.06, 43.26.N-(3,5-Bis(trifluoromethyl)phenyl)-2-(2,3-dioxoindolin-1-yl)acetamide (50)
Orange solid. Yield: 92%. 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.40–8.13 (m, 2H), 7.81 (s, 1H), 7.68 (td, J = 7.8, 1.4 Hz, 1H), 7.63 (d, J = 7.3 Hz, 1H), 7.25–7.13 (m, 2H), 4.65 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 183.28, 166.68, 162.86, 158.95, 151.10, 140.67, 138.95, 131.85, 131.52, 131.19, 130.86, 127.65, 125.07, 124.95, 124.15, 122.24, 119.77, 119.74, 119.71, 119.68, 119.53, 117.97, 117.22, 117.19, 117.16, 117.13, 111.54, 43.72.N-(2-Cyanophenyl)-2-(2,3-dioxoindolin-1-yl)acetamide (51)
Orange solid. Yield: 93%. 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H), 7.84 (dd, J = 7.9, 1.5 Hz, 1H), 7.68 (ddd, J = 9.3, 5.8, 1.6 Hz, 2H), 7.62 (d, J = 7.4 Hz, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.18 (t, J = 8.0 Hz, 2H), 4.65 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 183.36, 166.29, 158.85, 150.96, 139.82, 138.74, 134.45, 133.85, 126.88, 126.35, 124.95, 124.08, 118.10, 117.14, 111.53, 108.37, 43.36.N-(4-Bromo-3-chlorophenyl)-2-(2,3-dioxoindolin-1-yl)acetamide (52)
Orange solid. Yield: 95%. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 7.93 (d, J = 2.5 Hz, 1H), 7.75–7.58 (m, 3H), 7.41 (dd, J = 8.8, 2.5 Hz, 1H), 7.24–7.12 (m, 2H), 4.60 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.87, 165.52, 158.47, 150.72, 139.00, 138.41, 134.00, 133.12, 124.55, 123.60, 120.69, 119.75, 117.51, 115.07, 111.05, 43.23.2-(4-Bromo-2,3-dioxoindolin-1-yl)-N-(4-nitrophenyl)acetamide (53)
Orange solid. Yield: 84%. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (bs, 1H), 8.23 (d, J = 7.9 Hz, 2H), 7.81 (d, J = 7.9 Hz, 2H), 7.55 (d, J = 7.8 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.20 (d, J = 7.8 Hz, 1H) 4.67 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 180.18, 165.76, 157.70, 152.40, 144.38, 142.65, 138.87, 127.67, 125.02, 119.50, 119.31, 116.16, 110.31, 43.39.2-(7-Bromo-2,3-dioxoindolin-1-yl)-N-(4-nitrophenyl)acetamide (54)
Orange solid. Yield: 51%. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (bs, 1H), 8.24 (d, J = 8.1 Hz, 2H), 7.85–7.79 (m, 3H), 7.68 (d, J = 8.0 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H), 4.92 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 181.66, 166.50, 159.07, 147.01, 144.49, 142.98, 142.68, 125.41, 125.04, 124.22, 121.03, 119.40, 103.67, 44.73.N-(3,5-Bis(trifluoromethyl)phenyl)-2-(4-chloro-2,3-dioxoindolin-1-yl)acetamide (55)
Orange solid. Yield: 96%. 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.22 (bs, 2H), 7.95 (bs, 1H), 7.82 (bs, 1H), 7.65 (d, J = 7.1 Hz, 1H), 7.21–7.17 (m, 2H), 4.66 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 179.63, 166.03, 162.31, 157.77, 151.88, 140.13, 139.02, 131.35, 131.18, 131.03, 130.70, 130.38, 127.19, 124.61, 124.48, 121.77, 119.38, 119.35, 119.32, 119.29, 116.91, 116.88, 116.85, 116.82, 114.58, 109.92, 43.45.N-(3,5-Bis(trifluoromethyl)phenyl)-2-(6-chloro-2,3-dioxoindolin-1-yl)acetamide (56)
Orange solid. Yield: 87%. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (bs, 1H), 9.09 (t, J = 8.0 Hz, 1H), 8.33–8.30 (bs, 2H), 7.77 (s, 1H), 7.63 (d, J = 8.0 Hz, 1H), 6.63 (m, 1H), 4.20 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 200.35, 172.44, 169.08, 168.98, 151.24, 140.74, 140.57, 138.75, 136.45, 131.29, 130.97, 130.64, 130.32, 124.56, 121.86, 118.94, 118.92, 118.89, 118.86, 116.17, 116.13, 116.09, 116.06, 114.47, 113.93, 110.75, 46.11.N-(3,5-Bis(trifluoromethyl)phenyl)-2-(7-chloro-2,3-dioxoindolin-1-yl)acetamide (57)
Orange solid. Yield: 91%. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (bs, 1H), 8.21 (bs, 2H), 7.82 (s, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.21 (t, J = 8 Hz, 1H), 4.89 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 181.57, 169.91, 166.85, 158.86, 145.46, 140.22, 139.69, 134.88, 132.98, 131.41, 131.08, 130.75, 130.43, 125.14, 124.48, 123.81, 121.78, 120.72, 119.30, 119.27, 118.06, 116.96, 116.89, 116.85, 116.81, 116.25, 44.92.Synthesis of ethyl benzofuran-2-carboxylate (60)
Salicylaldehyde 58 (0.87 mL, 8.19 mmol) was dissolved in 30 mL of MeCN/DMF (4:1), followed by the addition of ethylbromoacetate (1.09 mL, 9.83 mmol) and cesium carbonate (5.34 g, 16.38 mmol). The mixture was refluxed overnight (TLC analysis: EtOAc:petroleum ether 1:1). The solvent was removed under reduced pressure, and the residue was washed with EtOAc. The filtrate was concentrated to dryness, washed with water (20 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were dried over anhydrous sodium sulfate, filtered, and evaporated to dryness, providing the desired product, which proved to be sufficiently pure to be directly used in the next reaction. White solid. Yield: 60%. 1H-NMR (400 MHz, DMSO-d6): 7.83–7.73 (m, 3H), 7.56–7.51 (m, 1H), 7.40–7.36 (m, 1H), 4.37 (q, 2H, J = 7.2 Hz), 1.35 (t, 3H, J = 7.2 Hz).
Synthesis of methyl benzo[b]thiophene-2-carboxylate (61)
2-Nitrobenzaldehyde 59 (1.00 g, 6.6 mmol) was dissolved in dry DMF (15 mL), followed by the addition of potassium carbonate (1.10 g, 8.0 mmol) and methylthioglycolate (600 μL, 6.6 mmol) at 0 °C. The resulting mixture was stirred at 0 °C for 30 min and then heated at 60 °C overnight (TLC analysis: EtOAc:petroleum ether 3:7). The reaction mixture was poured into ice and the obtained precipitate was filtered under reduced pressure, furnishing the desired product, which proved to be sufficiently pure to be directly used in the next reaction. Pale yellow solid. Yield: 77%. 1H-NMR (400 MHz, DMSO-d6): 8.23 (s, 1H), 8.08 (d, 1H, J = 8.0 Hz), 8.04 (d, 1H, J = 8.0 Hz), 7.57–7.47 (m, 2H), 3.90 (s, 3H).
General procedure for the synthesis of benzofuran-2-ylmethanol (62) and benzo[b]thiophen-2-ylmethanol (63)
LiAlH4 (106 mg, 2.8 mmol) was suspended in dry THF (4 mL), followed by the dropwise addition of the appropriate ester 60 or 61 (2.3 mmol), previously dissolved in 3 mL of the same solvent. The resulting mixture was stirred at room temperature for 2 h. The reaction was monitored with TLC until conversion was complete (TLC analysis: EtOAc/petroleum ether 1:1). A saturated aqueous potassium carbonate solution (0.5 mL) was added to the mixture, and the resulting suspension was filtered and washed with EtOAc. The organic phase was dried over anhydrous sodium sulfate, filtered, and evaporated to dryness, affording the desired products sufficiently pure to be directly used in the next step of the reaction.
Benzofuran-2-ylmethanol (62)
Brown oil. Yield: 80%. 1H-NMR (400 MHz, DMSO-d6): 7.59–7.52 (m, 2H), 7.29–7.20 (m, 2H), 6.75 (s, 1H), 5.49 (t, 1H, J = 5.8 Hz), 4.57 (d, 2H, J = 5.2 Hz).Benzo[b]thiophen-2-ylmethanol (63)
Reddish solid. Yield: 100%. 1H-NMR (400 MHz, DMSO-d6): 7.93–7.90 (m, 1H), 7.78–7.76 (m, 1H), 7.36–7.27 (m, 3H), 5.66 (t, 1H, J = 5.8 Hz), 4.75 (d, 2H, J = 5.6 Hz).General procedure for the synthesis of 2-(bromomethyl)benzofuran (64) and 2-(bromomethyl)benzo[b]thiophene (65)
A solution of benzofuran-2-ylmethanol 62 or benzo[b]thiophen-2-ylmethanol 63 (5.4 mmol) in dry THF was treated with PBr3 (0.51 mL, 5.4 mmol) at 0 °C. The resulting solution was allowed to react at room temperature for 3 h. The reaction was monitored with TLC until conversion was complete (TLC analysis: EtOAc:petroleum ether 6:4). The solution was washed with water and extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were dried over anhydrous sodium sulfate, filtered, and evaporated to dryness. The crude material was purified by flash chromatography (petroleum ether/EtOAc 9:1) affording pure derivatives 64 and 65.
2-(Bromomethyl)benzofuran (64)
Yellow oil. Yield: 59%. 1H-NMR (400 MHz, DMSO-d6): 7.64 (d, 1H, J = 7.6 Hz), 7.59 (d, 1H, J = 8.0 Hz), 7.36 (t, 1H, J = 7.6 Hz), 7.27 (t, 1H, J = 7.2 Hz), 7.04 (s, 1H), 4.93 (s, 2H).2-(Bromomethyl)benzo[b]thiophene (65)
Pale yellow solid. Yield: 60%. 1H-NMR (400 MHz, DMSO-d6): 7.96–7.94 (m, 1H), 7.84–7.82 (m, 1H), 7.56 (s, 1H), 7.39–7.37 (m, 2H), 5.11 (s, 2H).General procedure for the synthesis of N-alkylated isatins (66–70)
Isatin (196 mg, 1.33 mmol) was dissolved in dry DMF, followed by the addition of potassium carbonate (167 mg, 1.21 mmol). Concomitantly, the appropriate halide (64, 65, commercially available 2-(bromomethyl)naphthalene, 2-(chloromethyl)benzimidazole, 3-bromo-5-chloro-1-benzothiophene) (1.21 mmol) was dissolved in dry DMF followed by the addition of potassium iodide (201 mg, 1.21 mmol). After 30 min, the two mixtures were combined, and the resulting solution was stirred at room temperature for 5 h. The reaction was monitored with TLC until conversion was complete (TLC analysis: eluent mixture EtOAc/petroleum ether in different ratios). Then, the mixture was cautiously poured into ice and the obtained precipitate was filtered under reduced pressure. The crude material was purified by flash chromatography using EtOAc/petroleum ether in different ratios as the eluting system, to afford compounds 66–70.1-(Benzofuran-2-ylmethyl)indoline-2,3-dione (66)
Bright orange solid. Yield: 70%. 1H-NMR (400 MHz, DMSO-d6): 7.68–7.64 (m, 1H), 7.61–7.58 (m, 2H), 7.55–7.53 (m, 1H), 7.30–7.26 (m, 2H), 7.25–7.21 (m, 1H), 7.17–7.13 (m, 1H), 7.06–7.05 (m, 1H), 5.12 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.74, 157.96, 154.38, 151.69, 150.08, 138.08, 127.77, 124.55, 124.43, 123.50, 123.04, 121.13, 117.74, 111.13, 111.09, 105.55, 36.91.1-(Benzo[b]thiophen-2-ylmethyl)indoline-2,3-dione (67)
Yield: 100%. 1H-NMR (400 MHz, DMSO-d6): 7.92–7.90 (m, 1H), 7.79–7.77 (m, 1H), 7.67–7.58 (m, 3H), 7.38–7.31 (m, 2H), 7.24 (d, 1H, J = 8.0 Hz), 7.17–7.13 (m, 1H), 5.22 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.78, 157.92, 149.91, 139.14, 139.07, 138.80, 138.13, 124.63, 124.57, 124.51, 123.58, 123.56, 123.51, 122.52, 117.71, 111.07, 38.85.1-(Naphthalen-2-ylmethyl)indoline-2,3-dione (68)
Yield: 100%. 1H-NMR (400 MHz, DMSO-d6): 8.00 (s, 1H), 7.92–7.85 (m, 3H), 7.60–7.13 (m, 5H), 7.11 (t, 1H, J = 8.4 Hz), 6.99 (d, 1H, J = 8.0 Hz), 5.09 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 183.12, 158.43, 150.37, 137.93, 133.03, 132.88, 132.35, 128.36, 127.60, 127.58, 126.36, 126.03, 125.77, 125.49, 124.48, 123.31, 117.83, 111.08, 43.13.1-((1H-Benzo[d]imidazol-2-yl)methyl)indoline-2,3-dione (69)
Yield: 60%. 1H-NMR (400 MHz, DMSO-d6): 12.50 (bs, 1H), 7.64–7.57 (m, 3H), 7.43–7.41 (m, 1H), 7.17–7.13 (m, 3H), 6.97 (d, 1H, J = 8.0 Hz), 5.18 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.91, 158.19, 150.18, 148.91, 138.16, 124.52, 123.51, 121.87, 121.78, 120.42, 117.79, 110.84, 38.01.1-((5-Chlorobenzo[b]thiophen-2-yl)methyl)indoline-2,3-dione (70)
Yield: 90%. 1H-NMR (400 MHz, DMSO-d6): 8.11–8.02 (m, 3H), 7.62–7.58 (m, 2H), 7.45 (dd, 1H, J = 1.6 Hz, J = 8.4 Hz), 7.15–7.12 (m, 2H), 5.14 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.89, 158.32, 150.16, 138.73, 138.50, 137.82, 129.64, 129.44, 127.79, 124.74, 124.67, 124.44, 123.38, 121.77, 117.97, 111.11, 37.76.TK inhibition
For fixed-dose inhibition of ABL, CSK, EGFR, EPHA2, EPHB4, FGFR1, FLT3, IGF1R, ITK, JAK3, KDR, LCK, c-MET, PDGFRα, PYK2, SRC, SYK, TIE2, TRKA, and TYRO3 TKs and IC50 c-MET inhibition studies, the test compounds were dissolved in DMSO to achieve a 2 mM concentration. Then the solution was further diluted with assay buffer to make the final test compound solutions. Reference compounds for the assay control were prepared similarly. A description of the employed 20 kinases is reported in the ESI† (Table S1). An off-chip mobility shift assay (MSA) was then used to test the inhibition of the compounds against the selected TKs. Briefly, a 4× substrate/ATP/metal solution was prepared with kit buffer (20 mM HEPES, 0.01% Triton X-100, 5 mM DTT, pH 7.5), and a 2× kinase solution was prepared with assay buffer (20 mM HEPES, 0.01% Triton X-100, 5 mM DTT, pH 7.5). 5 μL of 4× compound solution, 5 μL of 4× substrate/ATP/metal solution, and 10 μL of 2× kinase solution were mixed and incubated in a well of a polypropylene 384 well microplate for 1 h at RT. 70 μL of termination buffer (QuickScout Screening Assist MSA; Carna Biosciences) was added to the well. The reaction mixture was applied to a LabChip™ system (Perkin Elmer), and the product and substrate peptide peaks were separated and quantified. The kinase reaction was evaluated by the product ratio calculated from the peak heights of product (P) and substrate (S) peptides (P/(P + S)). The readout value of the reaction control (complete reaction mixture) was set to 0% inhibition, and the readout value of the background (enzyme(−)) was set to 100% inhibition, then the percent inhibition of each test solution was calculated. The IC50 values were calculated from the concentration vs. % inhibition curves by fitting to a four parameter logistic curve. All experiments were run in triplicate and the results were expressed as averages ± SD.SMO radioligand binding assays
The binding experiments closely followed previously established procedures with only a few adjustments.55 In brief, 15 μg of HEK293T-WT SMO membranes (provided by Multispan Inc., Cod. MC1442; Kd = 20 nM; Bmax = 6.2 pmol mg−1 protein) were incubated for 4 hours at RT in a binding buffer composed of 50 mM HEPES, 5 mM MgCl2, and 0.02% BSA. This buffer contained 25 nM [3H]-cyclopamine (provided by American Radiolabeled Chemicals, Inc., Art. 1473, specific activity 20 Ci mmol−1) and varying concentrations of the compounds. Nonspecific binding was determined in the presence of 25 μM compound and the SMO antagonist vismodegib (2-chloro-N-[4-chloro-3-(2-pyridinyl)phenyl]-4-(methylsulfonyl)benzamide). The bound radioactivity was separated by rapid filtration through GF/C glass fiber filters pre-soaked for 2 hours in 0.3% polyethylenimine (pH 13.0) and washed three times with 4 mL of ice-cold phosphate-buffered saline with 0.01% Triton X-100 (pH 7.0). Radioactivity levels were measured using liquid scintillation spectrometry. For the active compounds, IC50 values were determined, and Ki values were calculated following the Cheng and Prusoff equation. Dose–response curves are provided in the ESI,† Fig. S4.Cell lines and treatments
The NCI-HCC827 and NCI-PC9 human lung adenocarcinoma cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The gefitinib-resistant HCC827 (HCC827/GR) and osimertinib-resistant PC9 (PC9/OR) cell lines were established in-house by exposing parental cell lines to increasing concentrations of TKIs. All cell lines were grown in RPMI 1640 (Gibco) medium supplemented with 10% fetal bovine serum (FBS), 100 IU mL−1 penicillin and 50 μg mL−1 streptomycin in a humidified incubator in 5% CO2 at 37 °C.Cell proliferation assay
Drug-induced toxicity was assessed by using the MTS assay (Cell Counting Kit-8) following established protocols.30 Briefly, cells were seeded in 96-well flat-bottomed plates at a density of 5000 cells per well and treated for 72 h with increasing concentrations (0.01–10 μM) of compounds. The number of viable cells was determined spectrophotometrically by measuring absorbance at 490 nm and expressed as the percentage of viable cells, considering the untreated control cells to be 100%. The concentration that inhibits 50% of cell growth (IC50) was determined by interpolation from the dose–response curves. At least three independent experiments were performed in quadruplicate and data were pooled.Western blot analysis
Protein lysates from NSCLC cells were obtained by homogenization in RIPA lysis buffer [0.1% sodium dodecyl sulfate (SDS), 0.5% deoxycholate, 1% Nonidet, 100 mmol L−1 NaCl, 10 mmol L−1 Tris-HCl (pH 7.4), 0.5 mmol L−1 dithiothreitol, and 0.5% phenylmethyl sulfonyl fluoride, protease inhibitor cocktail (Hoffmann-La Roche) and phosphatase inhibitor tablets (PhosSTOP; Roche Diagnostics)] and clarification by centrifugation at 15000 rpm for 20 min at 4 °C. Protein samples containing comparable amounts of proteins, estimated by a modified Bradford assay (Bio-Rad), were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels and electro-transferred onto 0.2 μm nitrocellulose membranes (Trans-Blot Turbo; BioRad). After blocking membranes for 90 min at room temperature, they were incubated overnight at 4 °C with primary antibodies, and then with a secondary antibody for 1 h at room temperature. Horseradish peroxidase-linked anti-rabbit (BioRad) and anti-mouse (BioRad) antibodies were used as secondary antibodies. Proteins were detected with Clarity Western ECL Substrate using the ChemiDoc (BioRad). Images were analysed using BioRad software Image Lab 3.0.1. Primary antibodies for western blot analysis against pAKT (Ser473) (D9E) (4060, 1:1000), AKT (9272, 1:1000), pMET (Tyr1234/1235) (3126, 1:1000), MET (D1C2) (8198, 1:1000), GLI1 (L42B10) (2643, 1:1000) and vimentin (D21H3) (5741, 1:1000) were purchased from Cell Signaling (Danvers, MA); the monoclonal anti-SMO (E-5) antibody (166685, 1:500) was from Santa Cruz Biotechnology and the monoclonal anti-α-tubulin antibody (T8203) was from Sigma Chemical Co.
Apoptosis assay
The gefitinib-resistant HCC827-GR and osimertinib-resistant PC9-OR cells were seeded in Petri dishes at a density of 500.000 cells per well and the day after treated for 24 h with test compounds at a 20 μM concentration. At the end of the incubation, the cells were collected and washed with phosphate-buffered saline. The cells were then stained with the annexin V-Alexa 488 conjugate and PI according to the manufacturer's protocol (“Invitrogen™ Alexa Fluor™ 488 annexin V/Dead Cell Apoptosis Kit”, cat.no V13241) and analyzed by flow cytometry using a BD Fortessa. Analysis was conducted using BD FACSDiva™ software (BD Biosciences).Statistical analysis and graphical quantification
Statistical analyses were performed using Prism 8 (GraphPad Software, San Diego, CA, USA). Two-way analysis of variance (ANOVA) was used to evaluate the statistical significance of the results. A P value <0.05 was considered to indicate statistical significance. Western blotting signals were quantified by morphodensitometric analysis using ImageJ software (NIH, Bethesda, MD, USA). Briefly, the product of the area and optical density of each band was determined and normalized to the same parameter derived from the equal loading used. The data are expressed as the relative protein levels of each treated sample compared with those of the corresponding vehicle-treated internal control.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
Stefano Tomassi, Emma Baglini, Elisabetta Barresi, Salvatore Di Maro, Federico Da Settimo, and Sabrina Taliani performed all the reported syntheses. Benito Natale, Michele Roggia, Giorgio Amendola, and Sandro Cosconati performed all the molecular modeling studies. Maria Letizia Trincavelli performed all the in vitro experiments. Luisa Amato, Caterina De Rosa, Carminia Maria Della Corte, Floriana Morgillo, and Fortunato Ciardiello performed all the cell-based experiments. Stefano Tomassi and Sandro Cosconati wrote the manuscript. Sabrina Taliani, Floriana Morgillo, and Sandro Cosconati conceived the project and supervised the work.Conflicts of interest
The authors affirm that there are no identifiable competing financial interests or personal relationships that could have been perceived to impact the findings presented in this paper.Acknowledgements
S. T. (Stefano Tomassi) acknowledges MUR-Ministero dell'Università e della Ricerca (Italian Ministry of University and Research), PON R&I 2014-2020-AIM (Attraction and International Mobility), and project AIM1873131 – 2, linea 1. G. A. was supported by an AIRC fellowship from Italy (Clementina Colombatti) for the project “Application of Advanced In Silico Methods to Fight Resistant Non-Small Cell Lung Cancer”. This research was funded by the University of Campania Luigi Vanvitelli (VALERE: Vanvitelli per la Ricerca, ANIMA, and VALEREPlus projects) and the University of Pisa (institutional funding).Notes and references
- Cancer Statistics Explorer Network, https://seer.cancer.gov/statistics-network/, (accessed 8 July 2024).
- Y. Cheng, T. Zhang and Q. Xu, MedComm, 2021, 2, 692–729 CrossRef PubMed.
- C. Goebel, C. L. Louden, R. McKenna, O. Onugha, A. Wachtel and T. Long, Cancer Genomics Proteomics, 2019, 16, 229 CrossRef CAS.
- D. S. Ettinger, W. Akerley, H. Borghaei, A. C. Chang, R. T. Cheney, L. R. Chirieac, T. A. D'Amico, T. L. Demmy, A. K. P. Ganti, R. Govindan, F. W. Grannis, L. Horn, T. M. Jahan, M. Jahanzeb, A. Kessinger, R. Komaki, F. M. Kong, M. G. Kris, L. M. Krug, I. T. Lennes, B. W. Loo, R. Martins, J. O'Malley, R. U. Osarogiagbon, G. A. Otterson, J. D. Patel, M. C. Pinder-Schenck, K. M. Pisters, K. Reckamp, G. J. Riely, E. Rohren, S. J. Swanson, D. E. Wood, S. C. Yang, M. Hughes and K. M. Gregory, J. Natl. Compr. Cancer Network, 2012, 10, 1236–1271 CrossRef CAS PubMed.
- Q. Guo, L. Liu, Z. Chen, Y. Fan, Y. Zhou, Z. Yuan and W. Zhang, Front. Oncol., 2022, 12, 945102 CrossRef CAS PubMed.
- A. Anighoro, J. Bajorath and G. Rastelli, J. Med. Chem., 2014, 57, 7874–7887 CrossRef CAS.
- A. Kabir and A. Muth, Pharmacol. Res., 2022, 176, 106055 CrossRef CAS PubMed.
- V. Shah, A. McNatty, L. Simpson, H. Ofori and F. Raheem, Biomedicines, 2023, 11(2), 950 CrossRef CAS PubMed.
- A. C. Tan, Thorac. Cancer, 2020, 11, 511–518 CrossRef.
- M. P. Curran, Drugs, 2012, 72, 99–107 CrossRef CAS.
- N. M. Raghavendra, D. Pingili, S. Kadasi, A. Mettu and S. V. U. M. Prasad, Eur. J. Med. Chem., 2018, 143 Search PubMed.
- S. A. Mani, W. Guo, M. J. Liao, E. N. Eaton, A. Ayyanan, A. Y. Zhou, M. Brooks, F. Reinhard, C. C. Zhang, M. Shipitsin, L. L. Campbell, K. Polyak, C. Brisken, J. Yang and R. A. Weinberg, Cell, 2008, 133, 704–715 CrossRef CAS PubMed.
- D. Ribatti, R. Tamma and T. Annese, Transl. Oncol., 2020, 13, 100773 CrossRef PubMed.
- L. A. Byers, L. Diao, J. Wang, P. Saintigny, L. Girard, M. Peyton, L. Shen, Y. Fan, U. Giri, P. K. Tumula, M. B. Nilsson, J. Gudikote, H. Tran, R. J. G. Cardnell, D. J. Bearss, S. L. Warner, J. M. Foulks, S. B. Kanner, V. Gandhi, N. Krett, S. T. Rosen, E. S. Kim, R. S. Herbst, G. R. Blumenschein, J. J. Lee, S. M. Lippman, K. K. Ang, G. B. Mills, W. K. Hong, J. N. Weinstein, I. I. Wistuba, K. R. Coombes, J. D. Minna and J. V. Heymach, Clin. Cancer Res., 2013, 19, 279–290 CrossRef CAS PubMed.
- T. Sowa, T. Menju, M. Sonobe, T. Nakanishi, K. Shikuma, N. Imamura, H. Motoyama, K. Hijiya, A. Aoyama, F. Chen, T. Sato, M. Kobayashi, A. Yoshizawa, H. Haga, T. Sozu and H. Date, Cancer Med., 2015, 4, 1853–1862 CrossRef CAS PubMed.
- K. R. Jakobsen, C. Demuth, B. S. Sorensen and A. L. Nielsen, Transl. Lung Cancer Res., 2016, 5, 172–182 CrossRef CAS.
- S. Boumahdi and F. J. de Sauvage, Nat. Rev. Drug Discovery, 2020, 19, 39–56 CrossRef CAS.
- N. Clere, S. Renault and I. Corre, Front. Cell Dev. Biol., 2020, 8, 747 CrossRef PubMed.
- T. K. Rimkus, R. L. Carpenter, S. Qasem, M. Chan and H. W. Lo, Cancers, 2016, 8(2), 22 CrossRef.
- Y. Zhang, M. Xia, K. Jin, S. Wang, H. Wei, C. Fan, Y. Wu, X. Li, X. Li, G. Li, Z. Zeng and W. Xiong, Mol. Cancer, 2018, 17(1), 45 CrossRef.
- S. Pilotto, L. Carbognin, N. Karachaliou, P. C. Ma, R. Rosell, G. Tortora and E. Bria, Cancer Treat. Rev., 2017, 60, 1–11 CrossRef CAS PubMed.
- L. Trusolino, A. Bertotti and P. M. Comoglio, Nat. Rev. Mol. Cell Biol., 2010, 11, 834–848 CrossRef CAS PubMed.
- S. F. Yeung, J. H. M. Tong, P. P. W. Law, L. Y. Chung, R. W. M. Lung, C. Y. K. Tong, C. Chow, A. W. H. Chan, I. Y. P. Wan, T. S. K. Mok and K. F. To, J. Thorac. Oncol., 2015, 10, 1292–1300 CrossRef CAS PubMed.
- G. M. Frampton, S. M. Ali, M. Rosenzweig, J. Chmielecki, X. Lu, T. M. Bauer, M. Akimov, J. A. Bufill, C. Lee, D. Jentz, R. Hoover, S. H. Ignatius Ou, R. Salgia, T. Brennan, Z. R. Chalmers, S. Jaeger, A. Huang, J. A. Elvin, R. Erlich, A. Fichtenholtz, K. A. Gowen, J. Greenbowe, A. Johnson, D. Khaira, C. McMahon, E. M. Sanford, S. Roels, J. White, J. Greshock, R. Schlegel, D. Lipson, R. Yelensky, D. Morosini, J. S. Ross, E. Collisson, M. Peters, P. J. Stephens and V. A. Miller, Cancer Discovery, 2015, 5, 850–860 CrossRef CAS.
- J. H. Tong, S. F. Yeung, A. W. H. Chan, L. Y. Chung, S. L. Chau, R. W. M. Lung, C. Y. Tong, C. Chow, E. K. Y. Tin, Y. H. Yu, H. Li, Y. Pan, W. P. Chak, C. S. H. Ng, T. S. K. Mok and K. F. To, Clin. Cancer Res., 2016, 22, 3048–3056 CrossRef CAS PubMed.
- B. Lutterbach, Q. Zeng, L. J. Davis, H. Hatch, G. Hang, N. E. Kohl, J. B. Gibbs and B. S. Pan, Cancer Res., 2007, 67, 2081–2088 CrossRef CAS PubMed.
- F. Wu, Y. Zhang, B. Sun, A. P. McMahon and Y. Wang, Cell Chem. Biol., 2017, 24, 252–280 CrossRef CAS.
- M. Y. Maitah, S. Ali, A. Ahmad, S. Gadgeel and F. H. Sarkar, PLoS One, 2011, 6(1), e16068 CrossRef CAS PubMed.
- A. S. Tsao, I. Wistuba, D. Xia, L. Byers, L. Diao, J. Wang, V. Papadimitrakopoulou, X. Tang, W. Lu, H. Kadara, D. N. Grigoryev, M. E. Selvan, Z. H. Gümüş, Z. Tan, S. Zhang, M. Nilsson and J. V. Heymach, JCO Precis. Oncol., 2017, 1–10 CrossRef.
- C. M. Della Corte, C. Bellevicine, G. Vicidomini, D. Vitagliano, U. Malapelle, M. Accardo, A. Fabozzi, A. Fiorelli, M. Fasano, F. Papaccio, E. Martinelli, T. Troiani, G. Troncone, M. Santini, R. Bianco, F. Ciardiello and F. Morgillo, Clin. Cancer Res., 2015, 21, 4686–4697 CrossRef CAS PubMed.
- F. Morgillo, G. Amendola, C. M. Della Corte, C. Giacomelli, L. Botta, S. Di Maro, A. Messere, V. Ciaramella, S. Taliani, L. Marinelli, M. L. Trincavelli, C. Martini, E. Novellino, F. Ciardiello and S. Cosconati, J. Med. Chem., 2017, 60(17), 7447–7458 CrossRef CAS PubMed.
- C. Kollmannsberger, H. Hurwitz, L. Bazhenova, B. C. Cho, D. Hong, K. Park, K. L. Reckamp, S. Sharma, H. Der-Torossian, J. G. Christensen, D. Faltaos, D. Potvin, V. Tassell, R. Chao and G. I. Shapiro, arget. Oncol., 2023, 18, 105–118 Search PubMed.
- Study Details|Phase 2 Study of Glesatinib, Sitravatinib or Mocetinostat in Combination With Nivolumab in Non-Small Cell Lung Cancer|ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT02954991, (accessed 18 January 2024).
- T. Guo and S. Ma, ChemMedChem, 2021, 16, 600–620 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2024, 200, 107059 CrossRef CAS PubMed.
- G. W. Collie, I. N. Michaelides, K. Embrey, C. J. Stubbs, U. Börjesson, I. L. Dale, A. Snijder, L. Barlind, K. Song, P. Khurana, C. Phillips and R. I. Storer, ACS Med. Chem. Lett., 2021, 12, 162–167 CrossRef CAS PubMed.
- S. Eathiraj, R. Palma, E. Volckova, M. Hirschi, D. S. France, M. A. Ashwell and T. C. K. Chan, J. Biol. Chem., 2011, 286, 20666–20676 CrossRef CAS PubMed.
- L. Xing, J. Klug-Mcleod, B. Rai and E. A. Lunney, Bioorg. Med. Chem., 2015, 23, 6520–6527 CrossRef CAS PubMed.
- Z. Liang, D. Zhang, J. Ai, L. Chen, H. Wang, X. Kong, M. Zheng, H. Liu, C. Luo, M. Geng, H. Jiang and K. Chen, Bioorg. Med. Chem. Lett., 2011, 21, 3749–3754 CrossRef CAS.
- J. Wang, R. A. Mook, J. Lu, D. M. Gooden, A. Ribeiro, A. Guo, L. S. Barak, H. Kim Lyerly and W. Chen, Bioorg. Med. Chem., 2012, 20, 6751–6757 CrossRef CAS PubMed.
- Z. Song, Y. Jin, Y. Ge, C. Wang, J. Zhang, Z. Tang, J. Peng, K. Liu, Y. Li and X. Ma, Bioorg. Med. Chem., 2016, 24, 5505–5512 CrossRef CAS PubMed.
- S. Usman, N. H. Waseem, T. K. N. Nguyen, S. Mohsin, A. Jamal, M. T. Teh and A. Waseem, Cancers, 2021, 13, 4985 CrossRef CAS PubMed.
- I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489–501 CrossRef CAS.
- I. Vermes, C. Haanen and C. Reutelingsperger, J. Immunol. Methods, 2000, 243, 167–190 CrossRef CAS.
- Schrödinger Release 2024-3, Maestro, Schrödinger, LLC, New York, NY, 2024.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- D. Santos-Martins, L. Solis-Vasquez, A. F. Tillack, M. F. Sanner, A. Koch and S. Forli, J. Chem. Theory Comput., 2021, 17, 1060–1073 CrossRef CAS.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CAS.
- W. G. Hoover, Phys. Rev. A, 1985, 31(3), 1695–1697 CrossRef PubMed.
- M. Milne, M. Lewis, N. McVicar, M. Suchy, R. Bartha and R. H. E. Hudson, RSC Adv., 2013, 4, 1666–1674 RSC.
- E. Hernández-Núñez, H. Tlahuext, R. Moo-Puc, H. Torres-Gómez, R. Reyes-Martínez, R. Cedillo-Rivera, C. Nava-Zuazo and G. Navarrete-Vazquez, Eur. J. Med. Chem., 2009, 44, 2975–2984 CrossRef.
- S. D. Tala, T. H. Ou, Y. W. Lin, K. S. Tala, S. H. Chao, M. H. Wu, T. H. Tsai, R. Kakadiya, S. Suman, C. H. Chen, T. C. Lee and T. L. Su, Eur. J. Med. Chem., 2014, 76, 155–169 CrossRef CAS PubMed.
- T. Qiu, G. P. A. Yap and J. Rosenthal, ACS Appl. Energy Mater., 2019, 2, 8560–8569 CrossRef CAS.
- Y. Kabri, A. Gellis and P. Vanelle, Green Chem., 2009, 11, 201–208 RSC.
- J. Wang, R. A. Mook, J. Lu, D. M. Gooden, A. Ribeiro, A. Guo, L. S. Barak, H. Kim Lyerly and W. Chen, Bioorg. Med. Chem., 2012, 20, 6751–6757 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00553h |
| ‡ ST and BN contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |