
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New anti-ovarian cancer quinolone derivatives acting by modulating microRNA processing machinery†
Tommaso
Felicetti‡
 a,
Nicola
Di Iacovo‡
b,
Maria Agnese
Della Fazia
b,
Danilo
Piobbico
b,
Stefania
Pieroni
b,
Martina
Pacetti
a,
Jialing
Yu
c,
Yilun
Sun
d,
Serena
Massari
a,
Maria Letizia
Barreca
a,
Stefano
Sabatini
a,
Oriana
Tabarrini
a,
Violetta
Cecchetti
a,
Fei
Wang
c,
Yves
Pommier
d,
Mariangela
Morlando§
*a,
Giuseppe
Servillo
*b and
Giuseppe
Manfroni
*a
a,
Nicola
Di Iacovo‡
b,
Maria Agnese
Della Fazia
b,
Danilo
Piobbico
b,
Stefania
Pieroni
b,
Martina
Pacetti
a,
Jialing
Yu
c,
Yilun
Sun
d,
Serena
Massari
a,
Maria Letizia
Barreca
a,
Stefano
Sabatini
a,
Oriana
Tabarrini
a,
Violetta
Cecchetti
a,
Fei
Wang
c,
Yves
Pommier
d,
Mariangela
Morlando§
*a,
Giuseppe
Servillo
*b and
Giuseppe
Manfroni
*a
aDepartment of Pharmaceutical Sciences, Section of Chemistry and Pharmaceutical Technology, University of Perugia, Via Del Liceo, 1-06123, Perugia, Italy. E-mail: mariangela.morlando@uniroma1.it; giuseppe.manfroni@unipg.it; Tel: +39 06 4991 2341 Tel: +39 075 585 5126
bDepartment of Medicine and Surgery, University of Perugia, Piazza L. Severi, 1/8-06132, Perugia, Italy. E-mail: giuseppe.servillo@unipg.it; Tel: +39 075 585 8110
cCenter for Natural Products Research, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China
dCenter for Cancer Research, Developmental Therapeutics Branch & Laboratory of Molecular Pharmacology, NCI, 31 Center Drive, Bethesda, MD 20892-4255, USA
First published on 27th September 2024
Abstract
MicroRNAs (miRNAs) play a crucial role in ovarian cancer (OC) pathogenesis and miRNA processing can be the object of pharmacological intervention. By exploiting our in-house quinolone library, we combined a cell-based screening with medicinal chemistry efforts, ultimately leading to derivative 33 with anti-OC activity against distinct cell lines (GI50 values 13.52–31.04 μM) and CC50 Wi-38 = 142.9 μM. Compound 33 retained anticancer activity against additional cancer cells and demonstrated a synergistic effect with cisplatin against cisplatin-resistant A2780 cells. Compound 33 bound TRBP by SPR (KD = 4.09 μM) and thermal shift assays and its activity was TRBP-dependent, leading to modulation of siRNA and miRNA maturation. Derivative 33 exhibited augmented potency against OC cells and a stronger binding affinity for TRBP compared to enoxacin, the sole quinolone identified as a modulator of miRNA maturation. Consequently, 33 represents a promising template for developing novel anti-OC agents with a distinctive mechanism of action.
Introduction
According to GLOBOCAN estimates, in 2020, ovarian cancer (OC) was diagnosed in 313![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 959 women worldwide, resulting in 207252 deaths.1 OC is the leading cause of death among women diagnosed with gynecologic cancers, making it the fifth most common cause of death in women.1–3 Pathologically, most OCs (approximately 90%) originate from the epithelial surface, while a smaller part arises from germ or stromal cells.4 Malignant epithelial OCs can be classified into high-grade serous ovarian carcinoma (HGSC), endometrioid carcinoma, clear cell carcinoma, mucinous carcinoma, and low-grade serous ovarian carcinoma.5 HGSC is considered to be the most lethal gynecological cancer6 and is frequently characterized by p53 mutations (96% of cases), which lead to p53 loss of function and induce HGSC development.7 Unfortunately, OCs are often associated with a poor prognosis due to diagnosis at advanced stages of disease and the development of chemotherapy resistance. The standard of care is cytoreductive surgery combined with adjuvant chemotherapy, but 70% of patients experience recurrence within 2 years of primary diagnosis, and the mortality rate reaches approximately 50% of women at five years.8,9 The strategy of adjuvant chemotherapy is mainly defined for patients who respond to platinum-based treatments, while approaches for platinum-resistant patients still seem to be poorly defined.10 Indeed, for platinum-refractory disease, doxorubicin, paclitaxel, gemcitabine and topotecan are used based on toxicity, treatment cost and accessibility; however, response rates range from 10% to 15% with an overall survival of about 1 year.11 In addition, patients with platinum-sensitive OCs become platinum-resistant at advanced stages and acquire chemoresistance through various mechanisms.12 Despite new treatments (i.e., olaparib monotherapy or in combination with bevacizumab) recently approved as maintenance therapy for advanced OCs,13 we are still far from satisfactory results and novel approaches are warranted.
959 women worldwide, resulting in 207252 deaths.1 OC is the leading cause of death among women diagnosed with gynecologic cancers, making it the fifth most common cause of death in women.1–3 Pathologically, most OCs (approximately 90%) originate from the epithelial surface, while a smaller part arises from germ or stromal cells.4 Malignant epithelial OCs can be classified into high-grade serous ovarian carcinoma (HGSC), endometrioid carcinoma, clear cell carcinoma, mucinous carcinoma, and low-grade serous ovarian carcinoma.5 HGSC is considered to be the most lethal gynecological cancer6 and is frequently characterized by p53 mutations (96% of cases), which lead to p53 loss of function and induce HGSC development.7 Unfortunately, OCs are often associated with a poor prognosis due to diagnosis at advanced stages of disease and the development of chemotherapy resistance. The standard of care is cytoreductive surgery combined with adjuvant chemotherapy, but 70% of patients experience recurrence within 2 years of primary diagnosis, and the mortality rate reaches approximately 50% of women at five years.8,9 The strategy of adjuvant chemotherapy is mainly defined for patients who respond to platinum-based treatments, while approaches for platinum-resistant patients still seem to be poorly defined.10 Indeed, for platinum-refractory disease, doxorubicin, paclitaxel, gemcitabine and topotecan are used based on toxicity, treatment cost and accessibility; however, response rates range from 10% to 15% with an overall survival of about 1 year.11 In addition, patients with platinum-sensitive OCs become platinum-resistant at advanced stages and acquire chemoresistance through various mechanisms.12 Despite new treatments (i.e., olaparib monotherapy or in combination with bevacizumab) recently approved as maintenance therapy for advanced OCs,13 we are still far from satisfactory results and novel approaches are warranted.
In recent years, some studies have shown that microRNAs (miRNAs), small non-coding RNAs of approximately 19–22 nucleotides that are involved in fine-tuning gene expression at the post-transcriptional level, play diverse roles in OC by acting as tumor suppressors or proto-oncogenes and also by controlling drug resistance mechanisms.14 Attempts to target miRNAs in cancer have been considered, with some small molecules acting as inhibitors of miRNA processing and function appearing to be more successful.15,16 Nevertheless, analysis of Dicer expression in invasive epithelial OC specimens from 111 patients has shown that low Dicer expression is significantly associated with an advanced tumor stage.17 Since Dicer is a key enzyme involved in the maturation of miRNAs,18 its reduced expression results in low levels of miRNAs, suggesting that a strategy aimed at their enhancement may have anticancer effects. In 2008, enoxacin, a fluoroquinolone antibacterial drug, was reported as a small molecule enhancer of miRNA (SMER) processing and exhibited anticancer activity characterized by a broad spectrum of action and an innovative mechanism.19–21 However, the SMER property appeared to be structure-dependent and not attributable to the whole class of quinolone molecules; in fact, within a set of approved quinolones and some synthesized enoxacin derivatives, none of the compounds showed activity except ciprofloxacin, which demonstrated to be only a very weak SMER.19 Subsequently, enoxacin was reported to be able to inhibit several cancer cell lines with a dose that inhibits the growth of cells by 50% (GI50) of 125 μM.20 Several groups have confirmed that the anticancer activity of enoxacin was dependent on the modulation of miRNA processing by restoring miRNA expression to physiological levels.21–25 Mechanistic studies have shown that enoxacin is able to enhance the transactivation response element RNA-binding protein-(TRBP)-mediated loading of pre-miRNAs on Dicer, by increasing the binding between pre-miRNAs and TRBP, thereby promoting Dicer cleavage at pre-miRNAs.19–21 The inactivity of enoxacin as an anticancer agent in TRBP knock-out (KO) cells further confirmed that its anticancer effect is dependent on TRBP.19–21
Until 2021, enoxacin remained the only molecule acting as a SMER compound. Then, Fei Wang and colleagues reported the natural product gomisin M1 as an anti-hepatocellular carcinoma agent due to its ability to modulate miRNA processing, thus extending this peculiar mode of action beyond enoxacin and renewing interest in SMER molecules.26
To the best of our knowledge, the anticancer activity of enoxacin has never been evaluated against OC cells. Therefore, we were interested to evaluate whether enoxacin (Table 1 for the chemical structure) could retain its anticancer activity against the OC cell line SKOV-3, which does not express p53 at protein and mRNA levels27 and is resistant to tumor necrosis factor and other cytotoxic drugs (i.e., diphtheria toxin, cisplatin, and adriamycin).28–30 As expected, enoxacin exhibited a GI50 value of 125 μM against the SKOV-3 cell line, thus extending its anticancer activity to OC. At this point, given the considerable number of quinolones present in our in-house library, previously synthesized as antibacterial, anti-mycobacterial, antiviral, and anticancer agents,31–40 we selected ten of them (compounds 1–10 in Fig. 1) for testing against proliferating SKOV-3 cells. The number of quinolones was restricted due to the inability to perform a high-throughput screening (HTS) campaign and the initial prudence in testing a multitude of quinolones, given the findings of Shan and colleagues that unambiguously demonstrated that the RNAi-enhancing activity of enoxacin was structure-dependent and not universal across all classes of quinolones.19 Therefore, the selection was primarily focused on 6-aminoquinolones (8 of the 10 selected), as they have been less extensively explored than 6-fluoro-quinolones. Consequently, it was deemed less probable that they were present in the library of quinolones tested by Shan et al.19 Notably, compound 10 (rufloxacin), the sole 6-fluoroquinolone in our preliminary screening, was selected due to its status as an approved drug and extensive availability in our in-house library, having been identified by some of us in previous years.38
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compd. | X | R1 | R6 | R7 | R8 | GI50 SKOV-3a (μM) | CC50 Wi-38b (μM) |
| a GI50: 50% cell growth inhibition. b CC50: 50% cytotoxicity concentration. c NT: not tested; CC50 was not determined for compounds showing GI50 values ≥25 μM. d ND: activity not dose dependent. | |||||||
| Enoxacin | N | –Et | –F |
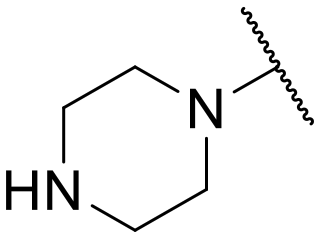
|
— | 125 | 216 ± 123.1 |
| 5 | C | –cPr | –NH2 |

|
–Me | 37.71 ± 3.38 | 38.51 ± 28.97 |
| 11 | C | –Et | –NH2 |
|
–Me | 13.14 ± 1.29 | 53.50 ± 20.64 |
| 12 | C | –cPr | –NH2 |

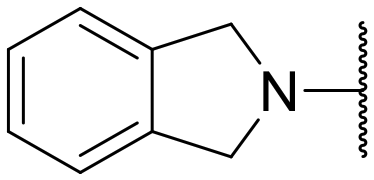
|
–Me | 17.14 ± 2.61 | 52.96 ± 14.91 |
| 13 | C | –cPr | –NH2 |
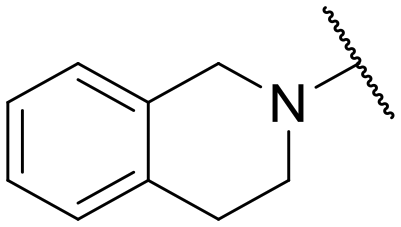
|
–Me | 28.77 ± 4.11 | NTc |
| 14 | C | –cPr | –NH2 |

|
–Me | 102.9 ± 21.58 | NT |
| 15 | C | –cPr | –NH2 |

|
–H | 13.2 ± 1.13 | 36.27 ± 8.86 |
| 16 | C | –Et | –NH2 |
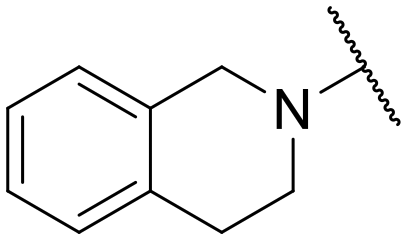
|
–H | 5.44 ± 0.13 | 8.19 ± 14.3 |
| 17 |

|
NDd | NT | ||||
| 18 | C | –cPr | –NH2 |

|
–H | 152.3 ± 3.05 | NT |
| 19 | C | –cPr | –NH2 |
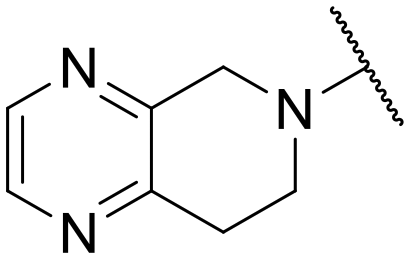
|
–H | 104.3 ± 2.03 | NT |
| 20 | C | –cPr | –NH2 |

|
–H | 65.42 ± 24.31 | NT |
| 21 | C | –cPr | –NH2 |

|
–H | 23.66 ± 1.02 | 103.4 ± 15.45 |
| 22 | C | –cPr | –NH2 |
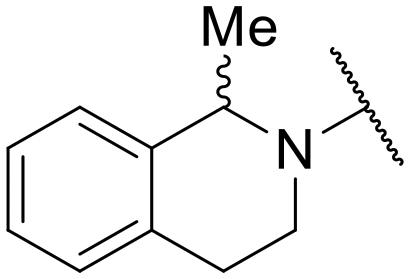
|
–H | 18.52 ± 1.71 | 59.87 ± 5.95 |
| 23 | C | –cPr | –NH2 |
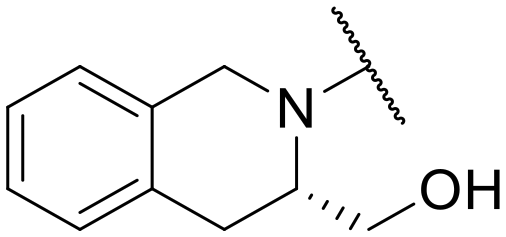
|
–H | 46.17 ± 8.38 | NT |
| 24 | C | –cPr | –NH2 |

|
–H | 9.19 ± 1.05 | 45 ± 13.37 |
| 25 | C | –cPr | –NH2 |

|
–H | 15.99 ± 2.94 | 79.74 ± 11.79 |
| 26 | C | –cPr | –NH2 |

|
–H | 33.93 ± 6.43 | NT |
| 27 | N | –Et | –F |

|
— | ND | NT |
| 28 | N | –cPr | –F |

|
— | 46.82 ± 7.44 | NT |
| 29 | N | –Et | –F |

|
— | 16.18 ± 1.03 | 25.50 ± 11.5 |
| 30 | N | –Et | –F |
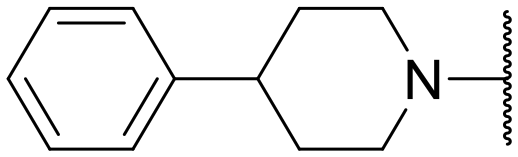
|
— | 8.34 ± 1.28 | 4.65 ± 5.96 |
| 31 | C | –Et | –F |

|
–Me | 14.89 ± 3.24 | 56.58 ± 37.36 |
| 32 | C | –cPr | –F |
|
–Me | 25 ± 6.04 | 90.73 ± 30.86 |
| 33 | C | –cPr | –F |

|
–H | 13.52 ± 1.45 | 142.9 ± 42.7 |

 | ||
| Fig. 1 Small set of selected in-house quinolones (1–10) and their antiproliferative activity against the SKOV-3 cell line expressed as GI50 values. | ||
The encouraging outcomes of this preliminary assessment prompted us to embark on a preliminary medicinal chemistry campaign around the quinolone core, with the objective of developing compounds that exhibit anticancer activity against OC cell lines and act through the modulation of miRNA maturation. The findings of this study led to the identification of novel quinolones with promising anticancer activity, which act as SMER agents by binding to TRBP.
Results and discussion
Preliminary screening and design of new quinolones
Preliminary screening of in-house quinolones (compounds 1–10 – Fig. 1) against the SKOV-3 cell line revealed the amino-quinolone 5, characterized by cyclopropyl, tetrahydroisoquinoline and methyl groups at C-1, C-7 and C-8 positions, respectively, as a promising anticancer compound with a GI50 of 38 μM (about 3-fold lower than enoxacin). Amino-quinolone derivatives 1–4, 6 and 7, which differ from 5 mainly by the different substituent at the C-7 position, were significantly less active, suggesting that non-aromatic or more polar moieties than tetrahydroisoquinoline at this position are not well-tolerated. The amino-quinolone 8 and the naphthyridone derivative 9, both sharing a thiazolyl-piperazine moiety at the C-7 position, also showed interesting GI50 values (50 μM). Finally, the approved drug 10 exhibited poor anticancer activity (GI50 = 100 μM), similar to the results obtained by Shan et al. who observed that ofloxacin (close analogue of 10) did not show any significant RNAi enhancer effect.19Since many quinolones have been reported in the literature as anticancer agents especially targeting topoisomerases, to exclude this already known mechanism, we evaluated the ability of the amino-quinolone 5 to target human topoisomerases (hTopI and hTopII). The results of cleavage assays conducted at 100 μM (Fig. S1A†) indicated that compound 5 did not trap human topoisomerases. This observation led us to conclude that the anti-OC activity of compound 5 was likely mediated by a topoisomerase-independent mechanism of action.
Encouraged by this evidence, we considered the possibility of testing additional quinolones from our in-house library. However, we reasoned that the data from the preliminary screening strongly indicated that the tetrahydroisoquinoline, as a C-7 substituent of the 6-aminoquinolone scaffold, played a pivotal role in conferring the anticancer activity observed in compound 5. Consequently, we concluded that it was not advantageous to pursue the selection of additional 6-aminoquinolones from our in-house library, as they lacked sufficient structural similarity to facilitate the investigation of a structure–activity relationship (SAR) surrounding compound 5. Conversely, we decided to use derivative 5 as a starting hit compound and started the design of a series of chemical modifications (Table 1 for chemical structures) by replacing the cyclopropyl at C-1 with an ethyl group (derivative 11) or the tetrahydroisoquinoline at the C-7 position with three different moieties (derivatives 12–14). Subsequently, removal of the C-8 methyl group of 5 and 11 led to the synthetically more accessible C-8 des-methyl analogues 15 and 16, respectively. After evaluating the role of the C-3 carboxyl function by designing the amide derivative 17, further efforts were focused on the C-7 substituent of 15, which was replaced with different groups, yielding derivatives 18–26. The resulting best C-7 substituents were then used to design close analogues of enoxacin (derivatives 27–30). Finally, since tetrahydroisoquinoline was found as the best moiety for the C-7 position, we planned the synthesis of fluoro-quinolones 31–33. Of note, compounds 32 and 33 were previously published by some of us as comparative analogues of antibacterial amino-quinolones.34 However, both compounds were no longer available in our in-house library, thus we synthesized them using optimized procedures different from those previously reported.34
Chemistry
Target compounds 11–14 were synthesized as reported in Scheme 1. Reaction of the acrylate intermediate 34 (ref. 33) with ethylamine in a mixture of Et2O and EtOH afforded derivative 35, which was cyclized in dry DMF in the presence of K2CO3 to give the quinolone intermediate 36. Subsequent hydrolysis of 36 with a mixture of 6 N HCl and EtOH afforded the quinolone acid derivative 37, which was reacted with tetrahydroisoquinoline in dry DMF in the presence of Et3N as a base to give compound 38. Under H2 flow, RANEY®/Ni catalyzed reduction in DMF afforded the target compound 11. Similarly, acid hydrolysis of quinolone ester 39 (ref. 33) afforded the corresponding acid 40,33 which was reacted with 4-phenylpiperidine or isoindoline in dry DMSO in the presence of Et3N as a base to afford C-7 substituted quinolone derivatives 41 and 42. Subsequently, nitro group reduction afforded the target compounds 12 and 13. On the other hand, starting from the ester intermediate 39 (ref. 33) and exploiting the good nucleophilic property of 2-amino-ethylpiperidine, the nitro derivative 43 was first obtained and then reduced to 44 and hydrolyzed under basic conditions to give the target compound 14. | ||
| Scheme 1 Reagents and conditions: i) ethylamine, Et2O/EtOH, rt, 15 min; ii) K2CO3, DMF, 100 °C, 1 h; iii) 6 N HCl, EtOH, reflux, 1 h; iv) amine, Et3N, dry DMF or DMSO, 80–100 °C, 1–2 h; v) RANEY®/Ni, H2, DMF, rt, 30 min–12 h; vi) 10% NaOH, MeOH, reflux, 1 h. | ||
Target compounds 15–26 were synthesized as reported in Scheme 2. Reaction of nitro-quinolone derivative 45 (ref. 31) with various amines in dry DMF or DMSO in the presence of Et3N as a base afforded C-7 substituted quinolone analogues 47–56. Subsequent hydrogenation under a catalytic amount of RANEY®/Ni of 47–50 and 52–56 afforded amino derivatives 57–65, which were then hydrolyzed to target compounds 15, 18–20 and 22–26. On the other hand, derivative 21 was obtained directly from the corresponding nitro ester derivative 51 by reaction with SnCl2 in EtOH at reflux. In parallel, the ester derivative 45 was also converted to the corresponding acid to give intermediate 66, which was reacted with SOCl2 and then treated with 7 N NH3 in MeOH and dry DMF to give the amide intermediate 67. Nucleophilic substitution of 67 with 1,2,3,4-tetrahydroisoquinoline in dry DMSO in the presence of Et3N gave the C-7 substituted intermediate 68, which was subjected to RANEY®/Ni catalyzed reduction of the nitro group to give the target compound 17. Starting from derivative 46,37 nucleophilic substitution with 1,2,3,4-tetrahydroisoquinoline in dry DMSO using Et3N as a base afforded derivative 69, which was first reduced to amino derivative 70 and then hydrolyzed under basic conditions to give target compound 16.
 | ||
| Scheme 2 Reagents and conditions: i) amine, Et3N, dry DMF or DMSO, 80–110 °C, 90 min–48 h; ii) RANEY®/Ni, H2, DMF, rt, 1–6 h; iii) 6 N HCl, EtOH, reflux, 9 h (for 15) or 10% NaOH, MeOH, reflux, 30 min–4 h; iv) SnCl2, EtOH, reflux, 2 h; v) SOCl2, reflux, 2 h; then, 7 N NH3 in MeOH, dry DMF, rt, 3 h. | ||
Target compounds 27–30 were synthesized as reported in Scheme 3. Starting from commercially available ethyl 3-(2,6-dichloro-5-fluoropyridin-3-yl)-3-oxopropanoate, naphthyridone derivatives 71 and 72 were synthesized by a one-pot procedure involving the initial reaction with DMF–DMA, a catalytic amount of AcOH and dry toluene, followed by the addition of alkylamines (ethylamine or cyclopropyl amine) and then tetrabutylammonium hydroxide. The reaction of 71 and 72 with different amines in dry DMF and in the presence of Et3N afforded the ester derivatives 73–76, which were hydrolyzed to give the target compounds 27–30.
 | ||
| Scheme 3 Reagents and conditions: i) DMF–DMA, AcOH, dry toluene, rt, 2 h; then, ethylamine or cyclopropylamine, rt, 30 min; then, 1.5 M Bu4NOH, rt, 5 min; ii) amine, Et3N, dry DMF, 90 °C, 2–3 h; iii) 6 N HCl, EtOH, reflux, 4–16 h or 10% NaOH, MeOH, reflux, 1 h (for 29). | ||
The synthesis of target compounds 31–33 is reported in Scheme 4. Although the synthetic procedure of analogue 32 has been reported previously,34 we encountered some problems related to the formation and handling of the difluoroborate complex envisaged in the published procedure;34 therefore, a different chemical synthesis was carried out to obtain both C-8 methyl derivatives 31 and 32. The new procedure involved the chlorination of commercial 2,4,5-trifluoro-3-methylbenzoic acid by oxalyl chloride, catalyzed by dry DMF, in dry CH2Cl2 to give the corresponding acyl chloride, which was immediately reacted with ethyl 3-(N,N-dimethylamino)acrylate in dry toluene to give intermediate 77. Reaction of 77 with ethylamine or cyclopropylamine in a mixture of Et2O and EtOH afforded derivatives 78 and 79, respectively. Cyclization of 78 and 79 was performed with NaH in dry DMF at 0 °C to give 6-fluoro quinolone intermediates 80 and 81,41 respectively. Since various attempts to introduce the tetrahydroisoquinoline moiety at the C-7 position of the 6-fluoro-8-methyl quinolone by nucleophilic substitution were unsatisfactory, we explored a different procedure by modifying a previously reported chemical synthesis.42 Accordingly, derivatives 80 and 81 were reacted with NaN3 in dry DMF to give derivatives 82 and 83, which were then reduced by H2 flow with a catalytic amount of Pd/C to give amino derivatives 84 and 85. Subsequently, a Sandmeyer reaction allowed the synthesis of bromo analogues 86 and 87, which were reacted with tetrahydroisoquinoline in a Buchwald–Hartwig reaction to give C-7 functionalized quinolone ester derivatives 88 and 89. Finally, acid hydrolysis afforded the target compounds 31 and 32.
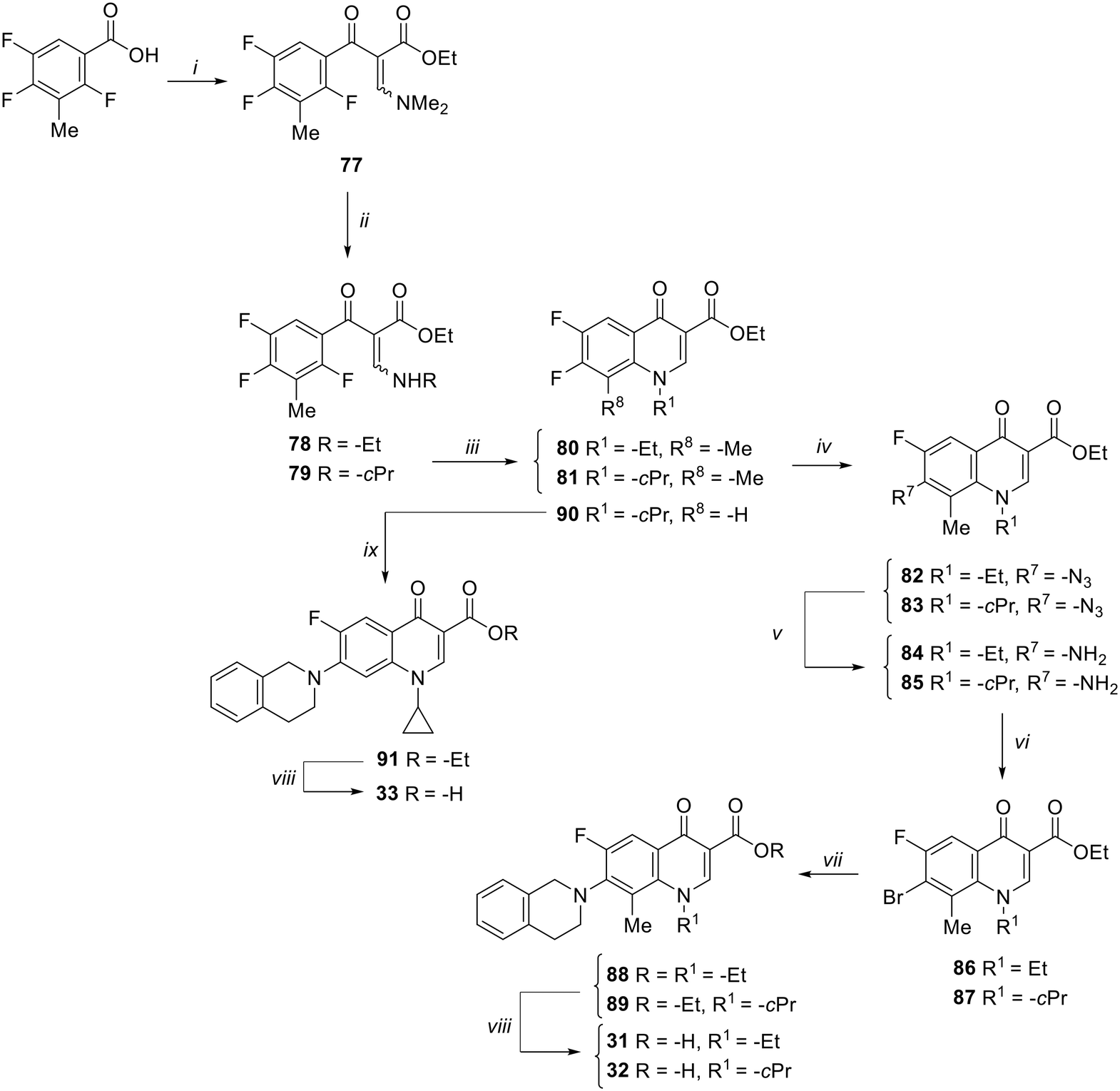 | ||
| Scheme 4 Reagents and conditions: i) C2O2Cl2, cat. dry DMF, dry CH2Cl2, rt, 4 h; then, ethyl 3-(N,N-dimethylamino)acrylate, Et3N, dry toluene, 90 °C, 4 h; ii) ethylamine or cyclopropylamine, Et2O, EtOH, rt, 1 h; iii) NaH, dry DMF, 0 °C, 3 h; iv) NaN3, dry DMF, 90 °C, 24 h; v) Pd/C, H2, DMF, rt, 3 h; vi) CuBr2, 5% HBr, 2 M NaNO2, 0 °C to rt, 30 min; vii) 1,2,3,4-tetrahydroisoquinoline, Pd2(dba)3, BINAP, Cs2CO3, dry toluene, reflux, 13 h; viii) 6 N HCl, EtOH, reflux, 1–4 h; ix) 1,2,3,4-tetrahydroisoquinoline, Et3N, dry DMSO, 100 °C, 4 h. | ||
On the other hand, the synthesis of C-8 des-methyl quinolone 33 was performed by a standard procedure starting with the nucleophilic substitution of derivative 90,43 with the 1,2,3,4-tetrahydroisoquinoline to give intermediate 91. Subsequent acid hydrolysis afforded the target compound 33.
Biological results
All the compounds were first tested for their ability to inhibit SKOV-3 proliferation at scalar concentrations, which allowed the determination of GI50 values (Table 1). In parallel, for compounds with GI50 ≤ 25 μM, CC50 values were determined in Wi-38 cells to observe any toxic effect against non-tumor cells (Table 1) and to evaluate any potential specific effect against tumor cells.Replacement of the cyclopropyl ring of 5 with an ethyl group resulted in 11 that showed an increase in antiproliferative activity (GI50 13.14 μM vs. SKOV-3) coupled with a CC50 value of 53.50 μM on Wi-38 cells, being approximately 4-fold more toxic against tumor cells. Notably, cytotoxicity experiments of the first hit compound 5, performed for comparison, showed a poor selectivity towards OC cell lines with respect to Wi-38 cells, with a CC50 value comparable to its GI50.
Similar to 11, good results were obtained when the tetrahydroisoquinoline of 5 was replaced by a phenylpiperidine moiety (compound 12) giving a GI50 of 17.14 μM against SKOV-3 and a CC50 in Wi-38 of 52.96 μM. On the other hand, when isoindoline and the more polar aminoethyl-piperidine moiety were used to replace the tetrahydroisoquinoline of 5, we obtained compounds 13 and 14, which showed a comparable or worse profile than compound 5. Removal of the C-8 methyl group of 5 led to the more potent compound 15 (37.71 μM vs. 13.2 μM), which achieved a modest selective anticancer effect. On the other hand, derivative 16, the C-8 des-methyl analogue of 11, showed an improvement in anticancer potency but also an increase in non-tumor cell toxicity compared to 11. Due to synthetic accessibility, the removal of the C-8 methyl group from the amino-quinolone scaffold led to an improvement in terms of yield and reaction time. Therefore, we decided to focus our efforts on 15 and evaluate the role of the carboxyl function by replacing it with an amide group (derivative 17), which was completely inactive. We then replaced the tetrahydroisoquinoline of 15 with more polar bicyclic heterocycles, thus obtaining compounds 18–20, which were less active, suggesting that the benzene ring of the tetrahydroisoquinoline seemed to be important to retain a potent anticancer effect, while the presence of a free NH moiety was detrimental. On the other hand, the bioisosteric replacement of the benzene ring of the tetrahydroisoquinoline of 15 with a thiophene moiety yielded the tetrahydrothienopyridine derivative 21, which retained a GI50 comparable to that of 15, while exhibiting a significantly higher CC50 value (103.4 μM vs. 36.27 μM), thus highlighting a good propensity to act preferentially on tumor cells. Modifications of the aliphatic part of the tetrahydroisoquinoline by introducing a methyl at the C-1′ position (compound 22) or a methyl alcohol at C-3′ (compound 23) did not lead to improvements in terms of anti-proliferative activity. Interestingly, good results were obtained when less rigid moieties were introduced at the quinolone C-7 position to replace the classical tetrahydroisoquinoline. Indeed, 24 and 25 showed comparable GI50 values to 15 coupled with favorable CC50s. However, when we tried to introduce a free alcoholic group to 25 in order to improve the polarity, we obtained compound 26, which showed a 2-fold increase in GI50 (33.93 μM vs. 15.99 μM). At this point, we considered introducing the best C-7 substituents identified so far for the quinolone scaffold in place of the piperazine ring of the naphthyridone nucleus of enoxacin. However, compounds 27–30 were less active than their respective quinolone analogues and in some cases very toxic when tested in Wi-38 cells (compounds 29 and 30). Ultimately, given the favorable outcomes observed with tetrahydroisoquinoline in the C-7 position when integrating the SAR data from amino-quinolone and naphthyridone derivatives, our research shifted its focus to fluoroquinolones. To this end, we synthesized three distinct molecules incorporating this moiety (compounds 31–33). Interestingly, compound 31, the close analogue of 11, showed a similar activity to 11 both in terms of anti-proliferative activity and cytotoxicity on non-tumor cells, suggesting a similar behavior for compounds possessing NH2 and F groups at the C-6 position. On the other hand, the replacement of the amino group of 5 with a fluorine yielded 32, which exhibited a GI50 of 25 μM and a CC50 of 90.73 μM, making it significantly less toxic to non-tumor cells than its amino analogue. Finally, the best results were obtained when the amino group of 15 was replaced by a fluorine to yield 33, which showed a comparable GI50 to 15 (13.52 μM), but a significant reduction in non-tumor cell toxicity (CC50 on Wi-38 = 142.9 μM). Compound 33 thus emerged as a promising fluoroquinolone worthy of further biological profiling.
Proliferation inhibitory effect of compound 33 against different OC cell lines
Based on the promising results of compound 33 regarding its anti-proliferative activity against SKOV-3 cell lines, coupled with a marked tendency to act preferentially on tumor cells, we decided to investigate whether its activity could be extended to two additional OC cell lines: OVCA433 and A2780 (Table 2).| Compd. | OC cell lines | |
|---|---|---|
| GI50 OVCA433 (μM) | GI50 A2780 (μM) | |
| 33 | 31.04 ± 6.75 | 18.13 ± 8.25 |
| Enoxacin | 117.80 ± 8.89 | 142.33 ± 8.73 |
Notably, 33 exhibited anti-proliferative activity against both OC cell lines, although it showed a slight increase in GI50 values. However, in both cases, 33 was significantly more potent than enoxacin, highlighting its potential as a “broad-spectrum” anti-OC agent.
Investigation on the mode of action of compound 33
As previously evaluated for compound 5, we assessed whether our best compound 33 acted by inhibiting or trapping hTopI and hTopII. As expected, 33 showed no inhibition or trapping activity of human topoisomerases when tested at 100 μM (Fig. S1B†), thus ruling out that the toxicity observed in SKOV-3 was due to topoisomerase inhibition/trapping.Subsequently, to evaluate whether compound 33 acted through an enoxacin-like mechanism, we performed SPR experiments on TRBP for compound 33 and the inactive amide derivative 17 as a negative control (Fig. 2 – enoxacin as a positive control). Interestingly, 33 bound TRBP with a KD value of 4.09 μM, whereas derivative 17 showed a very high KD value of 1.12 mM. It is also noteworthy that compound 33 exhibited a binding affinity approximately 40-fold stronger for TRBP than enoxacin.
 | ||
| Fig. 2 SPR analyses of enoxacin (ENX), 17 and 33 towards TRBP. | ||
A cellular thermal shift (Fig. 3A and B) further confirmed the involvement of TRBP in the mechanism of action of compound 33. In fact, treatment of TRBP with 50 μM of 33 resulted in a significant thermal shift when protein destabilization was compared with DMSO treatment (Fig. 3A). In addition, 33 was able to induce destabilization of TRBP in a concentration-dependent manner (Fig. 3B). Higher melting values are typical in thermal shift assays when a ligand binds to a protein. However, the destabilization of TRBP after treatment with compound 33 is similar to what has been observed with other TRBP binders, such as the SMER gomisin M1 (ref. 26) and the sulfonamide derivative CIB-3b, which was previously reported to inhibit miR-21 maturation.44
 | ||
| Fig. 3 Cellular thermal shift assays of TRBP in the presence of 33. A) Representative Western blot analysis showing the destabilization effect of 33 on TRBP at different temperatures. Graph showing the quantified levels of TRBP over GAPDH from three independent experiments. B) Western blot analysis showing the concentration dependency of the destabilization effect of 33 on TRBP at 55 °C. Graph showing the quantified levels of TRBP over GAPDH. Error bars represent SE. p-Value was calculated using paired two-tailed Student's t-test (* p < 0.05). | ||
To further confirm the role of TRBP in the mechanism of action of 33, we also examined its ability to promote shRNA to siRNA maturation (RNAi enhancing activity) (Fig. 4A). Notably, compound 33 at 50 μM exhibited a modest but significant RNAi enhancing effect by decreasing the amount of GAPDH mRNA, indirectly indicating an increase in siRNA maturation compared to DMSO. This effect was comparable to that of enoxacin tested at 50 μM. We also tested the ability of 33 to promote the maturation of endogenous miRNAs using a luciferase reporter construct carrying the miR-21-5p binding site downstream of Renilla cDNA (psi-R21). As shown in Fig. 4B, 33 produced significant positive effects on miR-21-5p biogenesis and this caused a decrease in Renilla luciferase activity. Notably, this effect parallels that obtained with the positive control enoxacin. The effect is specific since no significant variation was observed when using a control construct, psi-CTRL, which lacks the miR-21-5p binding site.
 | ||
| Fig. 4 Compound 33 promotes siRNA/miRNA maturation. A) RNAi enhancing effect of compound 33 and enoxacin at 50 μM. Values are shown as relative quantity with respect to sh-Scr/DMSO treated cells set to a value of 1. ATP5O is used as a reference gene. B) Left graph: Upregulation of miR-21 in HeLa cells upon 6 hours of treatment with 33 and enoxacin at 50 μM. Values are shown as relative quantity with respect to DMSO treated cells set to a value of 1. U6 is used as a reference gene. Upper: Schematic representation of the psi-R21 and psi-CTRL constructs. “miR-21 BS” depicts the binding site of the miRNA. Lower graph: miRNA enhancing effect of compound 33 and enoxacin at 50 μM. Values are shown as the ratio between Renilla (RLuc) and firefly (FLuc) luciferase signals. Error bars represent SE of at least three independent experiments. p-Values were calculated using paired two-tailed Student's t-test (* p < 0.05; ** p < 0.01; ns: not significant). | ||
To confirm that the anticancer activity of 33 was directly dependent on TRBP, we evaluated its anti-proliferative effect on HCCLM3 and SK-Hep-1 wild-type (WT) and KO cells for TRBP (enoxacin was used as a control – Fig. 5). Notably, after 48 hours, 33 at 30 μM showed no antiproliferative effect on both KO TRBP cells, while it significantly reduced (about 50%) the viability of WT HCCLM3 cells. On the other hand, the anti-proliferative effect of 33 at 30 μM on WT TRBP SK-Hep-1 cells was less evident and thus, it was also tested at 50 μM on both KO and WT cells. Of note, 33 at 50 μM significantly reduced the viability of WT SK-Hep-1 cells while not affecting KO TRBP SK-Hep-1 cells. Taken together, these data confirmed that 33 exerts its anti-proliferative activity by acting on TRBP and thus likely exhibits behavior similar to that of enoxacin. Furthermore, it can be stated that compound 33 retained anticancer activity against cancer cell lines other than those of ovarian origin, including HCCLM3 (hepatocellular carcinoma) and SK-Hep-1 (liver adenocarcinoma).
 | ||
| Fig. 5 Antiproliferative effect after 48 hours of enoxacin at 125 μM and compound 33 A) against WT and KO TRBP HCCLM3 cell lines (30 μM) and B) against WT and KO TRBP SK-Hep-1 cell lines (30 and 50 μM). T-Test results (** P ≤ 0.01, **** P ≤ 0.001). | ||
As previously stated in the introduction, this ability was observed for enoxacin as well, which retained anticancer activity against a wide panel of cancer cells with GI50 values of approximately 125 μM.20 This valuable broad-spectrum anticancer activity is to be attributed to its distinctive mechanism of action, which enables it to re-regulate dysregulated miRNAs in cancer cells. As one of the objectives of the study was to identify novel quinolones with an enoxacin-like mechanism, further investigation was conducted to ascertain whether compound 33 could exert any antiproliferative effect against three additional cancer cell lines: MCF-7 (breast cancer), A549 (lung carcinoma), and HCT-116 (colon cancer). It is noteworthy that all of these cell lines can be considered SMER-sensitive, as enoxacin has previously demonstrated efficacy against them.20 Interestingly, treatment with 33 (30 μM) for 72 hours resulted in a significant inhibitory effect on cell proliferation in all three cell lines (Fig. S2†), indicating that compound 33 possesses broad-spectrum anticancer potential.
Compound 33 induced A2780 cell death and sensitized cisplatin-resistant A2780 cells to cisplatin
We next tested whether the growth inhibition exerted by 33 on SKOV-3 and A2780 cells was accompanied by induction of cell death. Thus, we treated both cell lines with 33 at 30 μM for 72 hours and cells were counted using trypan blue solution in order to assess cell viability. Fig. 6A shows that after treatment, the total number of cells was greatly reduced for both cell lines, while only for A2780 was the percentage of dead cells increased to a great extent. This result was confirmed by the increased levels of cleaved PARP1 (c-PARP), a marker of apoptosis,45 and by the downregulation of the anti-apoptotic protein Bcl-XL46 in treated A2780 cells (Fig. 6B). No changes were observed in SKOV-3 cells, where compound 33 may exert a cytostatic effect as previously shown for cisplatin treatment.47 | ||
| Fig. 6 Compound 33 inhibited cell growth and induced apoptosis of A2780 cells and sensitized A2780 CIS to cisplatin. A) Left graph: Reduction of number of SKOV-3 and A2780 viable cells upon treatment with compound 33 at 30 μM for 72 hours. The number of viable cells is expressed as percentage with respect to DMSO condition set to a value of 100%; right graph: percentage of SKOV-3 and A2780 dead cells upon treatment described in “A”; B) representative Western blot analysis of protein extracts from SKOV-3 and A2780 cells treated as in “A”. β-Actin levels are used as a control for protein loading. C) Graph showing the number of viable cells of a culture of A2780 and A2780 CIS treated with 33 (30 μM), cisplatin (CIS, 1 μM) and a combination of the two compounds (33 + CIS). The number of viable cells is expressed as percentage with respect to DMSO condition set to a value of 100%. Error bars represent SE of at least three independent experiments. p-Values were calculated using paired two-tailed Student's t-test (* p < 0.05; ** p < 0.01). | ||
In consideration of the observed capacity of compound 33 to markedly elicit A2780 cell death, we proceeded to examine the potential synergistic effects of 33 in combination with cisplatin in view of a potential future combined treatment. Cisplatin is an effective first-line treatment for OC, but resistance is common. Although about 80% of OC patients are initially sensitive to cisplatin, many develop resistance, leading to high mortality within a few years. Various cellular processes contribute to platinum drug-resistance in OC including processes regulated by miRNAs. Indeed, it has been reported that modulation of the expression of specific miRNAs can reverse cisplatin resistance in human OC cells.48
Therefore, cisplatin-resistant A2780 cells (A2780 CIS)49 were treated with 33, cisplatin and with a combination of the two compounds for 48 hours. In parallel, the same experiments were performed using the A2780 cell line. Fig. 6C illustrates that, as expected, A2780 CIS exhibited significantly greater resistance to cisplatin compared to A2780, while both cell lines showed equal sensitivity to treatment with compound 33. Notably, the combination of cisplatin and compound 33 sensitized A2780 CIS to cisplatin and produced a more pronounced inhibitory effect on A2780 proliferation, suggesting a synergistic mechanism of action.
Conclusions
In this study, we aimed to identify new SMER compounds by exploiting our in-house library of quinolone derivatives. Preliminary evaluation of the antiproliferative activity of in-house derivatives against the OC SKOV-3 cell line led us to identify the 6-amino quinolone derivative 5 as a starting hit worthy of further exploration. Our medicinal chemistry efforts culminated in the identification of 33 as the best compound of the new series, exhibiting a GI50 of 13.52 μM against SKOV-3 cells, about 9-fold lower than the known SMER enoxacin and less toxic against non-tumor cells (CC50 on Wi-38 = 142.9 μM). Although featured by the presence of the most studied C-6 fluorine atom, like for enoxacin, compound 33 is characterized by an atypical base (tetrahydroisoquinoline moiety) at the C-7 position. Quinolone 33 demonstrated the ability to bind TRBP by both SPR and cellular thermal shift analyses, to modulate RNAi enhancing activity by promoting shRNA to siRNA maturation, and to enhance endogenous pre-miRNA processing. Furthermore, the anti-proliferative effect of 33 was also TRBP-dependent in the cell context; indeed, it lost its cytotoxic effect in TRBP KO SK-Hep-1 and HCCLM3 cells. Notably, in the A2780 cancer cell line, this anti-proliferative effect is also accompanied by the induction of the apoptotic process, as evidenced by the activation of PARP1 cleavage and the downregulation of Bcl-XL protein. In addition, compound 33 sensitized A2780 CIS to cisplatin and led to a more effective cisplatin inhibitory effect on A2780 proliferation, suggesting that, by enhancing the biogenesis of specific miRNAs, 33 may impact those cellular processes that promote proliferation and sustain cisplatin resistance. Finally, compound 33 exhibited antiproliferative activity against other cancer types, including breast, lung, and colon cancers, indicating a broader applicability of its anticancer properties.In conclusion, here, we demonstrate that enoxacin is not the sole member of the quinolone class to exert anticancer activity through the modulation of the miRNA/RNAi machinery (SMER activity). Furthermore, the novel quinolone derivative 33 exhibited enhanced potency and a more robust binding affinity for TRBP compared to enoxacin. These data provide further support for the innovative concept of targeting miRNA maturation by small molecules as a potential OC therapy. We are aware that compound 33 is not yet a viable preclinical candidate and further studies will be conducted to identify more potent and cancer-specific agents capable of interacting with TRBP. However, this represents the first study of medicinal chemistry to identify quinolone agents that act as antitumor agents through an innovative mechanism of action.
Materials and methods
Chemistry
Unless otherwise stated, all starting materials were commercially available. Reagents and solvents were purchased from common commercial suppliers and used as received. Organic solutions were dried over anhydrous Na2SO4 and concentrated under reduced pressure using a Büchi rotary evaporator. All reactions were routinely checked by TLC on silica gel 60F254 (Merck) and visualized by UV and iodine. Purifications were performed by flash column chromatography separations on Merck silica gel 60 (mesh 230–400) or BUCHI Reveleris-X2 Flash Chromatography. Yields are based on purified products and are not optimized. 1H NMR and 13C NMR spectra were recorded at 200 or 400 and 100 MHz, respectively, using Bruker Advance DRX-200 and DRX-400 instruments. Chemical shifts (δ) are reported in ppm relative to TMS and calibrated using residual undeuterated solvent as an internal reference. Coupling constants (J) are expressed in Hz. Spectra were acquired at 298 K. Data processing was performed using Bruker TopSpin 4.3.0 software, and the spectral data are consistent with the assigned structures. The purity (>95%) of the compounds was evaluated by HPLC analysis using an Agilent 1290 Infinity System instrument equipped with a DAD detector from 190 to 640 nm. Purity was determined at 254 nm using a Phenomenex AERIS Widepore C4, 4.6 mm, 100 mm (6.6 lm) at a flow rate of 0.85 mL min−1; acquisition time: 10 min; gradient: acetonitrile in water containing 0.1% formic acid (0–100% in 10 min); oven temperature, 30 °C. Peak retention time is given in minutes. HRMS detection was based on electrospray ionization (ESI) in positive polarity, using an Agilent 1290 Infinity System equipped with an Agilent 6540UHD Accurate Mass Q-TOF MS detector.:1, was obtained as a brown solid in 30% yield (0.30 g). 1H NMR (400 MHz, DMSO-d6): δ 0.83–0.86 (m, 2H, cyclopropyl-CH2), 1.15–1.20 (m, 2H, cyclopropyl-CH2), 1.80–1.85 (m, 4H, piperidine-CH2 ×2), 2.71–2.74 (m, 4H, piperidine-CH and CH3), 3.11–3.15 (m, 2H, piperidine-NCH2), 3.20–3.26 (m, 2H, piperidine-NCH2), 4.30–4.35 (m, 1H, CH), 7.17–7.20 (m, 1H, H-4′), 7.27–7.35 (m, 4H, Ar–H), 8.42 (s, 1H, H-2), 8.85 (s, 1H, H-5), 14.41 (bs, 1H, CO2H).
:2, was obtained as a brown solid in 20% yield (0.08 g). 1H NMR (400 MHz, DMSO-d6): δ 0.77–0.86 (m, 2H, cyclopropyl-CH2), 1.12–1.17 (m, 2H, cyclopropyl-CH2), 1.73–1.79 (m, 2H, piperidine-CH2), 1.87–1.96 (m, 2H, piperidine-CH2), 2.56 (s, 3H, CH3), 2.67 (t, J = 11.4 Hz, 1H, piperidine-CH), 3.10–3.17 (m, 2H, piperidine-NCH2), 3.22–3.37 (m, 2H, piperidine-NCH2), 4.25 (s, 1H, CH), 5.40 (s, 2H, NH2), 7.17 (t, J = 7.1 Hz, 1H, H-4′), 7.28 (t, J = 7.4 Hz, 2H, H-3′ and H-5′), 7.31–7.39 (m, 3H, H-5, H-2′ and H-6′), 8.59 (s, 1H, H-2), 15.68 (s, 1H, CO2H). 13C NMR (100 MHz, DMSO-d6): δ 10.76, 19.24, 34.46, 41.02, 42.48, 49.98, 104.08, 106.43, 124.39, 125.41, 126.44, 127.39, 128.67, 135.70, 144.41, 145.17, 146.93, 148.98, 166.91, 176.96. HPLC: rt 10.398 min. HRMS (ESI) calculated for C25H27N3O3 [M + H]+ 418.2130, found 418.21288.
:2 to 95:5, was obtained as a brown solid in 14% yield (0.05 g). 1H NMR (400 MHz, DMSO-d6): δ 0.85–0.91 (m, 2H, cyclopropyl-CH2), 1.13–1.19 (m, 2H, cyclopropyl-CH2), 2.53 (s, 3H, CH3), 4.18–4.29 (m, 1H, CH), 4.57 (s, 4H, isoindoline-CH2 ×2), 5.60 (s, 2H, NH2), 7.25–7.28 (m, 2H, Ar–H), 7.31–7.35 (m, 2H, Ar–H), 7.40 (s, 1H, H-5), 8.59 (s, 1H, H-2), 15.60 (s, 1H, CO2H). 13C NMR (100 MHz, DMSO-d6): δ 10.92, 18.12, 41.30, 55.70, 104.49, 106.14, 123.03, 125.70, 127.17, 128.82, 134.82, 139.60, 140.55, 146.57, 149.02, 166.87, 177.23. HPLC: rt 5.385 min. HRMS (ESI) calculated for C22H21N3O3 [M + H]+ 376.1661, found 376.16651.
:5, was obtained as an orange solid in 43% yield (0.08 g). 1H NMR (200 MHz, CDCl3): δ 0.85–0.88 (m, 2H, cyclopropyl-CH2), 1.08–1.13 (m, 2H, cyclopropyl-CH2), 1.36–1.59 (m, 9H, piperidine-CH2 ×3 and OCH2CH3), 2.38–2.41 (m, 6H, piperidine-NCH2 ×2 and NHCH2–), 2.66 (s, 3H, CH3), 3.21 (t, J = 5.0 Hz, 2H, –CH2N), 3.91–3.97 (m, 3H, CH and NH2), 4.35 (q, J = 7.1 Hz, 2H, OCH2CH3), 7.60 (s, 1H, H-5), 8.58 (s, 1H, H-2).
:3, was obtained as a light brown solid in 61% yield (0.57 g). 1H NMR (200 MHz, DMSO-d6): δ 0.99–1.15 (m, 2H, cyclopropyl-CH2), 1.23–1.27 (m, 5H, cyclopropyl-CH2, and OCH2CH3), 3.00 (t, J = 5.1 Hz, 2H, 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine-CH2), 3.60–3.65 (m, 3H, CH and 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine-CH2), 4.16 (q, J = 7.1 Hz, 2H, OCH2CH3), 4.47 (s, 2H, 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine-CH2), 7.57 (s, 1H, H-8), 8.40 (s, 1H, H-2), 8.51 (s, 1H, H-5), 8.65 (s, 1H, Ar–H), 8.94 (s, 1H, Ar–H).
:10, was obtained as a light brown solid in 78% yield (0.90 g). 1H NMR (200 MHz, DMSO-d6): δ 1.00–1.30 (m, 7H, cyclopropyl-CH2 ×2 and OCH2CH3), 3.00–3.10 (m, 2H, tetrahydropyrido[3,4-b]pyrazine-CH2), 3.48–3.70 (m, 3H, tetrahydropyrido[3,4-b]pyrazine-CH2 and CH), 4.20 (q, J = 7.0 Hz, 2H, OCH2CH3), 4.50 (s, 2H, tetrahydropyrido[3,4-b]pyrazine-CH2), 7.60 (s, 1H, H-8), 8.28–8.55 (m, 4H, H-2, H-5 and Ar–H).
:2 to 95:5, was obtained as a light brown solid in 34% yield (0.37 g). 1H NMR (400 MHz, DMSO-d6): δ 1.06–1.10 (m, 4H, cyclopropyl-CH2 ×2), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3), 3.30–3.33 (m, 2H, tetrahydroquinoxaline-CH2), 3.53–3.61 (m, 3H, tetrahydroquinoxaline-CH2 and cyclopropyl-CH), 4.17 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.04 (s, 1H, NH), 6.31 (t, J = 7.8 Hz, 1H, Ar–H), 6.55–6.60 (m, 2H, Ar–H), 6.68 (t, J = 7.5 Hz, 1H, Ar–H), 7.82 (s, 1H, H-8), 8.42 (s, 1H, H-2), 8.52 (s, 1H, H-5).
:1 to 98:2, was obtained as a yellow solid in 69% yield (0.48 g). 1H NMR (400 MHz, DMSO-d6): δ 1.05–1.09 (m, 2H, cyclopropyl-CH2), 1.19–1.28 (m, 5H, cyclopropyl-CH2 and OCH2CH3), 2.71–2.73 (m, 1H, tetrahydroisoquinoline-CH), 3.16–3.21 (m, 1H, ½ CH2OH), 3.42–3.46 (m, 2H, ½ CH2OH and cyclopropyl-CH), 3.59–3.63 (m, 2H, tetrahydroisoquinoline-CH2), 4.18 (q, J = 7.1 Hz, 2H, OCH2CH3), 4.31 (d, J = 16.2 Hz, 1H, tetrahydroisoquinoline-CH2 ×½), 4.62 (d, J = 16.1 Hz, 1H, tetrahydroisoquinoline-CH2 ×½), 4.75 (t, J = 5.2 Hz, 1H, OH), 7.18–7.23 (m, 4H, Ar–H), 7.58 (s, 1H, H-8), 8.42 (s, 1H, H-2), 8.49 (s, 1H, H-5).
:6, was obtained as a whitish solid in 40% yield (0.12 g). 1H NMR (200 MHz, DMSO-d6): δ 0.90–1.05 (m, 2H, cyclopropyl-CH2), 1.07–1.27 (m, 5H, cyclopropyl-CH2 and OCH2CH3), 3.00–3.15 (m, 2H, tetrahydropyrido[3,4-b]pyrazine-CH2), 3.39 (t, J = 5.3 Hz, 2H, tetrahydropyrido[3,4-b]pyrazine-CH2), 3.45–3.55 (m, 1H, CH), 4.12 (q, J = 7.1 Hz, 2H, OCH2CH3), 4.29 (s, 2H, tetrahydropyrido[3,4-b]pyrazine-CH2), 5.23 (s, 2H, NH2), 7.45 (s, 1H, H-8), 7.49 (s, 1H, H-5), 8.25 (s, 1H, H-2), 8.45 (d, J = 2.5 Hz, 1H, Ar–H), 8.49 (d, J = 2.5 Hz, 1H, Ar–H).
:1, was obtained as a whitish solid in 32% yield (0.06 g). 1H NMR (400 MHz, DMSO-d6): δ 0.94–1.06 (m, 4H, cyclopropyl-CH2 ×2), 1.22 (t, J = 7.0 Hz, 3H, OCH2CH3), 3.38–3.41 (m, 2H, dihydroquinoxaline-CH2), 3.47–3.50 (m, 3H, dihydroquinoxaline-CH2 and cyclopropyl-CH), 4.15 (q, J = 6.9 Hz, 2H, OCH2CH3), 5.17 (s, 2H, NH2), 5.74 (s, 1H, NH), 6.09 (d, J = 7.8 Hz, 1H, Ar–H), 6.29 (t, J = 5.5 Hz, 1H, Ar–H), 6.50–6.55 (m, 2H, Ar–H), 7.49 (s, 1H, H-5), 7.55 (s, 1H, H-8), 8.28 (s, 1H, H-2).
:7, was obtained as a pale yellow solid in 80% yield (0.14 g). 1H NMR (400 MHz, DMSO-d6): δ 1.00–1.30 (m, 4H, cyclopropyl-CH2 ×2), 2.98–3.18 (m, 2H, tetrahydropyrido[4,3-d]pyrimidine-CH2), 3.40–3.50 (m, 2H, tetrahydropyrido[4,3-d]pyrimidine-CH2), 3.70–3.78 (m, 1H, CH), 4.29 (s, 2H, tetrahydropyrido[4,3-d]pyrimidine-CH2), 5.50 (s, 2H, NH2), 7.50 (s, 1H, H-8), 7.62 (s, 1H, H-5), 8.50 (s, 1H, H-2), 8.60 (s, 1H, Ar–H), 9.00 (s, 1H, Ar–H), 15.00 (s, 1H, CO2H). 13C NMR (100 MHz, DMSO-d6): δ 7.84, 31.71, 35.99, 47.31, 49.10, 106.54, 107.36, 108.21, 122.61, 128.70, 134.26, 142.63, 145.18, 145.20, 155.08, 156.95, 163.63, 167.05, 176.92. HPLC rt: 3.5000 min. HRMS-ESI m/z [M + H]+ calcd. for C20H19N5O3, 378.1566, found: 378.1560.
:1, compound 21 was obtained as a yellow solid in 21% yield. 1H NMR (400 MHz, DMSO-d6): δ 1.03–1.09 (m, 2H, cyclopropyl-CH2), 1.12–1.18 (m, 2H, cyclopropyl-CH2), 2.91–2.97 (m, 2H, dihydrothienopyridine CH2), 3.39 (t, J = 5.4 Hz, 2H, dihydrothienopyridine CH2), 3.61–3.75 (m, 1H, CH), 4.23 (s, 2H, dihydrothienopyridine CH2), 5.45 (s, 2H, NH2), 6.92 (d, J = 5.1 Hz, 1H, Ar–H), 7.34 (d, J = 5.1 Hz, 1H, Ar–H), 7.52 (s, 1H, H-5), 7.60 (s, 1H, H-8), 8.46 (s, 1H, H-2), 15.86 (s, 1H, CO2H). 13C NMR (100 MHz, DMSO-d6): δ 7.71, 25.06, 35.91, 47.72, 50.09, 106.17, 106.98, 107.97, 121.98, 123.74, 125.76, 133.48, 133.62, 134.21, 142.58, 145.00, 145.36, 167.16, 176.66. HPLC: rt 4.900 min. HRMS (ESI) calculated for C20H19N3O3S [M + H]+ 382.1225, found 382.12217.
:3 to 95:5, was obtained as a yellow solid in 25% yield (0.07 g). 1H NMR (400 MHz, DMSO-d6): δ 0.91–1.01 (m, 2H, cyclopropyl-CH2), 1.12–1.16 (m, 2H, cyclopropyl-CH2), 2.95 (t, J = 5.5 Hz, 2H, tetrahydroisoquinoline-CH2), 3.33 (t, J = 5.7 Hz, 2H, tetrahydroisoquinoline-CH2), 3.52–3.62 (m, 1H, CH), 4.24 (s, 2H, tetrahydroisoquinoline-CH2), 5.20 (s, 2H, NH2), 7.13–7.17 (m, 4H, Ar–H), 7.30 (d, J = 4.8 Hz, 1H, CONH2 ×½), 7.51–7.54 (m, 2H, H-5 and H-8), 8.46 (s, 1H, H-2), 9.43 (d, J = 4.9 Hz, 1H, CONH2 ×½). 13C NMR (100 MHz, DMSO-d6): δ 7.67, 28.63, 34.97, 47.97, 52.66, 107.33, 108.23, 109.72, 124.07, 126.14, 126.70, 126.77, 129.12, 133.68, 134.52, 134.83, 141.36, 144.39, 144.81, 166.56, 174.96. HPLC: rt 4.557 min. HRMS (ESI) calculated for C22H22N4O2 [M + Na]+ 397.16407, found 397.16376.
:40), 2 M EtNH2 in THF (1.1 mL, 2.2 mmol) was added and the reaction mixture was stirred for additional 30 min at rt. Then, 3 mL of aq. 10% citric acid were added, and the two phases were separated; the organic phase was washed with water and aq. 1.5 M BuN4OH (1.5 mL, 2.2 mmol) was added. The reaction mixture was stirred for 5 min until the formation of a precipitate, then it was concentrated under vacuum to half volume and neutralized by 2 N HCl and the precipitate was filtered under vacuum to afford compound 71 as a white solid in 63% yield. 1H NMR (200 MHz, DMSO-d6): δ 1.19–1.38 (m, 6H, OCH2CH3, and NCH2CH3), 4.22 (q, J = 7.1 Hz, 2H, OCH2CH3), 4.40 (q, J = 7.1 Hz, 2H, NCH2CH3), 8.43 (d, J = 7.9 Hz, 1H, H-5), 8.85 (s, 1H, H-2).
:5, was obtained in 53% yield (0.72 g). 1H NMR (200 MHz, DMSO-d6): δ 0.93–1.06 (m, 2H, cyclopropyl-CH2), 1.09–1.16 (m, 2H, cyclopropyl-CH2), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 3.61–3.64 (m, 1H, CH), 4.20 (q, J = 7.1 Hz, 2H, OCH2CH3), 8.39 (d, J = 7.8 Hz, 1H, H-5), 8.53 (s, 1H, H-2).
:1) (300 mL) was hydrogenated over a catalytic amount of Pd/C (10% w/w) at rt for 3 h. Then, the reaction mixture was filtered over celite and the filtrate was evaporated to give, after purification by flash column chromatography eluting CHCl3/MeOH 95:5, compound 84 as a white solid in 53% yield. 1H NMR (200 MHz, DMSO-d6): δ 1.05–1.30 (m, 6H, OCH2CH3 and NCH2CH3), 2.25 (s, 3H, CH3), 4.10–4.40 (m, 4H, OCH2CH3, and NCH2CH3), 6.00 (s, 2H, NH2), 7.61 (d, J = 11.6 Hz, 1H, H-5), 8.50 (s, 1H, H-2).
:1, was obtained as an orange solid in 63% yield (0.60 g). 1H NMR (200 MHz, DMSO-d6): δ 0.75–0.80 (m, 2H, cyclopropyl-CH2), 1.00–1.30 (m, 5H, cyclopropyl-CH2 and OCH2CH3), 2.70 (s, 3H, CH3), 2.95 (t, J = 5.8 Hz, 2H, tetrahydroisoquinoline-CH2), 3.50 (t, J = 5.8 Hz, 2H, tetrahydroisoquinoline-CH2), 4.05–4.26 (m, 3H, CH and OCH2CH3), 4.30 (s, 2H, tetrahydroisoquinoline-CH2), 7.05–7.25 (m, 4H, Ar–H), 7.70 (d, J = 11.2 Hz, 1H, H-5), 8.60 (s, 1H, H-2).
Cell handling and treatment
SKOV-3, A2780 and A2780 CIS were cultured and maintained, as previously described,50 at 37 °C in RPMI 1640 medium (Euroclone) supplemented with 10% fetal bovine serum (FBS; Euroclone), L-glutamine and penicillin/streptomycin (SIGMA-ALDRICH). OVCA433, HEK-293T, HeLa, HCCLM3, SK-Hep-1, MCF-7, A549 and HCT-116 cells were maintained in DMEM medium (Euroclone) containing 10% FBS (Euroclone), L-glutamine and penicillin/streptomycin (SIGMA-ALDRICH). Wi-38 cells were cultured in DMEM medium (Euroclone) supplemented with 10% FBS (Euroclone), L-glutamine and penicillin/streptomycin (SIGMA-ALDRICH), and 1% non-essential amino acids (NEAA; Euroclone). The cultures were maintained in a constant temperature incubator at 37 °C with 5% CO2.For the evaluation of GI50 and CC50, cells were counted and plated in equal numbers, amounting to 100000 cells in 35 mm culture dish. Once cell adhesion had been promoted, the compounds were added to the culture medium. After 72 hours of treatment, the cells were counted, and the data analyzed by linear regression. Each experiment was carried out three times in triplicate and three separate cell counts were made for each sample.
HEK-293T_shSCR and HEK-293T_shGAPDH were treated with 50 μM of enoxacin and 33 for 48 hours before RNA extraction and expression analysis.
HeLa cells were transfected with 10 ng of psi-R21 or psi-CTRL using Lipofectamine™ 2000 (Thermo Fisher Scientific) and after 48 hours were treated with either enoxacin or 33 at 20 μM and 50 μM. After 6 hours Renilla luciferase (RLuc) and firefly luciferase (FLuc) activities were measured using the Dual-Luciferase® Reporter Assay System (Promega), following the manufacturer's instructions, and GloMax® Discover Microplate Reader (Promega).
HCCLM3 and SK-Hep-1 cells were seeded into 96-well plates at a density of 10000 cells per well and allowed to adhere overnight. Subsequently, the cells were treated with the following substances: 0.1% DMSO as the negative control, various concentrations of the test compounds, and 125 μM enoxacin as the positive control. After 48 h of incubation, the original culture medium was removed, and the cells were washed twice with PBS. Then, 100 μL of DMEM culture medium containing 10% CCK-8 (Bimake, USA) was added to each well. The plates were incubated for 30 minutes at 37 °C. To determine the fluorescence intensity, measurements were taken at a wavelength of 450 nm using a Varioskan from Thermo Fisher Scientific.
A2780 and SKOV-3 cells were treated with 50 μM of 33 for 72 h before RNA and protein extraction and expression analyses. A2780 and A2780 CIS were treated with 33 (30 μM) and/or cisplatin (1 μM, Sigma-Aldrich) for 48 hours while MCF-7, A549 and HCT-116 were treated with 33 (30 μM) for 72 hours. Total cell number and viability have been assessed using Trypan Blue Solution (Thermo Fisher Scientific) and Countess™ 3 Automated Cell Counter (Thermo Fisher Scientific).
Equation to evaluate GI50 and CC50
The formula suggested by GraphPad titled ‘Absolute IC50, X is the concentration’ was used to assess GI50 in SKOV-3, OVCA433 and A2780 cell lines and CC50 in Wi-38 cell lines.In detail, the formula is as follows:
| Fifty = (Top + Baseline)/2 |
| Y = Bottom + (Top − Bottom)/(1 + ((Top − Bottom)/(Fifty − Bottom) − 1) × (Absolute IC50/X)^HillSlope) |
| Considering: X = Compound concentration; Y = number of cells |
The confidence intervals (CI) of the parameters were calculated with a 95% confidence level. The goodness-of-fit was quantified by means of R squared and sum-of-squares.
Surface plasmon resonance (SPR) assay
Following the manufacturer's instructions, the BIAcore Sensor Chip (CM5) underwent initial activation using a mixture of NHS and EDC. Subsequently, recombinant human TRBP proteins at a concentration of 20 μg mL−1 (in 10 mM sodium acetate, pH 4.0) were injected over the CM5 chip. After reaching a coupling level of 12000 RU, a 1 M ethanolamine hydrochloride was injected to block the chip, preventing nonspecific binding. All the compounds were diluted in HBS-EP buffer (10 mM HEPES, pH 7.4, 3 mM EDTA, 150 mM NaCl, and 0.005% surfactant P-20) with 1% DMSO. Samples were injected at a rate of 10 μL min−1 for 120 seconds, followed by a dissociation phase of 90 seconds. After each cycle, a 50% DMSO solution was used to regenerate the chip surface and restore the sensorgram baseline. Binding analysis was performed using BIA evaluation version 3.2 software, determining the dissociation rate constant (KD).
Plasmid construction
The binding site for miR-21 was cloned into psiCHECK™-2 Vector (Promega), downstream the Renilla cDNA, by inverted PCR using the following oligonucleotides: 21MRE_FW CTGATAAGCTA AACCTAGAGCGGCCGCTGGC; 21MRE_REV ACTGATGTTGA GGCTCGAGCGATCGCCTAGAA. The resulting construct was named psi-R21.Lentivirus packaging and viral transduction
Lentiviral particles were produced by calcium phosphate transient transfection of HEK-293T cells with specific lentiviral plasmids (shGAPDH: TRCN0000445616 and shSCR: TRCN0000015937 from SIGMA-ALDRICH) together with the packaging plasmids (pLP1and pLP2) and the envelope plasmid pLP/VSVG. For each lentiviral plasmid one 150 mm plate of HEK293T cells was used for transfection. After 14–16 hours the medium was replaced with complete medium supplemented with 1 mM sodium butyrate (SIGMA-ALDRICH). The collection of medium containing lentiviral particles occurs 48 h and 72 h after transfection; complete medium supplemented with sodium butyrate was added after the first collection. Collected media were pulled and centrifuged at 1000 rpm for 5 min at room temperature. The supernatant was filtered through 0.45 μm pore nitrocellulose filters and then ultracentrifuged at 20000 rpm for 2 hours at 4 °C. The pellet containing lentiviral particles was then resuspended in 25 μL HBSS buffer (GIBCO ThermoFisher) and stored at −80 °C.
1 × 106 of HEK-293T cells were resuspended in 1 ml of serum-free and antibiotic-free medium supplemented with polybrene at 4 μg ml−1 and infected with 5 μL of the lentiviral particles in HBSS buffer. After 6 h one volume of medium with FBS 2% and penicillin/streptomycin 2× was added and after additional 24 h the medium was replaced with complete medium. 48 h after viral transduction, HEK-293T_shSCR and HEK-293T_shGAPDH transduced cells were selected using 3 μg mL−1 of puromycin (SIGMA-ALDRICH).
RNA extraction and analysis
Total RNA was extracted using the Direct-zol RNA MiniPrep kit (Zymo Research) with on-column DNase treatment, according to the manufacturer's instructions. Reverse transcription was carried out with SuperScript VILO cDNA Synthesis Kit (Life Technologies) and the cDNA samples were analyzed by qRT-PCR using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). For miR-21 expression analysis total RNA was retrotranscribed using miRCURY® LNA® RT Kit (Qiagen) and the cDNA was analyzed by using miRCURY® LNA® miRNA PCR Assay (Qiagen). The oligonucleotides used are the following: GAPDH_FW ACCCACTCCTCCACCTTTGA; GAPDH_REV TCCACCACCCTGTTGCTGTA; ATP5O_FW ACTCGGGTTTGACCTACAGC; ATP5O_REV GGTACTGAAGCATCGCACCT; hsa-miR-21-5p miRCURY LNA miRNA PCR Assay (YP00204230); U6 snRNA (v2) miRCURY LNA miRNA PCR Assay (YP02119464).Cell thermal shift assay
HEK-293T cells were harvested in PBS supplemented with protease inhibitor cocktail (complete, EDTA-free, Roche). Cell suspensions were freeze-thawed three times in liquid nitrogen and then centrifuged (20000g) for 10 min at 4 °C. The lysates were diluted at concentration of 2 μg μL−1 and then treated with DMSO and 50 μM of 33 or with increasing concentration of 33 for 30 min at 37 °C. After this incubation the lysates were divided into equal parts (50 μL) and heated individually at designated temperatures for 3 min. The heated samples were cooled at room temperature for 3 min and centrifuged (20000g) for 10 min at 4 °C. The supernatants were transferred to new tubes and analyzed by use for western blotting.
Western blotting
Lysates were loaded on 4–12% bis–tris-acrylamide gel (Life Technologies) and transferred to a nitrocellulose membrane. The membrane was blocked in 5% milk and hybridized with α-TRBP (15753-1-AP Proteintech), α-GAPDH (sc-25778, Santa Cruz Biotechnology), α-c-PARP (Bioss Antibodies, BSM-52408R), α-BCLXL (mAb #2764, Cell Signaling Technology) and α-β-actin (A3854, Sigma) antibodies. All the images were captured using the ChemiDocTM Touch Imaging System (Bio-Rad), and the densitometric analyses were performed using the associated Image Lab software (Bio-Rad).Detection of hTOP1 and hTOP2α cleavage complexes (TOP1ccs and TOP2ccs) in vivo
After compound treatments, 1 × 106 human cells in 35 mm dish per sample were washed with 1× phosphate-buffered saline (PBS) and lysed with 600 μL of DNAzol (Invitrogen), followed by precipitation with 300 μL of 200 proof ethanol. The nucleic acids were collected, washed with 75% ethanol, resuspended in 200 μL of TE buffer, and then heated at 65 °C for 15 min, followed by shearing with sonication (40% output for 10 s pulse and 10 s rest for four times). The samples were centrifuged at 15000 rpm for 5 min at 4 °C, and the supernatant were collected. 1 μL of each sample was removed for spectrophotometric measurement of absorbance at 260 nm to quantitate DNA content (NanoDrop). 2 μg of each sample was subjected to slot-blot for immunoblotting using anti-hTOP1 antibody (BD Pharmingen, 556597) and anti-hTOP2α antibody (Santa Cruz biotechnology, 365916).51
Abbreviation list
| GI50 | 50% growth inhibition |
| HGSC | High-grade serous ovarian carcinoma |
| KO | Knock out |
| OC | Ovarian cancer |
| SMER | Small-molecule enhancer of miRNA |
| SPR | Surface plasmon resonance |
| Tops | Topoisomerases |
| TRBP | TAR RNA-binding protein |
| WT | Wild type |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: T. F., N. D. I., M. M., G. S., G. M.; validation: T. F., N. D. I., M. A. D. F., S. P., J. Y., Y. S.; formal analysis and investigation: T. F., N. D. I., D. P.; S. P., M. P., J. Y., Y. S., S. M., M. L. B., S. S., O. T., V. C., M. M.; resources: M. M., G. S., G. M.; writing – original draft: T. F., N. D. I.; writing – review & editing: T. F., N. D. I., M. A. D. F., V. C., F. W., Y. P., M. M., G. S., G. M.; supervision: V. C., F. W., Y. P., M. M., G. S., G. M.; project administration: M. M., G. S., G. M.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We would like to thank Loretta Tuosto (Sapienza University of Rome) for providing A2780 and A2780 CIS cell lines and Marcella Marchioni (IBPM-CNR, Rome) for technical support. T. F. was supported by a FIRC-AIRC fellowship (2019–2020) for Italy and PON “Ricerca e innovazione” 2014–2020, Azione IV.4 (tematiche dell'innovazione) cod. J91B2100320006. M. M. was supported by DELPHI project – Departments of Excellence 2018–2022 (Italian Ministry of Universities and Research – MUR).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef.
- C. J. Cabasag, P. J. Fagan, J. Ferlay, J. Vignat, M. Laversanne, L. Liu, M. A. van der Aa, F. Bray and I. Soerjomataram, Int. J. Cancer, 2022, 151, 1535–1541 CrossRef CAS.
- T. Arora, S. Mullangi and M. R. Lekkala, in Ovarian Cancer, StatPearls Publishing, Treasure Island (FL), 2023 Search PubMed.
- B. M. Reid, J. B. Permuth and T. A. Sellers, Cancer Biol. Med., 2017, 14, 9 CAS.
- J. Prat, E. D'Angelo and I. Espinosa, Hum. Pathol., 2018, 80, 11–27 CrossRef CAS.
- F. Dao, B. A. Schlappe, J. Tseng, J. Lester, A. M. Nick, S. K. Lutgendorf, S. Mcmeekin, R. L. Coleman, K. N. Moore, B. Y. Karlan, A. K. Sood and D. A. Levine, Gynecol. Oncol., 2016, 141, 260–263 CrossRef PubMed.
- R. C. Bast, B. Hennessy and G. B. Mills, Nat. Rev. Cancer, 2009, 9, 415–428 CrossRef CAS.
- M. K. B. Parmar, J. A. Ledermann, N. Colombo, A. du Bois, J.-F. Delaloye, G. B. Kristensen, S. Wheeler, A. M. Swart, W. Qian, V. Torri, I. Floriani, G. Jayson, A. Lamont, C. Tropè and ICON and AGO Collaborators, Lancet, 2003, 361, 2099–2106 CrossRef CAS.
- C. Marchetti, F. De Felice, A. Romito, V. Iacobelli, C. M. Sassu, G. Corrado, C. Ricci, G. Scambia and A. Fagotti, Semin. Cancer Biol., 2021, 77, 144–166 CrossRef CAS PubMed.
- M. Markman, R. Rothman, T. Hakes, B. Reichman, W. Hoskins, S. Rubin, W. Jones, L. Almadrones and J. L. Lewis, J. Clin. Oncol., 1991, 9, 389–393 CrossRef CAS PubMed.
- D. Luvero, A. Milani and J. A. Ledermann, Ther. Adv. Med. Oncol., 2014, 6, 229 CrossRef CAS.
- A. M. Patch, E. L. Christie, D. Etemadmoghadam, D. W. Garsed, J. George, S. Fereday, K. Nones, P. Cowin, K. Alsop, P. J. Bailey, K. S. Kassahn, F. Newell, M. C. J. Quinn, S. Kazakoff, K. Quek, C. Wilhelm-Benartzi, E. Curry, H. S. Leong, A. Hamilton, L. Mileshkin, G. Au-Yeung, C. Kennedy, J. Hung, Y. E. Chiew, P. Harnett, M. Friedlander, M. Quinn, J. Pyman, S. Cordner, P. O'Brien, J. Leditschke, G. Young, K. Strachan, P. Waring, W. Azar, C. Mitchell, N. Traficante, J. Hendley, H. Thorne, M. Shackleton, D. K. Miller, G. M. Arnau, R. W. Tothill, T. P. Holloway, T. Semple, I. Harliwong, C. Nourse, E. Nourbakhsh, S. Manning, S. Idrisoglu, T. J. C. Bruxner, A. N. Christ, B. Poudel, O. Holmes, M. Anderson, C. Leonard, A. Lonie, N. Hall, S. Wood, D. F. Taylor, Q. Xu, J. Lynn Fink, N. Waddell, R. Drapkin, E. Stronach, H. Gabra, R. Brown, A. Jewell, S. H. Nagaraj, E. Markham, P. J. Wilson, J. Ellul, O. McNally, M. A. Doyle, R. Vedururu, C. Stewart, E. Lengyel, J. V. Pearson, N. Waddell, A. Defazio, S. M. Grimmond and D. D. L. Bowtell, Nature, 2015, 521, 489–494 CrossRef CAS PubMed.
- S. Arora, S. Balasubramaniam, H. Zhang, T. Berman, P. Narayan, D. Suzman, E. Bloomquist, S. Tang, Y. Gong, R. Sridhara, F. R. Turcu, D. Chatterjee, B. Saritas-Yildirim, S. Ghosh, R. Philip, A. Pathak, J. J. Gao, L. Amiri-Kordestani, R. Pazdur and J. A. Beaver, Onco Targets Ther, 2021, 26, e164–e172 CAS.
- S. N. Chen, R. Chang, L. Te Lin, C. U. Chern, H. W. Tsai, Z. H. Wen, Y. H. Li, C. J. Li and K. H. Tsui, Int. J. Environ. Res. Public Health, 2019, 16, 1510 CAS.
- K. Watashi, M. L. Yeung, M. F. Starost, R. S. Hosmane and K. T. Jeang, J. Biol. Chem., 2010, 285, 24707–24716 CAS.
- P. D. C. Monroig, L. Chen, S. Zhang and G. A. Calin, Adv. Drug Delivery Rev., 2015, 81, 104–116 CrossRef CAS PubMed.
- W. M. Merritt, Y. G. Lin, L. Y. Han, A. A. Kamat, W. A. Spannuth, R. Schmandt, D. Urbauer, L. A. Pennacchio, J.-F. Cheng, A. M. Nick, M. T. Deavers, A. Mourad-Zeidan, H. Wang, P. Mueller, M. E. Lenburg, J. W. Gray, S. Mok, M. J. Birrer, G. Lopez-Berestein, R. L. Coleman, M. Bar-Eli, A. K. Sood and N. Engl, J. Med., 2008, 359, 2641–2650 CAS.
- C. A. Vergani-Junior, G. Tonon-da-Silva, M. D. Inan and M. A. Mori, Biophys. Rev., 2021, 13, 1081 Search PubMed.
- G. Shan, Y. Li, J. Zhang, W. Li, K. E. Szulwach, R. Duan, M. A. Faghihi, A. M. Khalil, L. Lu, Z. Paroo, A. W. S. Chan, Z. Shi, Q. Liu, C. Wahlestedt, C. He and P. Jin, Nat. Biotechnol., 2008, 26, 933–940 CAS.
- S. Melo, A. Villanueva, C. Moutinho, V. Davalos, R. Spizzo, C. Ivan, S. Rossi, F. Setien, O. Casanovas, L. Simo-Riudalbas, J. Carmona, J. Carrere, A. Vidal, A. Aytes, S. Puertas, S. Ropero, R. Kalluri, C. M. Croce, G. A. Calin and M. Esteller, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4394–4399 CAS.
- T. Felicetti, V. Cecchetti and G. Manfroni, J. Med. Chem., 2020, 63, 12275–12289 CAS.
- E. J. Sousa, I. Graça, T. Baptista, F. Q. Vieira, C. Palmeira, R. Henrique and C. Jerónimo, Epigenetics, 2013, 8, 548–558 CAS.
- G. Valianatos, B. Valcikova, K. Growkova, A. Verlande, J. Mlcochova, L. Radova, M. Stetkova, M. Vyhnakova, O. Slaby and S. Uldrijan, PLoS One, 2017, 12, e0185801 CrossRef.
- T. C. Vracar, J. Zuo, J. S. Park, D. Azer, C. Mikhael, S. A. Holliday, D. Holsey, G. Han, L. VonMoss, J. K. Neubert, W. J. Rody, E. K. L. Chan and L. S. Holliday, Sci. Rep., 2018, 8, 16182 CrossRef.
- J. Ramírez-Moya, L. Wert-Lamas, G. Riesco-Eizaguirre and P. Santisteban, Oncogene, 2019, 38, 5486–5499 CrossRef.
- Z. Zhou, Y. Li, X. Ma, B. Cao, T. Peng, Y. Sheng, H. Peng, R. Li, Y. Cao, R. Xi, F. Li, M. Wang, H. Sun, G. Zhang, H. Zhang, K. Hu, W. Xiao and F. Wang, J. Med. Chem., 2021, 64, 7404–7421 CrossRef CAS.
- Y. Yaginuma and H. Westphal, Cancer Res., 1992, 52, 4196–4199 CAS.
- SK-OV-3: Human Ovarian Cancer Cell Line (ATCC HTB-77)|Memorial Sloan Kettering Cancer Center, https://www.mskcc.org/research-advantage/support/technology/tangible-material/human-ovarian-cell-line-sk-ov-3, (accessed 28 March 2023).
- L. da C. Braga, N. G. Gonçales, R. de S. Furtado, W. P. de Andrade, L. M. Silva and A. L. da S. Filho, Clinics, 2020, 75, e1492 CrossRef PubMed.
- P. Tudrej, M. Olbryt, E. Zembala-Nożyńska, K. A. Kujawa, A. J. Cortez, A. Fiszer-Kierzkowska, W. Pigłowski, B. Nikiel, M. Głowala-Kosińska, A. Bartkowska-Chrobok, A. Smagur, W. Fidyk and K. M. Lisowska, Int. J. Mol. Sci., 2018, 19, 2080 CrossRef PubMed.
- V. Cecchetti, S. Clementi, G. Cruciarti, A. Fravolini, P. G. Pagella, A. Savino and O. Tabarrini, J. Med. Chem., 1995, 38, 973–982 CrossRef CAS.
- V. Cecchetti, C. Parolin, S. Moro, T. Pecere, E. Filipponi, A. Calistri, O. Tabarrini, B. Gatto, M. Palumbo, A. Fravolini and G. Palu', J. Med. Chem., 2000, 43, 3799–3802 CrossRef CAS PubMed.
- V. Cecchetti, A. Fravolini, M. Palumbo, C. Sissi, O. Tabarrini, P. Terni and T. Xin, J. Med. Chem., 1996, 39, 4952–4957 CrossRef CAS PubMed.
- V. Cecchetti, A. Fravolini, M. C. Lorenzini, O. Tabarrini, P. Terni and T. Xin, J. Med. Chem., 1996, 39, 436–445 CrossRef CAS PubMed.
- V. Cecchetti, E. Filipponi, A. Fravolini, O. Tabarrini, D. Bonelli, M. Clementi, G. Cruciani and S. Clementi, J. Med. Chem., 1997, 40, 1698–1706 CrossRef CAS.
- O. Tabarrini, S. Sabatini, S. Massari, M. Pieroni, S. G. Franzblau and V. Cecchetti, Chem. Biol. Drug Des., 2012, 80, 781–786 CrossRef CAS PubMed.
- O. Tabarrini, M. Stevens, V. Cecchetti, S. Sabatini, M. Dell'Uomo, G. Manfroni, M. Palumbo, C. Pannecouque, E. De Clercq and A. Fravolini, J. Med. Chem., 2004, 47, 5567–5578 CrossRef CAS.
- V. Cecchetti, A. Fravolini, R. Fringuelli, G. Mascellani, P. Pagella, M. Palmioli, G. Segre and P. Terni, J. Med. Chem., 1997, 30, 465–473 CrossRef.
- S. Massari, D. Daelemans, M. L. Barreca, A. Knezevich, S. Sabatini, V. Cecchetti, A. Marcello, C. Pannecouque and O. Tabarrini, J. Med. Chem., 2010, 53, 641–648 CrossRef CAS.
- G. Franci, G. Manfroni, R. Cannalire, T. Felicetti, O. Tabarrini, A. Salvato, M. L. Barreca, L. Altucci and V. Cecchetti, Cell Proliferation, 2015, 48, 705–717 CrossRef CAS.
- H. Miyamoto, H. Ueda, T. Otsuka, S. Aki, K. Nakagawa, H. Tamaoka and M. Tominaga, Chem. Pharm. Bull., 1990, 38, 2472–2475 CrossRef CAS.
- S. W. Elmore, C. S. Cooper, C. C. Schultz, D. K. Hutchinson, P. L. Donner, B. E. Green, D. D. Anderson, Q. XieJurgen, D. Linda and M. Lynch, Quinoline and naphthyridine carboxylic acid antibacterials, US2002049223A1, 2002.
- F. Dubar, G. Anquetin, B. Pradines, D. Dive, J. Khalife and C. Biot, J. Med. Chem., 2009, 52, 7954–7957 CrossRef CAS.
- T. Peng, Y. He, T. Wang, J. Yu, X. Ma, Z. Zhou, Y. Sheng, L. Li, H. Peng, S. Li, J. Zou, Y. Yuan, Y. Zhao, H. Shi, F. Li, W. Liu, K. Hu, X. Lu, G. Zhang and F. Wang, J. Med. Chem., 2022, 65, 11010–11033 Search PubMed.
- S. H. Kaufmann, S. Desnoyers, Y. Ottaviano, N. E. Davidson and G. G. Poirier, Cancer Res., 1993, 53, 3976–3985 Search PubMed.
- M. Li, D. Wang, J. He, L. Chen and H. Li, Pharmacol. Res., 2020, 151, 104547 CrossRef PubMed.
- M. E. Pisanu, A. Ricci, L. Paris, E. Surrentino, L. Liliac, M. Bagnoli, S. Canevari, D. Mezzanzanica, F. Podo, E. Iorio and R. Canese, Br. J. Cancer, 2013, 110, 625–635 Search PubMed.
- M. Moghbeli, J. Ovarian Res., 2021, 14, 1–16 Search PubMed.
- B. C. Behrens, T. C. Hamilton, H. Masuda, K. R. Grotzinger, J. Whang-Peng, K. G. Louie, T. Knutsen, W. M. McKoy, R. C. Young and R. F. Ozols, Cancer Res., 1987, 47, 414–418 Search PubMed.
- M. Castelli, S. Pieroni, C. Brunacci, D. Piobbico, D. Bartoli, M. M. Bellet, E. Colombo, P. G. Pelicci, M. A. Della Fazia and G. Servillo, Oncogene, 2013, 32, 3350–3358 Search PubMed.
- Y. Sun, J. Visualized Exp., 2023, 194, e65315 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Figure of TOP1 and TOP2α cleavage assay of compounds 5 and 33. Figures of 1H NMR and 13C NMR spectra of compounds 11–33. See DOI: https://doi.org/10.1039/d4md00649f |
| ‡ Authors contributed equally to this work. |
| § Current address: Department of Biology and Biotechnologies “C. Darwin”, Sapienza University of Rome, P.le Aldo Moro 5, 00185, Rome, Italy. |
| This journal is © The Royal Society of Chemistry 2025 |