
High-yield electrochemical hydrogen peroxide production from an enhanced two-electron oxygen reduction pathway by mesoporous nitrogen-doped carbon and manganese hybrid electrocatalysts†
Ayeong
Byeon
ab,
Jinwon
Cho
ai,
Jong Min
Kim
c,
Keun Hwa
Chae
 d,
Hee-Young
Park
a,
Seok Won
Hong
e,
Hyung Chul
Ham
f,
Seung Woo
Lee
g,
Ki Ro
Yoon
*h and
Jin Young
Kim
*a
d,
Hee-Young
Park
a,
Seok Won
Hong
e,
Hyung Chul
Ham
f,
Seung Woo
Lee
g,
Ki Ro
Yoon
*h and
Jin Young
Kim
*a
aCenter for Hydrogen and Fuel Cell Research, Korea Institute of Science and Technology (KIST), Hwarangno 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: jinykim@kist.re.kr
bAdvanced Materials R&D, LG Chemical Research Park, 188 Munji-ro, Yuseong-gu, Daejeon 34122, Republic of Korea
cMaterials Architecturing Research Center, Korea Institute of Science and Technology, Hwarangno 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea
dAdvanced Analysis Center, KIST, Seoul, 02792, Republic of Korea
eWater Cycle Research Center, KIST, Seoul 02792, Republic of Korea
fChemical Engineering, Inha University, Incheon, 22212, Republic of Korea
gG. W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
hTechnical Textile and Materials R&D Group, Korea Institute of Industrial Technology (KITECH), 143, Hanggaul-ro, Sangnok-gu, Ansan-si, Gyeonggi-do 15588, Republic of Korea. E-mail: kryoon@kitech.re.kr
iSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332-0245, USA
First published on 5th February 2020
Abstract
Electrochemical hydrogen peroxide (H2O2) production by the direct two-electron (2e−) oxygen reduction reaction (ORR) has received much attention as a promising alternative to the industrially developed anthraquinone fabrication process. Transition metal (M) and nitrogen doped carbon (M–N–C, M = Fe or Co) catalysts are known to be active for four electron ORR pathways via two + two electron transfer, where the former is for the ORR and the latter for the peroxide reduction reaction (PRR). Here, we report mesoporous N-doped carbon/manganese hybrid electrocatalysts composed of MnO and Mn–Nx coupled with N-doped carbons (Mn–O/N@NCs), which have led to the development of electrocatalysis towards the 2e− ORR route. Based on the structural and electrochemical characterization, the number of transferred electrons during the ORR on the Mn–O/N@NCs was found to be close to the theoretical value of the 2e− process, indicating their high activity toward H2O2. The favored ORR process arose due to the increased number of Mn–Nx sites within the mesoporous N-doped carbon materials. Furthermore, there was a strong indication that the PRR is significantly suppressed by adjacent MnO species, demonstrating its highly selective production of H2O2 (>80%) from the oxygen electrochemical process. The results of a real fuel cell device test demonstrated that an Mn–O/N@NC catalyst sustains a very stable current, and we attributed its outstanding activity to a combination of site-dependent facilitation of 2e− transfer and a favorable porosity for mass transport.
New conceptsThis work demonstrates a selective and efficient non-precious electrocatalyst composed of MnO and Mn–Nx co-doped carbon nanostructures (Mn–O/N@NCs), to form hydrogen peroxide from oxygen via electrochemical oxygen reduction reaction. For the catalyst optimization, we sought to achieve the efficient two electron oxygen reduction process while blocking the subsequent peroxide reduction reaction. Combined study of structural and electrochemical characterization provide strong evidence that co-doping of MnO and Mn–Nx into porous carbon nanostructures are the most active and stable sites for peroxide production, providing new insight into potential optimization for these non-precious electrocatalysts for effective peroxide production. Furthermore, this work highlights the prospective opportunities for the practical application with the first demonstration for the peroxide formation in a real fuel cell device. |
Introduction
Hydrogen peroxide (H2O2) is a valuable chemical of rapidly growing demand for a wide range of applications in industry, medicine, and environmental purification, and it also exhibits potential as an energy carrier. Although the sequential hydrogenation/oxidation of an anthraquinone has been widely used for the production of H2O2, this is an indirect and energy-intensive process with organic byproducts that cannot be easily recycled, and so is suitable only for the large scale production of H2O2.1 Furthermore, the transportation and storage costs of concentrated H2O2, and the safety issues arising from its instability, mean that a clean, sustainable, and cost-effective process to generate H2O2 on-site and on a reduced scale are required.Recently, the direct synthesis of H2O2 has been studied using an oxygen electrochemical process in both fuel cell and water electrolysis modes.2,3 In fuel cell systems, the electrochemical reduction of oxygen gas, i.e., the so-called oxygen reduction reaction (ORR), occurs spontaneously to produce electrical energy. A water oxidation reaction generating H2O2 is also theoretically possible, but little research has been carried out in this area due to the difficulty of catalyzing this reaction.4 Furthermore, considering the safety issues associated with possible explosions, the physical separation of H2 and O2 gases using a polymeric electrolyte membrane in a fuel cell configuration is highly favorable compared to water electrolysis systems.
Currently, Pt deposited on carbon supports is broadly adopted as a catalyst for the ORR in fuel cells due to the high electrical energy generation efficiency achieved via the four-electron (4e−) pathway (Eqn 1). However, the final product of this pathway is H2O, and so it is unsuitable for H2O2 production through the two-electron (2e−) pathway expressed in eqn (2).
| O2 + 4H+ + 4e− → H2O, E0 = 1.23 V | (1) |
| O2 + 2H+ + 2e− → H2O2, E0 = 0.70 V | (2) |
Recently, studies into the development of Pt-based electrocatalysts have focused on the effect of surface poisoning with less active metals that generate H2O2via electrocatalysis, whereby the ORR active sites are incorporated into an inactive species such as Au, Hg, or S to eliminate adjacent active metal sites.5–7 The partial coverage of Pt particles could therefore modulate the adsorption of O2 (the so called ‘end-on’ configuration), thereby facilitating the 2e− ORR by preventing the further electrochemical reduction of H2O2, and in turn resulting in a considerably enhanced selectivity towards H2O2 production.8 In addition, the alloying Pd with a transition metals, such as Hg and Au, has also been reported to show a significant increase in H2O2 production by tailoring of the adsorption energy for reaction intermediates such as O2* and OOH*.6,9–12 However, the use of precious metals as sacrificial electrodes remains undesirable in the context of wide implementation.
Numerous efforts to develop nonprecious catalysts with high activities and stabilities have been carried out to date. For example, heteroatom-doped carbon-based materials offer an alternative as ORR cathode catalysts in fuel cells.13–15 In particular, transition metals (Ms; or their nitrides, oxides, and carbides) and nitrogen (N)-doped carbon hybrid catalysts (i.e., M–N–C, where M = Fe or Co) have been studied as promising ORR catalysts, exhibiting high durabilities and selectivities towards the 4e− ORR following the 2e− + 2e− pathway in acidic media.16–21 In addition, Choi et al. reported that indirect ORR pathways involving an H2O2 intermediary (O2 → H2O2 → H2O) through the 2e− + 2e− mechanism may occur at different sites of Fe–N–C catalysts, i.e., one site for the reduction of O2 to H2O2 and another site for the peroxide reduction reaction (PRR; eqn (3)).22 To establish viable M–N–C catalysts that exhibit highly selective H2O2 formation, the suppression of H2O2 reduction by either the 2e− ORR or the PRR is also pivotal. However, due to the complexity of multiple ORR pathways and the heterogeneity of active sites on M–N–C catalysts, the catalytic functions and synergistic behavior of M–N–C composites, including bulk metal (or their oxides), N-doped carbon materials, and M–Nx moieties, are elusive and not yet clearly understood.
| H2O2 + 2H+ + 2e− → 2H2O, E° = 1.76 V | (3) |
Thus, we herein report mesoporous carbon spheres doped with MnO nanoparticles and Mn–Nx moieties (Mn–O/N@NCs) and their application in the 2e− ORR. The hybrid structures are synthesized via the silica-assisted polymerization of aniline and a metal impregnation process, whereby the effects of the specific ratios of the precursors and the annealing temperature are examined in terms of formation of the unique hybrid structure. The structural configurations of the obtained product are then investigated to identify the optimal composition and to promote the 2e− ORR while suppressing the PRR. The synergistic effects of the hierarchical porosity, the Mn–Nx ratio, the MnO active sites, and the hybridization with N-doped carbon spheres are also examined for the formation of Mn–O/N@NCs hybrids exhibiting efficient and stable two-electron oxygen electrochemical performances.
Results and discussions
Initially, we employed density functional theory (DFT) calculations to identify promising candidates for the M–N–C catalysts (Fig. S1a, ESI†) and to enhance the electrochemical synthesis of H2O2. As described in the ESI,† the electronic structures and binding energies of the intermediate species in the 2e− ORR pathways (e.g., O2* and OOH*) on the M–N–C catalysts are modulated when Fe, Mn, and Cr, as representative M candidates, are incorporated into the N-doped carbon composites. As a result, changes in the binding energy in the H2O2 synthetic pathways result in all stages being exothermic, although an endothermic reaction occurs during the final stage (i.e., OOH + H* → H2O2*) in the cases of the Fe–N–C and Cr–N–C catalysts (Fig. S1b, ESI†). To demonstrate the catalytic activities of the given M–N–C catalysts, we further investigated the electronic structures of Mn, Fe, and Cr. More specifically, the lowest filling of the d-band near the Fermi level and the d-band center were observed in Mn (Fig. S1c and d, ESI†), which are known to be a descriptor for H2O2 production.23–25 Upon the optimization of key reaction parameters, the most promising conversion routes for producing H2O2 were observed in the case of Mn–N–C, and so an experimental study was carried out for electrochemical estimation and optimization of the parameters for H2O2 production.The synthetic procedures employed to obtain the Mn–N–C model catalysts are illustrated schematically in Fig. 1a. More specifically, for preparation of the mesoporous NCs as starting materials, the polymerization of aniline with APS was initially carried out at 2 °C for 24 h in the presence of various-sized (i.e., 5, 12, and 20 nm) silica nanoparticles. Subsequently, PANI/silica composite spheres were carbonized in an Ar-filled tube furnace at 800 °C, and the silica nanoparticles were leached out by storing them in 1 M NaOH for 24 h, followed by washing with deionized (DI) water several times. The resulting highly porous NCs with various pore sizes were then mixed with various concentrations of (CH3COO)2Mn·4H2O using relative mass ratios of 5–70 against the NCs. Finally, the mixtures were heated at 800 °C under an Ar atmosphere, and the resulting materials were designated as Mn–O/N@NCs, which consist of mesoporous NCs and several inorganic Mn moieties and particles (Mn–Nx, MnO).
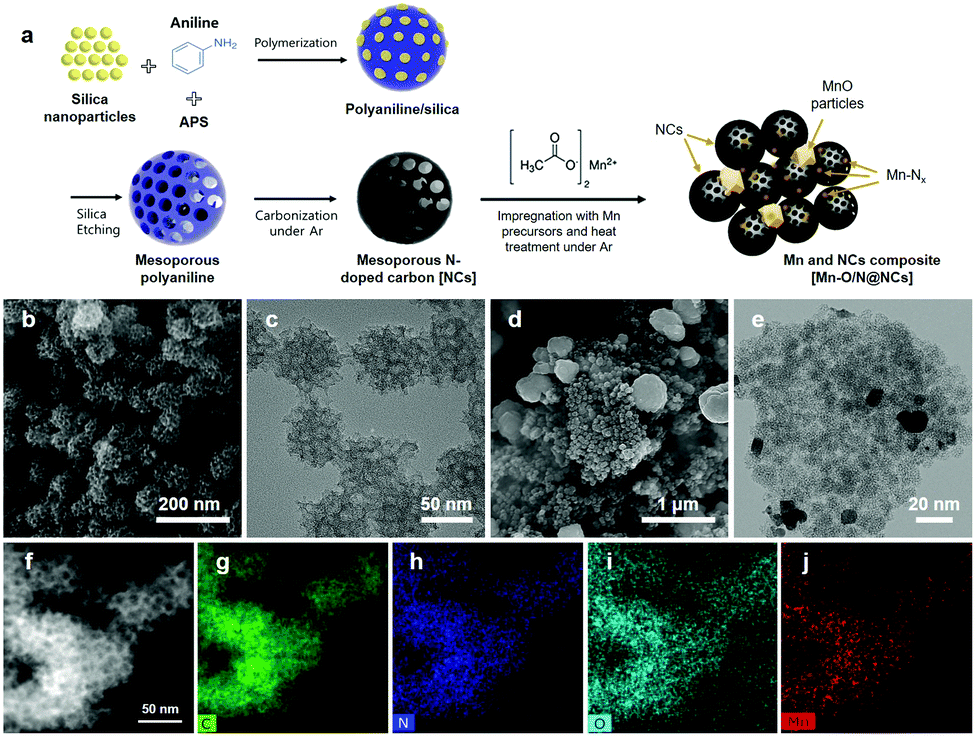 | ||
| Fig. 1 (a) Schematic illustration of the preparation of polyaniline-derived N-doped carbon spheres (NCs) and Mn–O/N@NC catalysts. (b) Scanning electron microscopy (SEM) and (c) transmission electron microscopy (TEM) images of the NCs. (d) SEM and (e) TEM images of the Mn–O/N@NC catalyst. (f) Scanning transmission electron microscopy (STEM) image of the Mn–O/N@NC catalyst and corresponding elemental mapping images for (g) C, (h) N, (i) O, and (j) Mn. | ||
The microstructural morphologies of the NCs and Mn–O/N@NCs were then investigated by SEM and TEM. As shown in Fig. 1b, highly porous carbon spheres of uniform size (∼60 nm) were clearly observed. The TEM images further confirmed the presence of micropores in the NCs, which may facilitate mass transfer during catalytic reactions.26,27 The pore size could be tuned using different-sized of template-silica particles (Fig. S2, ESI†), and according to the Barrett–Joyner–Halenda (BJH) method, the average pore diameter (DBJH) was closely affected by the sizes of the silica particles, which were 5.15, 12.93, and 16.07 nm in the cases where the Mn–O/N@NCs containing silica nanoparticles measuring 5, 12, and 20 nm, respectively (Table 1). The corresponding Brunauer–Emmett–Teller (BET) surface areas were 71.58, 43.65, and 32.33 m2 g−1, and the pore volumes were 0.09, 0.14, and 0.12 cm3 g−1, respectively. It is noted that the presence of mesopores (2–50 nm) in carbon materials is known to be more beneficial for the production of H2O2 than micropores (<2 nm), since the H2O2 generated at the active sites may be stored in micropores for longer; this facilitates further electrochemical reduction (eqn (3)) or chemical disproportionation (H2O2 → H2O + 1/2O2).28 We therefore carried out preliminary electrochemical tests to optimize the pore sizes of the NCs, as shown in Fig. S3 (ESI†), and 12 nm-sized silica-driven Mn–O/N@NCs were selected as a candidate for further characterization and electrochemical analysis.
| Sample (size of silica nanoparticles) | D BJH (nm) | S BET (m2 g−1) | V t (cm3 g−1) |
|---|---|---|---|
| Mn–O/N@NCs-50 (5 nm) | 5.15 | 71.58 | 0.09 |
| Mn–O/N@NCs-50 (12 nm) | 12.93 | 43.65 | 0.14 |
| Mn–O/N@NCs-50 (20 nm) | 16.07 | 32.33 | 0.12 |
For the prepared Mn–O/N@NCs, several spherical and smooth particles with sizes of ∼200 nm were placed in the mesoporous NC clusters (Fig. 1d and e), which we considered as derivatives of the Mn species. We further investigated the atomic distribution of mesoporous carbon species in the Mn–O/N@NCs using an elemental mapping process. As shown in Fig. 1f–i, nitrogen and oxygen dopants were spread over the carbon supports. More importantly, Mn species were also uniformly detected in the overall carbon area (Fig. 1j), indicating that Mn atomically doped the NCs during the impregnation and post-treatment steps.
The compositions and morphologies of the Mn–O/N@NCs were controlled by varying the mass ratios of the Mn precursors from 5 to 70 with respect to the NCs (designated as Mn–O/N@NCs-5, Mn–O/N@NCs-20, Mn–O/N@NCs-50, and Mn–O/N@NCs-70); the resulting SEM images are shown in Fig. 2a–d. It was found that upon increasing the amount of Mn species, the inorganic particles became more aggregated, and almost no NCs were detected in the case of Mn–O/N@NCs-70 according to our SEM observations (Fig. 2d). The TEM images of NCs, Mn–O/N@NCs-5, Mn–O/N@NCs-20, and Mn–O/N@NCs-50 are shown in Fig. S4 (ESI†).
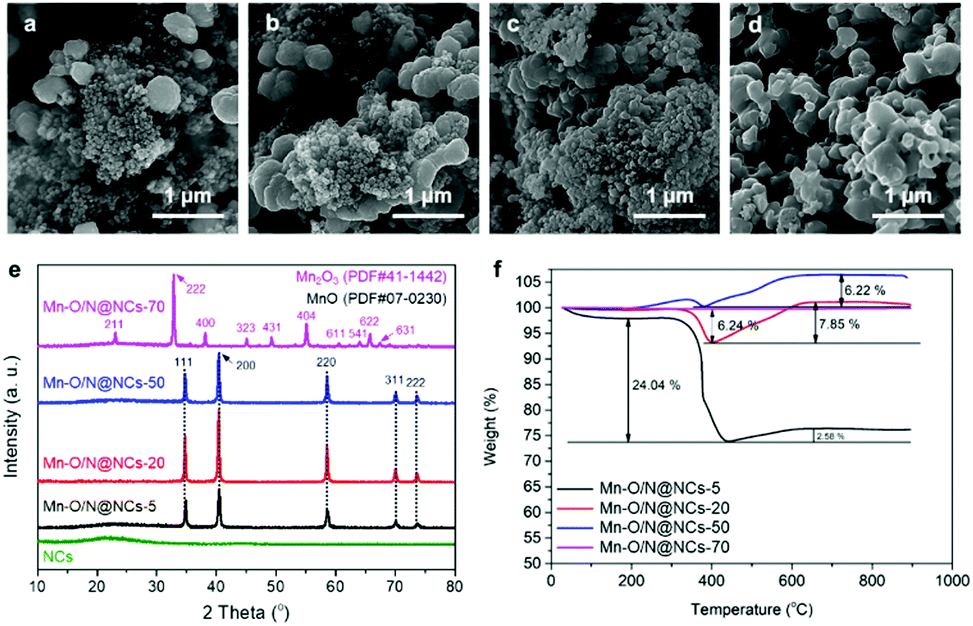 | ||
| Fig. 2 SEM images of (a) Mn–O/N@NCs-5, (b) Mn–O/N@NCs-20, (c) Mn–O/N@NCs-50, and (d) Mn–O/N@NCs-70. (e) X-ray diffraction (XRD) and (f) thermogravimetric analysis (TGA) spectra of the same samples. | ||
Fig. 2e shows the XRD patterns of the NCs, Mn–O/N@NCs-5, Mn–O/N@NCs-20, Mn–O/N@NCs-50, and Mn–O/N@NCs-70. More specifically, the XRD spectra of the NCs show a broad peak at 20°, which corresponds to amorphous carbon, without specific crystalline-related peaks being observed.29 Although the synthetic procedure employed was similar for all Mn–O/N@NCs samples, (with the exception of the amount of manganese precursor), different crystalline structures were obtained. The majority of peaks in Mn–O/N@NCs-5, Mn–O/N@NCs-20, and Mn–O/N@NCs-50 were attributed to MnO (JCPDS PDF#07-0230), while those of Mn–O/N@NCs-70 corresponded to Mn2O3 (JCPDS PDF#41-1442).30 We therefore speculated that the higher oxygen contents of the Mn precursors ((CH3COO)2Mn·4H2O) in Mn–O/N@NCs-70 could lead to further oxidation of MnO into Mn2O3, rather than CO2.
Thermogravimetric analysis (TGA) of the Mn–O/N@NCs-x samples was then carried out in an air atmosphere to investigate this further. In general, carbon decomposition can occur up to a certain heating temperature (<400 °C). The carbon contents determined by TGA were 24.04 and 6.24% for Mn–O/N@NCs-5 and Mn–O/N@NCs-20, respectively, and negligible contents were detected for Mn–O/N@NCs-50 and Mn–O/N@NCs-70 (Fig. 2f). Interestingly, the weights of Mn–O/N@NCs-5 (2.58%), Mn–O/N@NCs-20 (7.85%), and Mn–O/N@NCs-50 (6.22%) increased at high temperature (>400 °C). This phenomenon was attributed to the further oxidation of MnO to form Mn3O4 or Mn2O3 in Mn–O/N@NCs-5, Mn–O/N@NCs-20, and Mn–O/N@NCs-50, in turn due to the presence of sufficient oxygen species in the ambient air.7,31
The surface chemical compositions of the prepared samples were then investigated by X-ray photoelectron spectroscopy (XPS) and the deconvolution results for the Mn 2p, O 1s, and N 1s spectra are summarized in Fig. S5 and Table S1 (ESI†). The main valence state of Mn 2p was Mn2+ for Mn–O/N@NCs-5 (63.92 at%), Mn–O/N@NCs-20 (75.57 at%), and Mn–O/N@NCs-50 (73.85 at%), while it was Mn3+ (71.49 at%) for Mn–O/N@NCs-70. This is in good agreement with the XRD results. In addition, the increased oxidation state of manganese was evident from the deconvoluted XPS O 1s peaks, whereby the amount of Mn–O bonding was significantly higher in Mn–O/N@NCs-20 (0.77 at%), Mn–O/N@NCs-50 (0.79 at%), and Mn–O/N@NCs-70 (0.78 at%) than in Mn–O/N@NCs-5 (0.48 at%). For N 1s, pristine NCs have only pyridinic and pyrrolic N, whereas the formation of Mn–Nx was found in Mn–O/N@NCs. Interestingly, the ratio of Mn–Nx gradually increased from 4.37 at% (Mn–O/N@NCs-5) to 27.80 at% (Mn–O/N@NCs-70) with decreasing pyridinic N from 31.55 at% (Mn–O/N@NCs-5) to 4.83 at% (Mn–O/N@NCs-70). The result indicates that the added Mn atoms are readily incorporated into the pyridinic N sites of Mn–O/N@NCs. Furthermore, the graphitic N appeared after the addition of Mn components, implying that doping contributes to induce the graphitization in the Mn–O/N@NCs.
We then sought to assess the electrochemical half-cell measurements using the rotating-ring disk electrode (RRDE) technique to identify the optimal composition of the Mn–O/N@NCs for H2O2 production, i.e., to obtain the most efficient catalyst. Thus, Fig. 3a shows the disk current densities of the NCs, Mn–O/N@NCs-5, Mn–O/N@NCs-20, Mn–O/N@NCs-50, and Mn–O/N@NCs-70, using an O2-purged 0.1 M HClO4 solution and a three-electrode cell system. A decreased current density was observed after introducing the Mn species, and the onset potentials for the ORR shifted negatively from 0.78 to 0.47 V upon increasing the Mn content. A negligible current density was observed for Mn–O/N@NCs-70, implying that the MnOx components exhibit almost no ORR activity. As the thermodynamic electrode potential of the 2e− ORR is 0.70 V (eqn (2)),32 we tentatively concluded that Mn–O/N@NCs-5, Mn–O/N@NCs-20, and Mn–O/N@NCs-50 primarily followed the 2e− ORR pathway, while the NCs followed the 4e− ORR pathway.
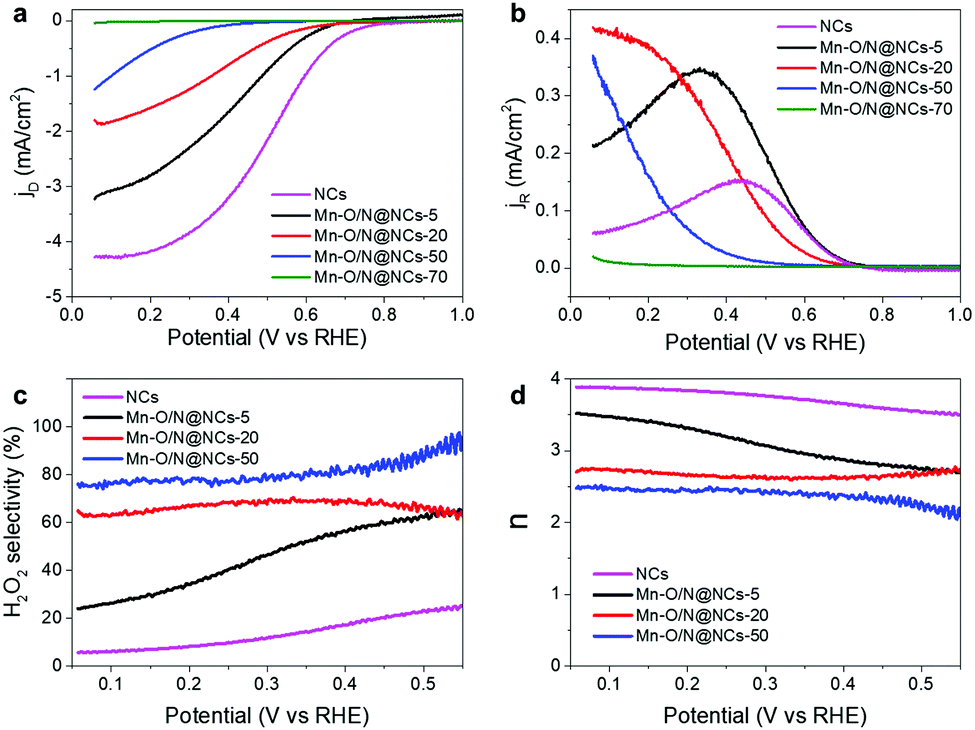 | ||
| Fig. 3 Electrochemical performances of the NCs, Mn–O/N@NCs-5, Mn–O/N@NCs-20, Mn–O/N@NCs-50, and Mn–O/N@NCs-70. (a) Disk and (b) ring current densities, (c) calculated H2O2 selectivities, and (d) electron transfer numbers (n) of the prepared samples as a function of the applied potential. All electrochemical measurements were performed in O2 purged 0.1 M HClO4 solution at 900 rpm and ring potential was constant at 1.2 VRHE. | ||
The ring currents induced by oxidation of the H2O2 generated on the disk electrode were then measured under a positive bias (1.2 V; Fig. 3b), providing a basis for the calculation of the H2O2 selectivity and the electron transfer number (n), as outlined in Fig. 3c and d. It was found that the H2O2 selectivity tended to decrease as the potential became more negative, which is the opposite behavior to that of Pt-based catalysts.8,33 As expected, the NCs exhibited less than 20% selectivity for H2O2 production, indicating a direct 4e− ORR pathway (Fig. 3c). In contrast, the H2O2 selectivity increased gradually over almost the entire potential range with increasing Mn content from Mn–O/N@NCs-5 to Mn–O/N@NCs-50. The H2O2 selectivity reached to 74% at 0.2 V in the case of Mn–O/N@NCs-50, which is comparable value to the previously reported non-precious carbon-based catalysts (Table S2, ESI†). Mn–O/N@NCs-70 was excluded from the selectivity calculations because it exhibited essentially no ORR activity. The improved selectivity for H2O2 production was also evaluated based on the decrements in the n values (Fig. 3d), whereby the n values of the samples at the potentials tested decreased ranged from 3.9 (NCs) to 2.5 (Mn–O/N@NCs-50). Furthermore, a Koutecký–Levich (K–L) plot (measured at 0.05, 0.1, and 0.2 V; Fig. S6, ESI†) was employed to calculate the average n value of Mn–O/N@NCs-50; this was found to be 2.09, thereby confirming the improved 2e− ORR for H2O2 production.
Considering these preliminary results, we hypothesized that the presence of Mn–Nx species is closely related to the 2e− ORR activity. It has been reported that pyridinic N acts as a bonding site for Ms in carbon supports when they form M-doped N–C (M–Nx/C).34–36 Although the amount of pyridinic N increased significantly after the incorporation of manganese species (i.e., from 25.42 at% for the NCs to 32.17, 38.02, and 30.91 at% for Mn–O/N@NCs-5, Mn–O/N@NCs-20, and Mn–O/N@NCs-50, respectively; Fig. S5, ESI†), these amounts were not proportional to the 2e− ORR activity. Furthermore, the additional NH3-treated catalysts (Mn–O/N@NCs-50-NH3) exhibited reduced H2O2 selectivities (from 76 to 51% at 0.1 V), implying that the incorporation of additional N doping or Mn–Nx bonds is simply not effective for the 2e− ORR (Fig. S7, ESI†). Hence, it is reasonable to suspect that the other components, such as the MnO species, exhibit a specific function that increases the H2O2 production selectivity. We should also consider the active sites for further reduction of the generated H2O2, which may occur via the re-adsorption of H2O2 on the M–N–C catalysts, thereby resulting in a 2e− + 2e− pathway.37
To investigate the role of the MnOx species, we prepared a control sample by leaching out inorganic MnO particles in a 1 M H2SO4 solution overnight under magnetic stirring. The obtained catalyst, Mn–O/N@NCs-50-acid, contained almost no crystalline peaks in its diffraction patterns (Fig. S8, ESI†), indicating that bulk MnO particles were completely removed. Fig. 4a shows the direct PRR results of the NCs, Mn–O/N@NCs-50, and Mn–O/N@NCs-50-acid, which were evaluated in the presence of 10 mM H2O2 in a 0.1 M HClO4 electrolyte solution at 900 rpm. Considering the standard reduction potentials of the PRR (E0 = 1.76 V; see eqn (3)), the generated H2O2 is prone to be reduced to H2O under the potentials used at pre-existing catalytic sites (i.e., Mn–Nx or N-doped C). Interestingly, Mn–O/N@NCs-50 exhibited almost no current density, whereas the NCs and Mn–O/N@NCs-50-acid showed negatively increased current densities, thereby indicating PRR reactivity. From the perspective of the catalytic components, we deduced that the MnO species present in Mn–O/N@NCs-50 prevented further reduction (or the PRR), thereby increasing the selectivity towards the first 2e− reduction (i.e., H2O2 production), as indicated in Fig. 4b. It should also be noted that Mn–O/N@NCs-50-acid exhibited a higher H2O2 selectivity than the NCs, implying that the PRR is most likely to occur on N-doped C sites rather than Mn–Nx species.
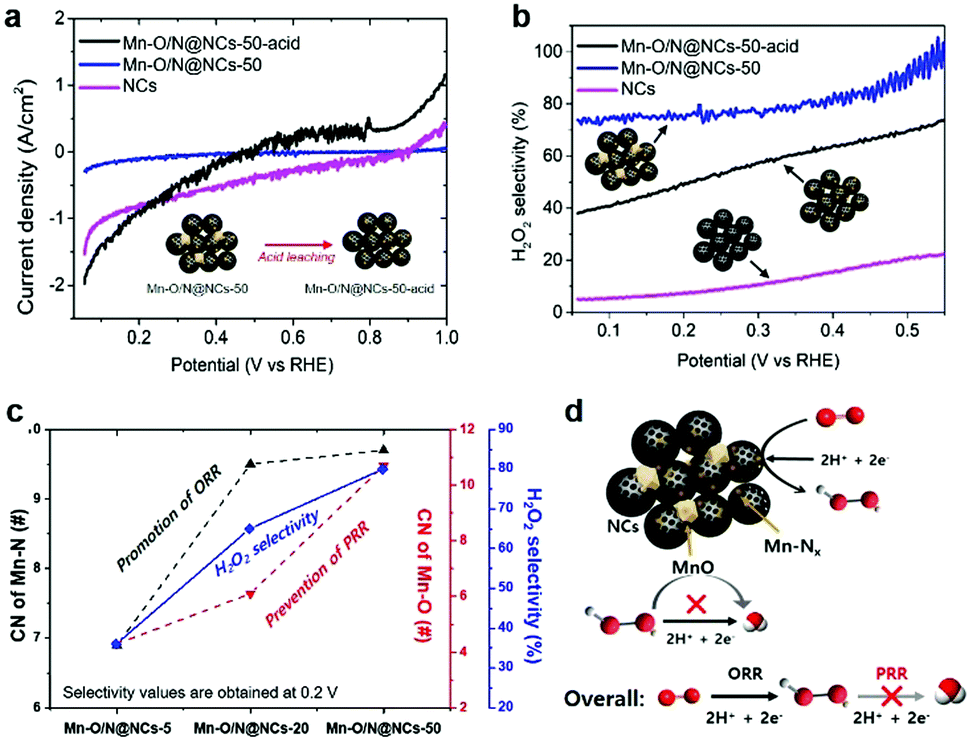 | ||
| Fig. 4 Electrochemical analysis to investigate the active sites. (a) Peroxide reduction reaction (PRR) activities, (b) Hydrogen peroxide selectivities of the NCs, Mn–O/N@NCs-50, and Mn–O/N@NCs-50-acid, (c) H2O2 selectivity versus the coordination number (CN) of Mn–N. All measurements were performed in 10 mM H2O2 dissolved in a 0.1 M HClO4 solution at 900 rpm. (d) Schematic illustration of the overall reaction mechanism on Mn–O/N@NCs-50. | ||
We also conducted RRDE tests on the composite sample (MnCl2–NCs), comprising a mechanical mixture of manganese chloride (MnCl2; Sigma-Aldrich) and NCs (Fig. S9, ESI†), which possessed Mn 2p bonds with no Mn–Nx species. Consequently, MnCl2–NCs exhibited a reduced H2O2 selectivity compared to Mn–O/N@NCs-50-acid (Fig. S9e, ESI†). Based on these comprehensive results, we can infer that the major active site for the 2e− ORR is Mn–Nx, while MnO prevents any further reduction of H2O2 in the 2e− + 2e− pathway, thereby increasing the likelihood of the 2e− pathway occurring. Furthermore, Mn–O/N@NCs-50 exhibited a high ORR durability, with no significant performance degradation with only ∼30 mV half-wave potential changes after 6000 cycles of an accelerated degradation test (ADT) (Fig. S10, ESI†). Based on these electrochemical results, the prepared Mn–O/N@NCs-50 catalyst may exhibit a high H2O2 selectivity due to the synergistic functionality of the components involved (i.e., Mn–Nx and MnO on mesoporous NCs), in addition to its long-term stability.
We also carried out extended X-ray absorption fine structure (EXAFS) analyses of Mn–O/N@NCs-5, Mn–O/N@NCs-20, Mn–O/N@NCs-50 to evaluate the relationship between the localized atomic structure and the catalytic activity. The coordination number (CN) values and radial distances (R) of the Mn–N, Mn–O, and Mn–Mn bonds were obtained by fitting of the Fourier-transformed EXAFS data (Fig. S11, ESI†). One noticeable finding was that the CN of the Mn–N bonds increased significantly from Mn–O/N@NCs-5 (6.9) to Mn–O/N@NCs-50 (9.7) (Fig. 4c), but no large differences were observed between Mn–O/N@NCs-20 (9.5) and Mn–O/N@NCs-50. Thus, we can infer that the superior H2O2 selectivity of Mn–O/N@NCs-50 was due to the higher CN of the Mn–O bonds in Mn–O/N@NCs-50 (10.7) compared to those of Mn–O/N@NCs-20 (6.1), which contained mainly inorganic MnO particles. The proposed mechanism of H2O2 production on Mn–O/N@NCs-50 is depicted schematically in Fig. 4d. More specifically, the Mn–Nx sites dispersed in the composites can facilitate the 2e− ORR for the production of H2O2, but the PRR mainly occurs at N-doped C sites. At this point, the pre-existing MnO particles close to the ORR sites can prevent the further reduction of H2O2 or the PRR taking place, thereby resulting in an increased selectivity for H2O2 production.
Finally, to demonstrate the electrochemical generation of H2O2 using the developed catalysts, we carried out single cell tests using membrane electrode assemblies (MEAs) composed of a commercial Nafion membrane, Pt/C, and Mn–O/N@NCs-50 as the electrolyte, anode, and cathode materials, respectively. The MEA prepared using the Mn–O/N@NCs-50 catalyst exhibited polarization behavior according to the power density in a single cell, as shown in Fig. 5a. More importantly, the production of H2O2 at an almost linear rate of ca. 20 μmol h−1 cm−2 was observed (Fig. 5b). This confirms that the simultaneous generation of electrical energy and useful chemicals, i.e., H2O2, is possible using our sustainable and efficient concept, by employing a widely adopted fuel cell system and selecting adequate cathode catalyst materials.
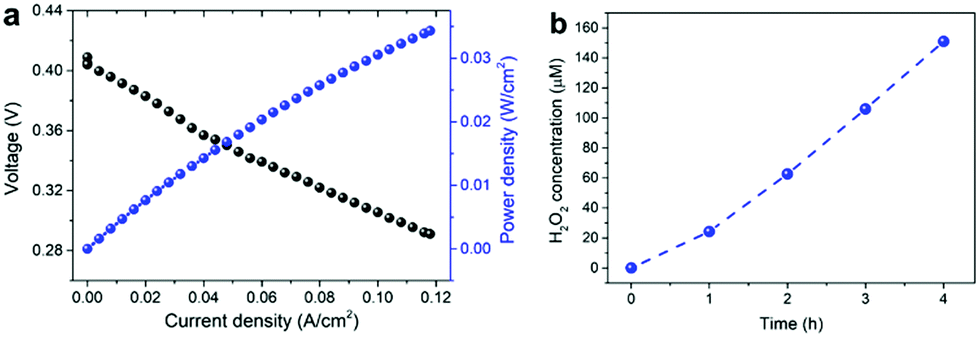 | ||
| Fig. 5 (a) Polarization curves of single cells based on Mn–O/N@NCs-50, and (b) H2O2 accumulation in a single cell based on Mn–O/N@NCs-50. | ||
Conclusions
Non-precious manganese-based composite catalysts, composed of inorganic MnO particles and Mn–Nx moieties with mesoporous N-doped carbon spheres (Mn–O/N@NCs), were synthesized for the electrochemical production of H2O2. We optimized the compositions of the heterogeneous catalysts by adjusting the Mn contents during their preparation, and this was demonstrated to have a significant effect on the ORR activity. The developed Mn–O/N@NCs exhibited an outstanding 2e− ORR activity, an extremely high (>80%) selectivity, and an outstanding durability. We demonstrated that an even distribution of Mn–Nx sites on the NCs promotes the ORR via the 2e− pathway, while N-doped C without Mn components can facilitate the 4e− ORR. More importantly, the presence of MnO particles in the composites effectively mitigates the further reduction of H2O2, which is the most frequently encountered issue in electrochemical H2O2 production. This comprehensive study extends our understanding of M–N–C hybrid catalysts in terms of their synergistic functions in H2O2 synthesis, via a system that was especially devised for use in sustainable and energy efficient fuel cell systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the KIST Institutional Program 2E30380. This work was supported by the Global Frontier R&D Program on Center for Multiscale Energy System funded by the Nation Research Foundation under the Ministry of Science, ICT & Future Planning, Korea (2016M3A6A7945505). This research was supported by the Technology Development Program to Solve Climate Changes of the National Research Foundation (NRF) funded by the Ministry of Science, ICT, & Future Planning (NRF-2015M1A2A2056690). This work was supported by the Korea Ministry of Environment as a “Global Top Project” (Project No. 2016002190003). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2019R1F1A1044908). This research was also supported by Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (Grant No. 20188550000440). This research was also supported by the Hydrogen Energy Innovation Technology Development Program of the National Research Foundation of Korea (NRF) funded by the Korean government (Ministry of Science and ICT(MSIT)) (No. 2019R1A2C2006117).Notes and references
- J. K. Edwards, B. Solsona, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 1801909 CrossRef.
- E. Lobyntseva, T. Kallio, N. Alexeyeva, K. Tammeveski and K. Kontturi, Electrochim. Acta, 2007, 52, 7262–7269 CrossRef CAS.
- M. Melchionna, P. Fornasiero and M. Prato, Adv. Mater., 2019, 31, 1802920 CrossRef PubMed.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. S. Stephens, I. E. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- C. H. Choi, H. C. Kwon, S. Yook, H. Shin, H. Kim and M. Choi, J. Phys. Chem. C, 2014, 118, 30063–30070 CrossRef CAS.
- H. Jing, Q. Zhang, N. Large, C. Yu, D. A. Blom, P. Nordlander and H. Wang, Nano Lett., 2014, 14, 3674–3682 CrossRef CAS PubMed.
- E. Pizzutilo, O. Kasian, C. H. Choi, S. Cherevko, G. J. Hutchings, K. J. J. Mayrhofer and S. J. Freakley, Chem. Phys. Lett., 2017, 683, 436–442 CrossRef CAS.
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef PubMed.
- E. Pizzutilo, S. J. Freakley, S. Cherevko, S. Venkatesan, G. J. Hutchings, C. H. Liebscher, G. Dehm and K. J. J. Mayrhofer, ACS Catal., 2017, 7, 5699–5705 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- I. Monte-Pérez, S. Kundu, A. Chandra, K. E. Craigo, P. Chernev, U. Kuhlmann, H. Dau, P. Hildebrandt, C. Greco, C. Van Stappen, N. Lehnert and K. Ray, J. Am. Chem. Soc., 2017, 139, 15033–15042 CrossRef.
- Y.-H. Wang, M. L. Pegis, J. M. Mayer and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 16458–16461 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- P. Gouérec and M. Savy, Electrochim. Acta, 1999, 44, 2653–2661 CrossRef.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS.
- I. Roche, E. Chaînet, A. M. Chatenet and J. Vondrák, J. Phys. Chem. C, 2006, 111, 1434–1443 CrossRef.
- M. Suk, M. W. Chung, M. H. Han, H.-S. Oh and C. H. Choi, Catal. Today DOI:10.1016/j.cattod.2019.05.034.
- R. B. Valim, M. C. Santos, M. R. V. Lanza, S. A. S. Machado, F. H. B. Lima and M. L. Calegaro, Electrochim. Acta, 2012, 85, 423–431 CrossRef CAS.
- C. H. Choi, W. S. Choi, O. Kasian, A. K. Mechler, M. T. Sougrati, S. Brüller, K. Strickland, Q. Jia, S. Mukerjee, K. J. J. Mayrhofer and F. Jaouen, Angew. Chem., Int. Ed., 2017, 56, 8809–8812 CrossRef CAS.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef.
- M. Nugraha, M.-C. Tsai, W.-N. Su, H.-L. Chou and B. J. Hwang, Mol. Syst. Des. Eng., 2018, 3, 896–907 RSC.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2014, 54, 588–593 Search PubMed.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
- J. Park, Y. Nabae, T. Hayakawa and M. Kakimoto, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS.
- K. R. Yoon, K. Shin, J. Park, S.-H. Cho, C. Kim, J.-W. Jung, J. Y. Cheong, H. R. Byon, H. M. Lee and I.-D. Kim, ACS Nano, 2018, 12, 128–139 CrossRef CAS PubMed.
- K. R. Yoon, G. Y. Lee, J.-W. Jung, N.-H. Kim, S. O. Kim and I.-D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef CAS PubMed.
- D. Sun, Y. Tang, D. Ye, J. Yan, H. Zhou and H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 5254–5262 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, USA, 2001 Search PubMed.
- S. Marcotte, D. Villers, N. Guillet, L. Roué and J. P. Dodelet, Electrochim. Acta, 2004, 50, 179–188 CrossRef CAS.
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421–3428 CrossRef CAS.
- H. R. Byon, J. Suntivich, E. J. Crumlin and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2011, 13, 21437–21445 RSC.
- P. Xu, W. Chen, Q. Wang, T. Zhu, M. Wu, J. Qiao, Z. Chen and J. Zhang, RSC Adv., 2015, 5, 6195–6206 RSC.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, DFT calculation details, additional characterizations such as BET, TEM, XPS, XRD, and EXAFS, and electrochemical results. See DOI: 10.1039/c9nh00783k |
| This journal is © The Royal Society of Chemistry 2020 |