
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring color space: an investigation of heteroaryl-substituted benzobis[1,2-d:4,5-d′]oxazoles and their application in organic light-emitting diodes†
David L.
Wheeler
a,
Shambhavi
Tannir
 a,
Hadar R.
Yakir
b,
Or
Dishi
b,
Ori
Gidron
b and
Malika
Jeffries-EL
*ac
a,
Hadar R.
Yakir
b,
Or
Dishi
b,
Ori
Gidron
b and
Malika
Jeffries-EL
*ac
aDepartment of Chemistry, Boston University, 590 Commonwealth Ave, Boston, MA 02215, USA. E-mail: malikaj@bu.edu
bInstitute of Chemistry, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Jerusalem, Israel
cDivision of Materials Science and Engineering, Boston University, 15 St. Mary Street, Boston MA02215, USA
First published on 22nd February 2024
Abstract
This study investigates the use of heterocycles to design electrochemically stable, multi-colored dopants for use in organic light-emitting diodes (OLEDs). To this end, 24 furan- and thiophene-based semiconductors were synthesized using the novel benzobis[1,2-d:4,5-d′]oxazole (BBO) core. The optical and electronic properties of these materials were predicted using computational tools and verified using electrochemical and spectroscopic data. The fluorescence quantum yields for the BBOs ranged between 33–98%, while all relaxation lifetimes occured in <5 ns. Solution-processed OLEDs were formed using the BBO materials, producing a series of dopants that achieved deep blue-to-orange electroluminescence. As a result, four deep blue devices and a color temperature tunable white OLED were formed using various BBO dopants with brightness values suitable for display technology.
Introduction
Since their inception, organic semiconductors (OSC) have become one of the most widely researched class of materials due to their versatility for use in various electronic applications.1 These unique small molecules and polymers are covalently structured using the σ-framework while the π-conjugated system(s) allows for control over the optical and electronic properties.2 Depending upon the extent of π-conjugation, one can tune the HOMO–LUMO gap and optical band gap (Eoptg) of these materials to produce electrical current when initiated by photoexcitation or when an electric potential is applied.3,4 As a result, OSCs have shown remarkable achievements in photovoltaics,5–8 field-effect transistors,9,10 electrochromics,11–13 and, in particular, light-emitting diodes (OLED).14,15Due to the increased energy-efficiency and low-cost fabrication of OLEDs, this technology has recently begun to enter the consumer market in the form of smartphone displays and televisions.16 OLEDs emit less thermal radiation than inorganic LEDs, due to their ability to theoretically convert all injected charges into photons via tuning of the emissive material's singlet and triplet excited states.17 Furthermore, this technology does not require the use of backlighting units like those used in liquid crystal displays, making it thin and light-weight. OLED displays also have a higher contrast ratio compared to LED displays due to the ability to achieve zero luminescence or “absolute” black.18 In comparison to heavy metal complexes, OSCs can be functionalized with solubilizing chains, which allows OLED fabrication to be conducted using a variety of solution-processing techniques. This capability reduces the cost and time necessary for production while eliminating the current problem of size-limited fabrication current LED technology faces.19 Because these processing techniques form amorphous films, OLED devices can be made on flexible substrates for the production of bendable displays and lighting.20 While all of these qualities are ideal for commercialization, OLEDs still suffer from lower lifetimes in contrast to their inorganic kin. Thus, research efforts towards realizing an electrochemically stable palette of emissive materials (dopants) for OLED display and solid-state lighting applications are currently being pursued.21
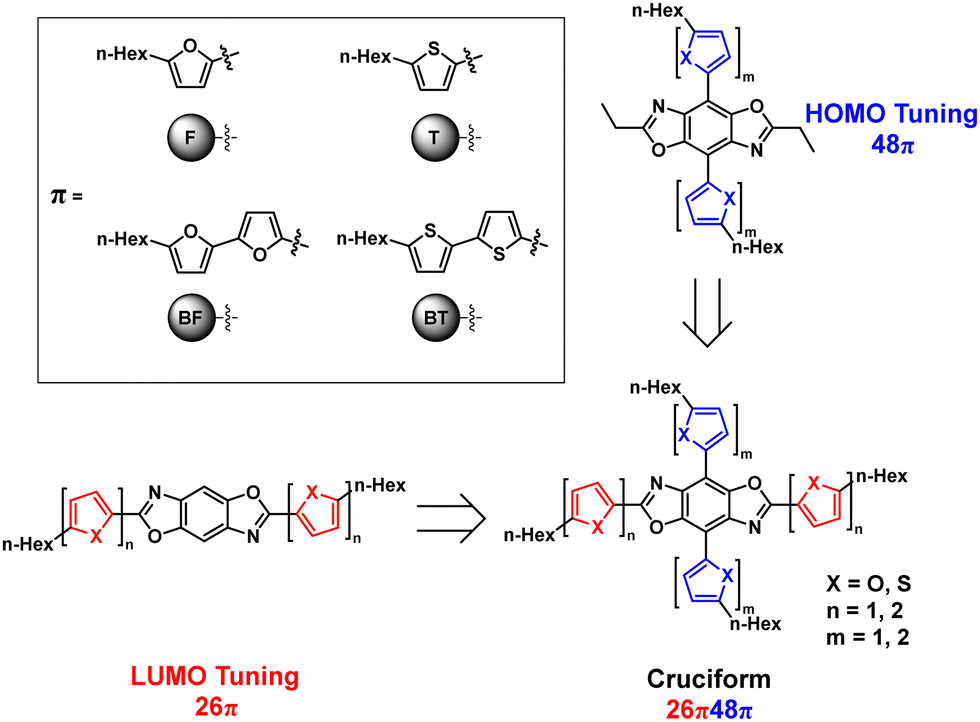
One class of materials that show great promise are those which incorporate the heterocyclic core benzobis[1,2-d:4,5-d′]oxazole (BBO), Fig. 1. As a result of their unique, cross-conjugated structure, these compounds possess segregated frontier molecular orbitals. This allows for semi-autonomous tuning of the HOMO and LUMO levels via aryl substitution along the 4,8- or 2,6-axis, respectively.22–27 In the solid-state, the energy levels can be further altered using steric hindrance to disrupt intermolecular interactions without changing the Eoptg.27 Additionally, materials based on BBO are known to exhibit high thermal stabilities (>200 °C) and possess excellent oxidative stability.28–31 To date, only a limited number of solution-processed host–guest OLEDs have been made using BBO dopants, and most of them exhibit deep-blue electroluminescence (EL).23,24,32 Although the development of stable blue-light emitting materials is a major challenge in the field,33 there is a need for green, yellow, orange, and red OLED dopants for full color displays and solid-state lighting. Accordingly, we seek to design tunable BBO materials that emit across the range of the color spectrum.
 | ||
| Fig. 1 Linear (top right and bottom left) and cross-conjugated (bottom right) BBO structures investigated. | ||
The synthesis of OSCs composed from alternating electron-accepting (A) moieties, like BBO, and electron-donating (D) arenes, like thiophene or furan, is an effective way to tune the energy levels and band gaps.34–36 As a result of the wide commercial availability and ease of functionalization, thiophene and its derivatives have been widely studied in OSCs. Exemplified by regio-regular poly-3-alkylthiophene, materials based on thiophene are known for having high charge carrier mobilities due to the strong intermolecular interactions resulting from polarizability of the sulfur atoms.37 Furan, the oxygen analog of thiophene, has not been as widely explored as an active material in organic semiconductors. This is due in part to the known photo-oxidation and perceived instability of furan in comparison to its more aromatic counterpart.38 However, furan, oligofuran39–41 and furan derivatives9,42 have been shown to be thermally stable with some exhibiting strong fluorescence.43 Throughout literature, there are several examples where furan containing OSCs show comparable efficiencies to their thiophene counterparts.38,44–46 Furan based OSCs have also shown promise in the design of emissive materials, although the reports are limited in scope and number.43 The use of furan-based BBOs could lead to significant quantum yield enhancements and, thus, highly emissive materials.
The electronic energy levels and Eoptg, of an OSC can be further modulated by extending conjugation. Therefore, we sought to investigate the collective impact of heteroatom substitution and extended conjugation on the optical and electronic properties of a series of BBOs designed using the molecular heredity principle based on all possible combinations of furan, thiophene, 2,2′-bifuran, and 2,2′-bithiophene.27,47 In total, eight linear BBO parents (those conjugated through only one axis) and 16 cross-conjugated BBO children (conjugated through both axes) were synthesized and evaluated. Herein, we report the synthesis and electrochemical/spectroscopic characterization of these materials, including a detailed analysis of structure–property relationships. Following characterization, each material was solution-processed into an active OLED to determine their electroluminescent (EL) properties.
Results and discussion
Synthesis
The synthetic steps used to form the final molecules of interest are shown in Scheme 1. The linear BBOs were synthesized using either a condensation reaction for substitution along the 2,6-axis or a cross-coupling reaction for substitution along the 4,8-axis, Scheme 1. The cross-conjugated BBOs were prepared via a sequential two-step process: first, the formation of the substituted BBO core via a condensation reaction followed by 4,8-functionalization via Stille cross-coupling. The synthesis of all precursor stannanes and orthoesters is described in the supplemental information. Using the precursor bis(hydrochloride) 2,5-diamino-p-hydroquinone (1), the cyclization to form the linearly conjugated 2,6-small molecules produced four compounds with the following yields: 26F, 30%; 26BF, 11%; 26T, 16%; and 26BT, 13% (Fig. 2). Using the same conditions, the cruciform precursors were formed in higher yields relative to their non-halogenated kin (26F48Br, 77%; 26BF48Br, 33%; 26T48Br, 56%; 26BT48Br, 34%). Using previous condensation methods to form 3 from 2,26 the addition of 4,8-aryl substituents was achieved in moderate to good yields using 3 and the corresponding aryl stannane. With traditional Stille cross-coupling methods, the 4,8-parents 48F (63%), 48BF (67%), 48T (83%), and 48BT (73%) were produced. Using the same stannane synthons, the cruciforms were produced in yields between 25–81%. All compounds (Fig. 2) show moderate solubility in chlorinated solvents and were characterized by 1H and 13C (where applicable) NMR spectroscopy and high-resolution mass spectrometry. The cruciforms showed high thermal stability with Tg ranging from 360–440 °C. Details of the thermogravimetric analysis and differential scanning calorimetry can be found in the ESI† (Table S01 and Fig. S102).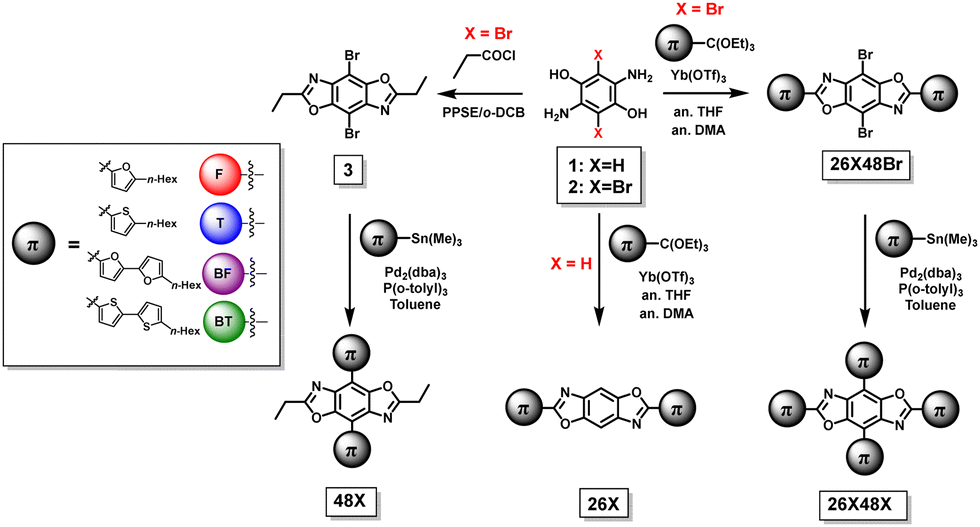 | ||
| Scheme 1 General synthesis of the BBO cruciforms. | ||
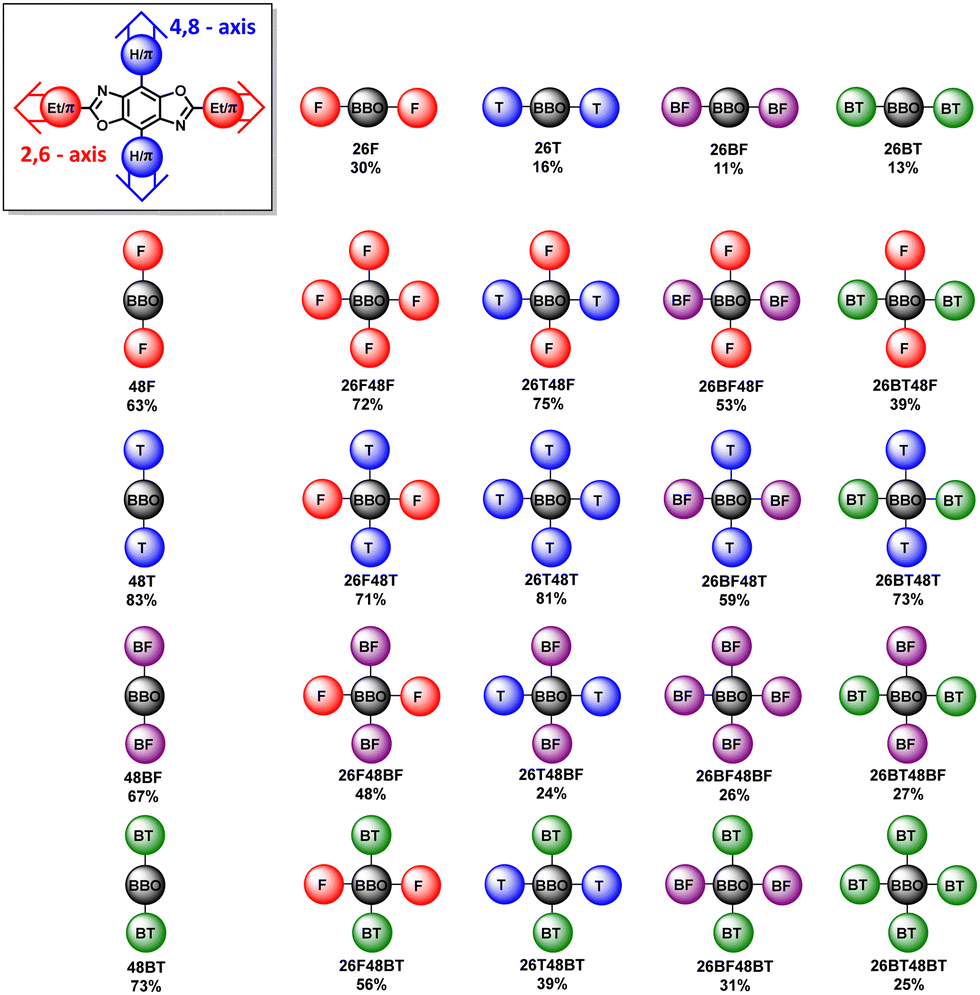 | ||
| Fig. 2 General representation of the 24 BBOs in the series. Yields are shown in below each name. | ||
Electronic properties
The calculated (DFT/B3LYP/6-31G(d)) and experimental electronic properties of all 24 compounds are listed in Table 1.48,49 The experimental HOMO energies were measured in solution and as thin films and calculated from the onset of oxidation determined from cyclic voltammetry experiments as referenced to a Fc/Fc+ redox as standard. The optical gap (Eoptg) was determined from the onset of each UV-Vis spectrum.| BBO | HOMO (eV) | LUMO (eV) | E CVg (eV) | E optg (eV) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sola | Filma | DFTe | Solb | Filmb | DFTe | Solc | Filmc | Sold | Filmd | |
| a Calculated from onset of oxidation: HOMO = −4.8 + (EFcox − ESampleox). b LUMO calculated from onset of reduction: LUMO = −4.8 + (EFcox − ESamplered). c Electrochemical gap calculated from difference of HOMO and LUMO of corresponding physical state. d Optical gap calculated from onset of absorption in UV-Vis spectra. e Determined from the first excited-state output from TD-DFT calculations; B3LYP/6-31G(d). Details are described in the computational section. | ||||||||||
| 48F | −5.27 | −5.39 | −4.75 | −2.88 | −2.55 | −1.40 | 2.39 | 2.84 | 3.09 | 2.81 |
| 26F | −5.70 | −5.85 | −5.25 | −2.99 | −2.61 | −1.62 | 2.71 | 3.24 | 3.19 | 3.00 |
| 48T | −5.39 | −5.54 | −5.13 | −2.88 | −2.60 | −1.14 | 2.51 | 2.94 | 3.04 | 2.89 |
| 26T | −5.71 | −5.69 | −5.05 | −2.87 | −2.74 | −2.05 | 2.84 | 2.95 | 3.08 | 2.60 |
| 48BF | −5.08 | −5.34 | −4.50 | −2.91 | −2.58 | −1.66 | 2.17 | 2.76 | 2.63 | 2.53 |
| 26BF | −5.48 | −5.55 | −4.94 | −2.91 | −2.63 | −1.84 | 2.57 | 2.92 | 2.78 | 2.65 |
| 48BT | −5.16 | −5.32 | −4.96 | −2.76 | −2.64 | −1.23 | 2.40 | 2.68 | 2.56 | 2.37 |
| 26BT | −5.72 | −5.67 | −4.85 | −3.27 | −2.99 | −2.35 | 2.45 | 2.68 | 2.66 | 2.52 |
| 26F48F | −5.25 | −5.43 | −4.67 | −2.88 | −2.56 | −1.72 | 2.37 | 2.87 | 2.72 | 2.58 |
| 26F48T | −5.36 | −5.52 | −4.83 | −2.98 | −2.78 | −1.87 | 2.38 | 2.74 | 2.75 | 2.74 |
| 26T48F | −5.21 | −5.69 | −4.72 | −2.91 | −2.66 | −1.88 | 2.30 | 3.03 | 2.66 | 2.42 |
| 26T48T | −5.30 | −5.55 | −5.05 | −2.81 | −3.14 | −1.36 | 2.49 | 2.41 | 2.68 | 2.53 |
| 26F48BF | −5.06 | −5.21 | −4.45 | −2.87 | −2.59 | −1.88 | 2.19 | 2.62 | 2.44 | 2.31 |
| 26F48BT | −5.09 | −5.27 | −4.64 | −2.89 | −2.70 | −2.09 | 2.20 | 2.57 | 2.74 | 2.18 |
| 26T48BF | −5.01 | −5.48 | −4.49 | −2.90 | −2.58 | −2.00 | 2.11 | 2.90 | 2.41 | 2.25 |
| 26T48BT | −5.05 | −5.35 | −4.90 | −2.89 | −2.60 | −1.43 | 2.16 | 2.75 | 2.42 | 2.24 |
| 26BF48F | −5.21 | −5.56 | −4.64 | −2.96 | −2.56 | −1.87 | 2.25 | 3.00 | 2.58 | 2.45 |
| 26BF48T | −5.32 | −5.52 | −4.78 | −2.88 | −2.58 | −1.99 | 2.44 | 2.94 | 2.61 | 2.39 |
| 26BT48F | −5.17 | −5.56 | −4.71 | −2.88 | −2.65 | −2.14 | 2.29 | 2.91 | 2.40 | 2.19 |
| 26BT48T | −5.30 | −5.66 | −5.03 | −2.89 | −3.26 | −1.40 | 2.41 | 2.40 | 2.45 | 2.31 |
| 26BF48BF | −5.00 | −5.35 | −4.44 | −2.90 | −2.58 | −1.99 | 2.10 | 2.77 | 2.35 | 2.23 |
| 26BF48BT | −5.05 | −5.37 | −4.62 | −2.94 | −2.68 | −2.16 | 2.11 | 2.69 | 2.35 | 2.13 |
| 26BT48BF | −4.90 | −5.28 | −4.49 | −2.92 | −3.14 | −2.21 | 1.98 | 2.14 | 2.31 | 2.19 |
| 26BT48BT | −4.97 | −5.40 | −5.24 | −3.31 | −3.23 | −1.73 | 1.66 | 2.17 | 2.29 | 2.17 |
In solution, the HOMO energies are affected by the chalcogen present in the aryl substituents as well as the conjugation length and position. Starting with the linear 4,8-conjugated materials, 48T has the lowest HOMO energy of the set. When the chalcogen is changed from sulfur to oxygen (48F), an increase in energy by 0.12 eV is observed. This same effect is observed comparing 48BT to 48BF. As conjugation length of the aryl substituent is increased, the HOMO energy is increased. In contrast to the linear 4,8-parents, the HOMO energy for all linear 2,6-conjugated materials is decreased. The observation is a common phenomenon in linear BBO small molecules. For the 2,6-parents, all aforementioned trends are observed, with the exception of increased conjugation between 26T and 26BT yielding the same HOMO energy. However, this anomaly could be due to the poor solubility of 26BT. In all cases, the HOMO energy of the cruciforms is inherited by the 4,8-parent with a minor increase due to the extended conjugation provided by the 2,6-aryl substituents. This trend follows previous findings for similar BBO systems and is further supported by the density functional theory (DFT) calculations on all molecules where a clear localization of the HOMO is found along the 4,8-axis (Fig. S103–105, ESI†).23,24,26 The HOMO energy level for all cruciforms in solution range from −5.36 eV to −4.90 eV. Trends between the LUMO energies for the 24 molecules were not clear. This finding correlates to the DFT calculation, where the LUMO is observed to be delocalized along the entire molecule.
We next analyzed the Eoptg in solution of the parent molecules. In each case, the 4,8-parents have a smaller Eoptg than their 2,6-counterparts. When the conjugation axis is kept constant, switching the chalcogen from oxygen to sulfur further decreases Eoptg. We attribute this finding to the impact of thiophene's higher aromaticity on stabilizing each compound's LUMO energy. While the HOMO and LUMO energies are inherited from a specific parent due to frontier molecular orbital (FMO) similarities, the Eoptg for the children is not inherited from a single parent and is a product of an extended π-system. Therefore, the Eoptg for all 24 compounds ranges from 2.29–3.26 eV in solution. However, the cruciforms with similar conjugation lengths produce relatively narrow Eoptg ranges with minor shifts based upon the aforementioned trends. Upon film formation, all compounds undergo a decrease in the Eoptg (2.13–3.00 eV), indicating significant intermolecular interactions. We attribute the differences in the ECVg solution measurements being lower than their optical counterpart due to solvent stabilization effects lowering the LUMO energy50 while the trendless differences in the solid-state measurements could be a feature of differences in sample preparation between the two measurements, altering the intermolecular packing of molecules, thereby changing the density of states that are probed from each measurement. However, the variations in the solid-state ECVg and Eoptg could also be due to the intrinsic differences of how the HOMO and LUMO states are probed between the two techniques as well.50
Optical properties
The UV-Visible spectra for the BBOs were experimentally measured in chloroform (Fig. 3) and as thin-films (Fig. S90, ESI†). All relevant data is shown in Table 2. BBOs have been shown to have tunable absorption based on linear or cruciform forms. This depends on the types of groups attached at the 2,4,6, and 8 positions, and the extension of conjugation along single or both axes.48,51,52 In solution, the parent BBOs containing furan and bifuran substituents all show electronic transitions with small vibronic shoulders. When the chalcogen in these compounds is changed to sulfur, a bathochromic shift is observed for the absorbance profiles. The band is sharp, with the 0-0 and 0-1 transitions clearly observed for all parent compounds with either furan or thiophene substituents. However, parents with bithiophenes in either the 2,6 or 4,8 positions (26BT and 48BT) display significantly broader absorption peaks, whereas the bifuran analogs (26BF and 48BF) remain sharp. Unlike previous studies with benzene-based systems,27 the cruciforms formed from furan and thiophene-based substituents do not retain spectral properties similar to their parents. All cruciforms that have the same extent of conjugation along both axes retain nearly identical absorbance profiles, only varying by relative transition intensity. However, the fine structure in these compounds is lost. Compounds with the structure 26X48X have peak maxima centered at ∼400 nm with π–π* transitions between 350–450 nm in the visible region. Extending the conjugation through the 4,8-axis, cruciforms with the structure 26X48BX span a nearly uniform absorbance range of 350–500 nm. When the axis of conjugation is flipped (compounds 26BX48X), a significant narrowing of the absorbance is observed (375–480 nm). Compounds with the general formula 26BX48BX exhibit peak maxima at ∼445 nm and span from 350–550 nm. Due to the slightly acidic nature of chloroform, our studies show that after several hours of solution preparation, certain compounds undergo oxidation when left at room temperature but can be avoided when stored at −20 °C. We noticed compounds that have furan units along the 4,8-axis or bifuran units along either axis seem to undergo this degradation. However, this transformation is not observed in the solid state.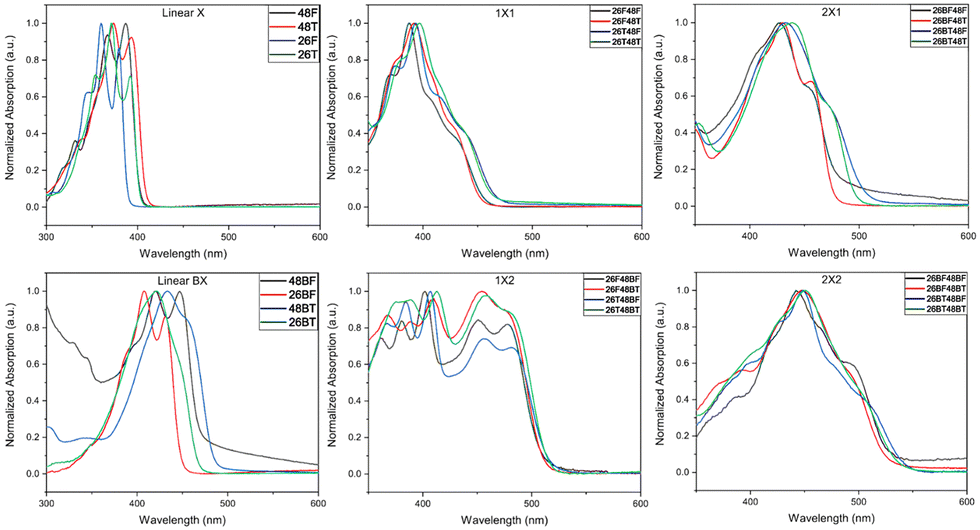 | ||
| Fig. 3 UV-Vis spectra of each BBO dissolved in CHCl3. | ||
| BBO | Solution | Film | |||||
|---|---|---|---|---|---|---|---|
| abs λ max (nm) | em λ max (nm) | Stokes shift (eV) | QY (%) | Lifetimes (ns) | abs λ max (nm) | em λ max (nm) | |
| 48F | 387, 367 | 424, 402 | 0.12 | 59 | 2.7 | 402, 379, 354 | 538 |
| 26F | 380, 369 | 428, 409, 383 | 0.03 | 97 | 2.2 | 386, 362*, 343*, 315 | 470 |
| 48T | 393, 373 | 458, 428, 409 | 0.12 | 33 | 1.4 | 401, 378, 358 | 494, 459 |
| 26T | 392, 372, 354 | 446, 421, 398 | 0.05 | 70 | 1.7 | 403, 358*, 340*, 326 | 502 |
| 48BF | 447, 420, 393* | 539*,499, 466 | 0.11 | 75 | 3.7 | 460, 427, 403 | 570 |
| 26BF | 431, 408 | 476, 447 | 0.10 | 98 | 1.5 | 438, 360 | 570 |
| 48BT | 434 | 558*, 517, 488 | 0.30 | 67 | 1.4 | 487, 451, 426 | 616 |
| 26BT | 422 | 533*, 497, 468 | 0.23 | 94 | 1.4 | 370 | 593 |
| 26F48F | 430*, 408*, 387, 371 | 485, 463 | 0.53 | 54 | 4.2 | 431, 406, 392* | 558 |
| 26F48T | 428*, 392, 375* | 481, 456 | 0.18 | 37 | 1.0 | 438, 412, 338, 323 | 600, 562, 517, 479 |
| 26T48F | 438*, 416*, 392, 375 | 500, 478 | 0.24 | 39 | 4.9 | 437*, 400, 379, 342 | 587 |
| 26T48T | 436*, 397, 379* | 492, 470 | 0.21 | 48 | 3.1 | 451, 406, 388, 345, 333* | 559, 526, 493 |
| 26F48BF | 487, 451, 402, 380, 361 | 538, 519 | 0.16 | 70 | 3.5 | 494, 458, 411, 390, 372* | 615 |
| 26F48BT | 475*, 452, 410, 389, 368 | 541, 509 | 0.31 | 33 | 3.9 | 518, 488, 413, 388 | 716, 600 |
| 26T48BF | 481, 456, 407, 384, 367 | 550, 524 | 0.21 | 58 | 4.2 | 499, 463, 414, 393, 305 | 639 |
| 26T48BT | 481*, 459, 413, 389, 375 | 550, 515 | 0.17 | 42 | 1.8 | 518, 480, 449*, 424, 390 | 627 |
| 26BF48F | 453*, 428 | 492 | 0.22 | 70 | 4.1 | 472, 439, 418, 347* | 598 |
| 26BF48T | 456*, 430 | 504, 471 | 0.09 | 48 | 2.5 | 483, 445*, 423, 352 | 632 |
| 26BT48F | 472*, 432 | 492 | 0.11 | 50 | 2.5 | 444, 430* | 672 |
| 26BT48T | 470*, 439, 353 | 526, 498 | 0.15 | 52 | 2.3 | 504, 469, 443, 363, 350 | 606 |
| 26BF48BF | 491, 442, 389* | 536 | 0.21 | 45 | 3.1 | 502, 463, 321 | 625 |
| 26BF48BT | 491*, 447, 392 | 561, 526 | 0.17 | 38 | 2.5 | 514*, 436, 410*, 331 | 723 |
| 26BT48BF | 449 | 549 | 0.50 | 58 | 4.5 | 512*, 448, 409*, 340 | 672 |
| 26BT48BT | 451 | 526, 498 | 0.26 | 52 | 2.9 | 528, 487, 469* | 672 |
In the solid state, each material undergoes a bathochromic shift, indicated by the decrease in Eoptg. As expected, extending the conjugation from one furan or thiophene unit to either bifuran or bithiophene also results in a bathochromic shift, with the thiophene parents continuing to experience a similar bathochromic shift relative to their furan counterparts. All 2,6-parents are hypsochromically shifted compared to their 4,8-conjugated equivalents. The fine structure for all 4,8-parents is still well-resolved, although slight broadening around each electronic transition is observed. However, the fine structure for the 2,6-parents is lost, most likely due to the newly accessible intermolecular transitions. Cross-conjugated molecules of identical conjugation length experience very similar spectral profiles. Cruciforms with formula 26X48X exhibit two absorption bands, one ranging from 300–350 nm and another from 350–475 nm. Extending the conjugation at the 4,8-axis (26X48BX) results in a shift of these bands to 350–450 nm and 450–550 nm, respectively. When the conjugation axes are inverted (26BX48X), the two bands merge together, creating one extensive absorption band from 400–535 nm.
Finally, for the 26BX48BX cruciforms, we see a similar pattern towards the creation of broadly absorbing materials (350–600 nm).
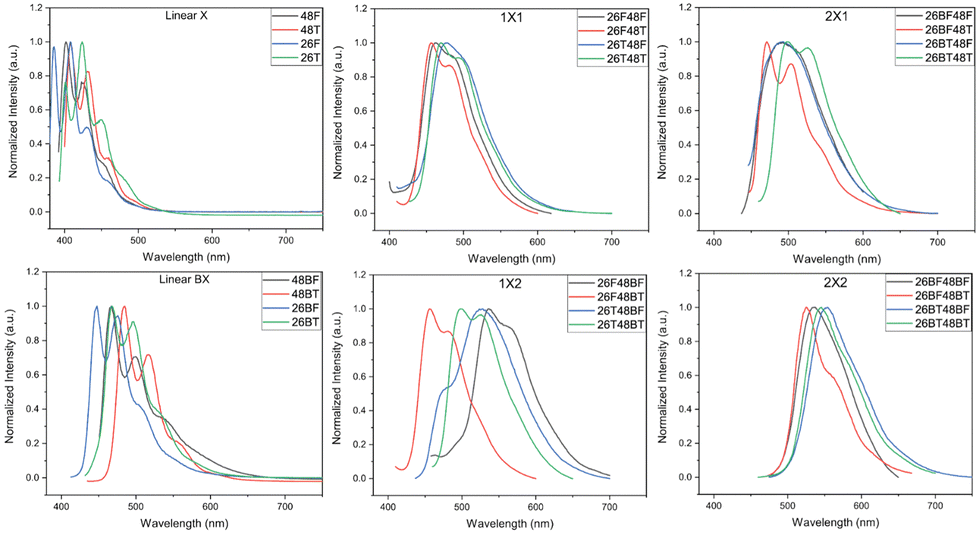
The photoluminescence (PL) spectra for the BBOs were experimentally measured in chloroform (Fig. 4) and as thin films (Fig. S91, ESI†). All relevant data is shown in Table 2. The solution-state PL of each parent reveals small Stokes shifts (0.03–0.12 eV) for compounds that do not contain the bithiophene unit. The fine structure in these spectra is well-resolved and distinct. Relative to the PL of the furan parents, the emission is red-shifted when the chalcogen is exchanged for sulfur and when the axis of conjugation is switched from the 2,6- to the 4,8-axis. Following previous Eoptg observations, extending the conjugation from F to BF or T to BT results in a bathochromic shift. Much like the absorption spectra, the 26X48X cruciforms with similar conjugation along each axis exhibit similar PL profiles and show Stokes shifts between 0.18–0.53 eV. Moreover, these cruciforms undergo minor blue-shifts when the chalcogen along the 2,6-axis is oxygen compared to their sulfur counterparts. This same observation is noted for the 26X48BX children with a smaller Stokes shifts range (0.16–0.31 eV). However, the 26BX48X cruciform series starts to deviate from this trend where shifts as much as ∼25 nm can be seen between spectra. This effect is further exacerbated in the 26BX48BX series where shifts between peak maxima can range up to 0.50 eV. Therefore, while similar conjugation between cruciforms may provide very similar absorption profiles, the chalcogen effect and placement of aryl substituents provides substantial changes to the PL of each system.
 | ||
| Fig. 4 PL spectra of each BBO dissolved in CHCl3. | ||
In film, we begin to see a divergence from several trends found in solution. Namely, chalcogen identity and axis of conjugation effects seem to be lost when probing these materials in the solid-state, which is believed to be a direct consequence of the different intermolecular interactions resulting from the packing arrangement adopted by each material. For the furan parents, the emission peak maxima rank as 26F > 48F > 48BF ≈ 26BF while the thiophene parents rank as 48T ≈ 26T > 26BT > 48BT. The parents experience dramatic Stokes shifts, but do provide a collective array of colored emission, ranging from 470–616 nm. Upon cross-conjugation, these emission wavelengths are further, red-shifted due to the increased conjugation of the systems. Much like the parent spectra, the 26X48X series all have unique emission profiles with emission maxima ranging from 526–600 nm. While cruciforms with furan through the 4,8 give nearly monochromatic emission, changing the aryl substituent to thiophene broadens the spectral profile and produces multiple emission peaks. Increasing the conjugation along either the 4,8-axis (26X48BX series) or 2,6-axis (26BX48X series) results in further redshift of emission of 615–716 nm and 598–672 nm, respectively. The cruciforms with the highest degree of conjugation (26BX48BX) emit between 625 and 723 nm. Much like the parents, all cruciforms experience a larger Stokes shift in the solid phase, except for 26T48T.
Quantum yields and fluorescent lifetimes
For the parent molecules, all compounds were found to exhibit photoluminescent lifetimes in the nanosecond regime. The PLQYs of each 2,6-parent was found to be very efficient, ranging from 70–98%. However, by changing the conjugation axis, a suppression of the QY for each material is observed (33–75%). In each case, changing the chalcogen from oxygen to sulfur also reduces the QY and the fluorescent lifetimes, which is the result of more flexible thiophene backbone, and a heavy-atom effect of the sulfur, enabling intersystem crossing pathway. In most cases, increasing the conjugation of each heterocyclic unit supplies a dramatic enhancement of the QY. The opposite effect is noticed for the lifetimes where extending conjugation lowers the lifetime in nearly all cases.The cruciforms displayed several QY and PL lifetime trends with regards to chalcogen identity, axis of conjugation, and conjugation extent. It is observed that cruciforms with only thiophene-based substituents have a QY between 42–52%. By keeping the conjugation length of each cruciform constant and replacing the chalcogens along the 2,6-axis with oxygen, we see a QY reduction for all compounds. Alternatively, when the chalcogens are replaced along the 4,8-axis, those containing a bifuran unit experience a QY enhancement while those with only furan show a slight reduction. When all chalcogens are replaced with oxygen (apart from 26BF48BF) an increase in the QY and radiative lifetimes is observed. Furan has been shown to have higher fluorescence quantum yields attributed to their more planar structure allowing for better charge transfer as compared to thienyl substituents. While we do not see a generalized pattern of PLQY and molecular structure, the BBOs functionalized with only furanyl groups show the highest quantum yield. Those functionalized with a combination of furanyl and thienyl substituents show intermediate PLQYs and those with only thienyl show the lowest PLQYs.43,48,53 A separate set of trends is observed when conjugation length is altered along a specific axis. For the 26X48X series, cruciforms with only one aryl-substituent type (only furan or thiophene) have the highest quantum yields and could be a result of higher locally excited character. When the conjugation is extended along the 4,8-axis, cruciforms with bifuran are noted to have slightly higher quantum yields while those with bithiophene are reduced. Alternatively, when the conjugation of the 26X48X series is extended through the 2,6-axis, an overall increase is observed in the QY, regardless of the aryl substituent used. However, when only one type of substituent is used, these children have the highest quantum yields of the set. Finally, extending conjugation along both axes simultaneously increases the PLQY for all compounds except for 26BF48BF.
Device properties
Having thoroughly investigated the electrochemical and spectral properties of each BBO, we next used these materials in the active layers of OLEDs to assess their potential for organic emitters. The device characteristics of each BBO were evaluated as dopants (5% w/w) in a mixed-host matrix: ITO (100 nm)/PEDOT:PSS (35 nm)/[1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt/wt] 26DCzPPY:TCTA (5% BBO)/TmPyPB (35 nm)/LiF (1 nm)/Al (100 nm). The device efficiencies and electroluminescent (EL) properties are listed in Table 3. Overall, the efficiencies of each device were poor, indicating a need for improved device architecture. Assuming pure fluorescence and a QY of 100%, the maximum EQE that could be obtained from these devices would be ∼5% when considering the out-coupling factor of OLEDs fabricated on glass substrates. However, in most cases, the devices produced EQEs less than 1%, current efficiencies less than 1.0 cd A−1, and maximum luminous efficacies of 0.7 lm W−1. These observations have been noted and future studies are in place to understand methods of increasing the efficiencies of these materials, which will be reported in due course. While the performance parameters can undoubtedly be improved through extensive optimization, we achieved our goal of obtaining a palette of electroactive luminophores using a BBO core.
:1 wt/wt] 26DCzPPY:TCTA (5% BBO)/TmPyPB (35 nm)/LiF (1 nm)/Al (100 nm). The device efficiencies and electroluminescent (EL) properties are listed in Table 3. Overall, the efficiencies of each device were poor, indicating a need for improved device architecture. Assuming pure fluorescence and a QY of 100%, the maximum EQE that could be obtained from these devices would be ∼5% when considering the out-coupling factor of OLEDs fabricated on glass substrates. However, in most cases, the devices produced EQEs less than 1%, current efficiencies less than 1.0 cd A−1, and maximum luminous efficacies of 0.7 lm W−1. These observations have been noted and future studies are in place to understand methods of increasing the efficiencies of these materials, which will be reported in due course. While the performance parameters can undoubtedly be improved through extensive optimization, we achieved our goal of obtaining a palette of electroactive luminophores using a BBO core.
| BBO | λ EL | CIE (x,y) | V 100 | L max (cd m−2) | CEMax (cd A−1) | PEMax (lm W−1) | EQEMax (%) |
|---|---|---|---|---|---|---|---|
| L = luminance. CE = current efficiency. PE = power efficiency. EQE = external quantum efficiency. Peaks marked with * indicate TCTA emission. | |||||||
| 48F | 433, 411 | 0.16, 0.07 | 8.0 | 174.8 | 0.053 | 0.030 | 0.204 |
| 26F | 437, 418, 394 | 0.16, 0.06 | 7.7 | 171.8 | 0.087 | 0.056 | 0.243 |
| 48T | 440, 417 | 0.16, 0.10 | 5.2 | 293.6 | 0.155 | 0.105 | 0.239 |
| 26T | 433, 410 | 0.16, 0.07 | 4.3 | 277.8 | 0.326 | 0.256 | 0.463 |
| 48BF | 506, 476 | 0.17, 0.37 | 4.6 | 1403 | 0.437 | 0.305 | 0.201 |
| 26BF | 484, 457 | 0.15, 0.23 | 5.6 | 372.0 | 0.117 | 0.067 | 0.182 |
| 48BT | 531, 497 | 0.27, 0.57 | 4.9 | 1953 | 0.272 | 0.109 | 0.173 |
| 26BT | 471, 393* | 0.17, 0.23 | 4.1 | 826.0 | 0.954 | 0.666 | 0.478 |
| 26F48F | 481 | 0.17, 0.33 | 7.1 | 491.4 | 0.094 | 0.038 | 0.074 |
| 26F48T | 496 | 0.18, 0.31 | 4.9 | 762.9 | 0.426 | 0.276 | 0.220 |
| 26T48F | 498 | 0.22, 0.42 | 6.2 | 517.7 | 0.114 | 0.046 | 0.041 |
| 26T48T | 466 | 0.21, 0.44 | 6.0 | 1030 | 0.147 | 0.049 | 0.214 |
| 26F48BF | 517 | 0.33, 0.62 | 5.8 | 931.4 | 0.283 | 0.132 | 0.082 |
| 26F48BT | 524 | 0.35, 0.58 | 5.0 | 1208 | 0.348 | 0.182 | 0.105 |
| 26T48BF | 532 | 0.38, 0.57 | 7.1 | 749.5 | 0.153 | 0.048 | 0.045 |
| 26T48BT | 542 | 0.42, 0.58 | 6.1 | 1194 | 0.148 | 0.053 | 0.061 |
| 26BF48F | 507 | 0.26, 0.55 | 6.0 | 660.6 | 0.278 | 0.159 | 0.250 |
| 26BF48T | 519, 487 | 0.28, 0.53 | 5.5 | 575.2 | 0.206 | 0.109 | 0.067 |
| 26BT48F | 532 | 0.36, 0.60 | 4.8 | 1352 | 0.399 | 0.228 | 0.932 |
| 26BT48T | 549 | 0.38, 0.58 | 6.0 | 923.8 | 0.323 | 0.169 | 0.444 |
| 26BF48BF | 530, 393* | 0.30, 0.39 | 3.9 | 1000 | 1.011 | 0.705 | 0.987 |
| 26BF48BT | 558 | 0.46, 0.54 | 6.6 | 459.0 | 0.056 | 0.021 | 0.063 |
| 26BT48BF | 580 | 0.51, 0.47 | 6.2 | 487.3 | 0.085 | 0.037 | 0.249 |
| 26BT48BT | 600 | 0.55, 0.43 | 4.9 | 654.9 | 0.081 | 0.037 | 0.152 |
In general, the EL of each BBO device more closely matched their solution-state PL emission, with moderate bathochromic shifts due to π–π stacking interactions with host materials. By controlling the conjugation length at each axis, we successfully produced a variety of colored dopants with luminance output useful for display technology in unoptimized devices. For all parent molecules containing either furan or thiophene, these materials produced deep-blue emission (CIEy < 0.10) and luminance between 175–294 nits. Moreover, BBO parents using bifuran and bithiophene provide cyan-to-sea green EL and more intense luminance (372–1953 nits). The cruciform families all provide relatively similar emission profiles due to their similar conjugation, producing luminance between 459–1352 nits. Akin to the bithiophene and bifuran parent molecules, the 26X48X family emits cyan/sea-green light. Extending conjugation through the 4,8-axis (26X48BX) or the 2,6-axis (26BX48X) red-shifts the emission profiles and gives rise to green dopants. When conjugation is extended through both axes (26BX48BX), it forms yellow and orange dopants. Of the 24 dopants, the EL profile of 26BT and 26BF48BF were quite interesting as these devices produced dual-band emission which was not seen in the PL. Due to the poor solubility of each compound, we found that the high-energy emission band is an attribute of the co-host TCTA, thereby providing a greenish-white device. With a variety of electroluminescent dopants available, we combined two BBO dopants (blue and yellow) that would give rise to a functioning, solution-processed white-OLED (WOLED).
From the single-dopant devices, four compounds were observed to give deep blue EL (48F, 26F, 48T, 26T) while three produced yellow-to-orange EL (26BF48BT, 26BT48BF, 26BT48BT). We created a dual-dopant active layer solution by combining volumetric ratios of the solutions used for the single dopant devices. This provided a constant 5% w/w dopant concentration for the active layer and the ability to tune the ratio of the two dopants used. With this control, we hoped to create functional WOLEDs but also adjust the temperature of the light, producing cool (primary blue emission) to warm (primary yellow emission) lighting. From the various combinations, we found that using 48T as the blue dopant and 26BT48BT as the yellow dopant provides the best combinations for white light emission (Fig. 5). We can confirm the high-energy emission in all devices is produced by 48T rather than TCTA as the peak maximum matches the EL of the corresponding single dopant device. At 1:1 (48T:26BT48BT) volumetric ratios, the EL from 48T is not observed, thereby producing a device like the 26BT48BT-only device. Reducing the volume of the 26BF48BT solution to a 3:1 ratio allows the blue emission to be introduced, creating a warm-light WOLED. We can control the color temperature by varying the volumetric ratio of 48T:26BT48BT from 4:1 to 9:1.
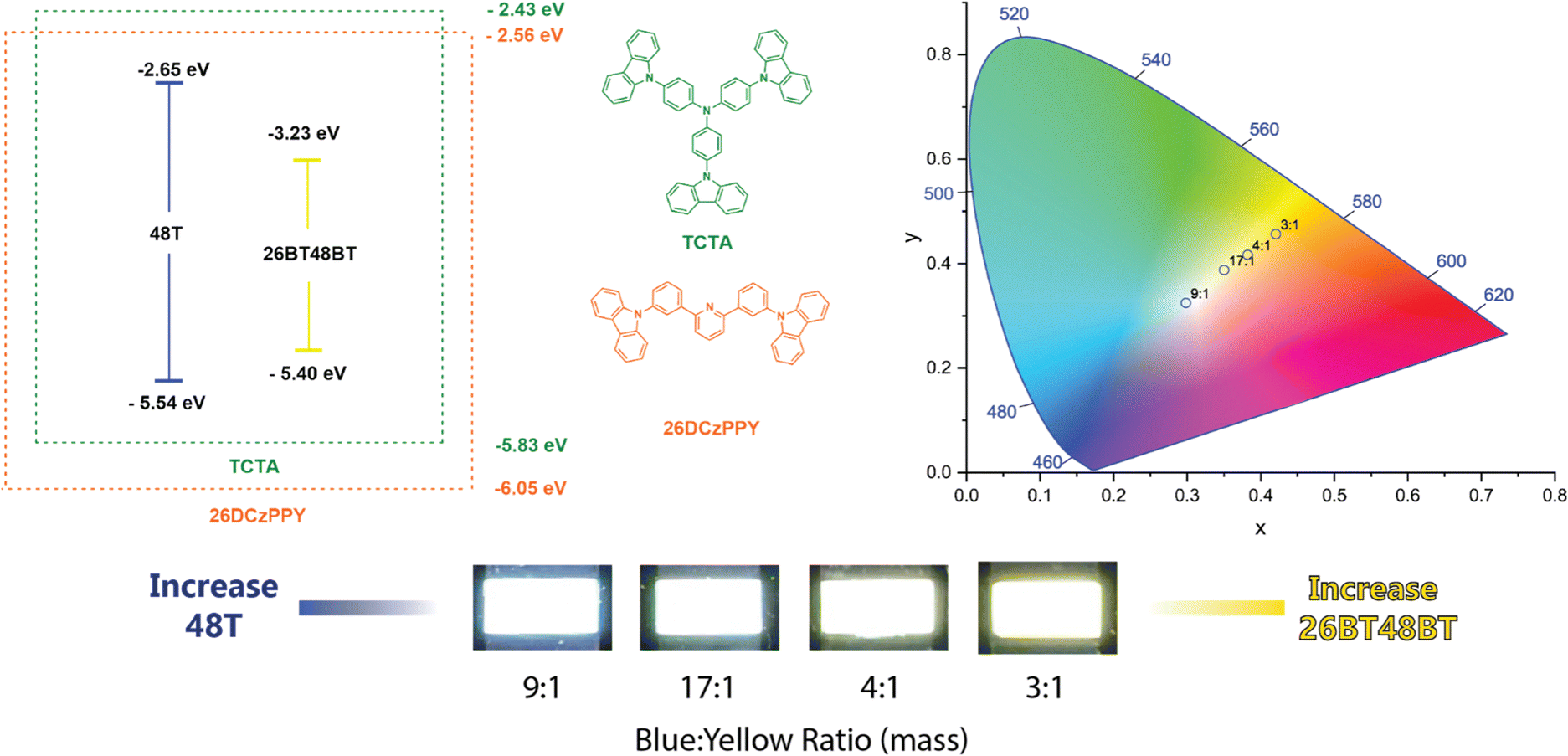 | ||
| Fig. 5 (Left) Band diagram and host molecules used to construct WOLEDs. (Right) CIE 1931 coordinate diagram of WOLED devices. (Bottom) Pictures of WOLED devices with varying volumetric ratios of 48T:26BT48BT. | ||
To further expand on the optimization of the 48T:26BT48BT device, we selected the 4:1 combination as our model system and systematically fabricated devices using a variety of host materials in the active layer, while keeping the fabrication process consistent (Table 4). From our optimization motifs, the most efficient and brightest device uses a 1:1 26DCzPPy:TCTA mixed host system with an EQE of 0.80.%. In general, we can see a clear trend that implementing a dual-ion transfer system introduces higher quantum efficiencies and lower power requirements to activate the device. This is hypothesized to be a contribution of better positive and negative charge injection in the active layer, resulting in higher levels of recombination. This hypothesis is supported when considering the outputs of the hole-only, mixed-host system TAPC:TCTA where this device had the lowest EQE of the set. Of the mixed host systems used, we found that the ambipolar isomers of DCzPPy provided the best performance when paired with TCTA. When replacing 26DCzPPy with 35DCzPPy, the device results in comparable performance for both 1:1 DCzPPy:TCTA ratios. The PE and CE curves show higher efficiencies for the DCzPPy:TCTA host systems and the most stability at higher voltages (Fig. 6b and c). Additionally, the mixed host systems comprising 2,6- or 3,5-DCzPPy and TCTA produced the most temperature neutral white emission based on the CIE coordinates. Recent literature has examples of WOLEDs based on mixing two TADF based emitters, or TADF and phosphorescent emitters.54–56 Consequently, their reported EQE's are generally >10%. Here we describe the generation of white emission by mixing two BBO materials with prompt fluorescence, which by rule will have lower EQEs. Unfortunately, our devices exhibit low EQE performances even for fluorescent materials. We believe that BBO-based materials are a promising and tunable platform for producing EL materials. However, this study indicates that heteroatom substitution was not helpful as these materials underperformed relative to our previously published ones.23
:26BT48BT (4:1) as the dopant (5%). Top part shows individual emitter performance with Host = 1:1:26DCzPPy:TCTA. Bold values indicate emission peak of highest intensity
| BBO in 26DCzPPy:TCTA (1:1) |
V on (V) | V 100 (V) | L max (cd m−2) | EQEmax (%) | CIE (x,y) |
|---|---|---|---|---|---|
| 48T | — | 5.2 | 293.6 | 0.239 | 0.16, 0.10 |
| 26BT48BT | — | 4.9 | 654.9 | 0.152 | 0.55, 0.43 |
| Host system | V on (V) | V 100 (V) | L max (cd m−2) | EQEmax (%) | CIE (x,y) |
|---|---|---|---|---|---|
| 26DCzPPy:TCTA (1:1) |
3.5 | 6.5 | 1537 | 0.8 | 0.29, 0.33 |
| 35DCzPPy:TCTA (1:1) |
4.0 | 6.0 | 1354 | 0.72 | 0.32, 0.36 |
| 26DCzPPy:TAPC (1:1) |
3.5 | 5.5 | 515 | 0.17 | 0.43, 0.46 |
| 35DCzPPy:TAPC (1:1) |
5.0 | 7.0 | 719 | 0.12 | 0.42, 0.44 |
| TAPC:TCTA (1:1) |
3.5 | 5.5 | 485 | 0.06 | 0.34, 0.33 |
| TmPyPb:TCTA (1:1) |
5.0 | 7.0 | 757 | 0.6 | 0.42, 0.44 |
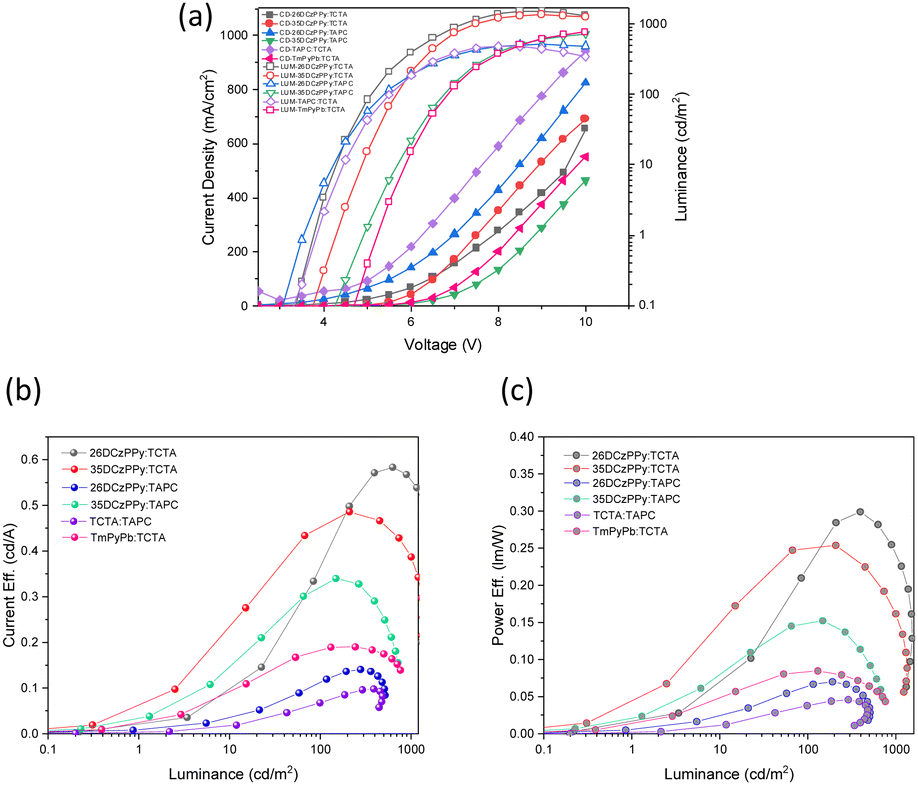 | ||
| Fig. 6 (a) JVL (current density–voltage-luminance) (b) current efficiency vs. luminance (c) power efficiency vs. luminance curves for the different host combinations in the active layer and 5% 48T:26BT48BT (4:1). | ||
Conclusions
In summary, we designed a series of 24 BBO molecules to investigate opto-electronic trends based on aryl substituent identity and conjugation length and assess their application in solution-processed OLEDs. Our results demonstrate that the BBO platform offers a versatile template for constructing OLED dopants of desired EL. The Eoptg of BBO systems can be primarily tuned via conjugation length at each axis to produce desired emission while the HOMO and LUMO levels can be secondarily adjusted via substituent/chalcogen identity at the 4,8-axis and 2,6-axis, respectively. By combining these various trends, a series of materials were created producing violet-to-yellow PL. Due to their good-to-moderate solubilities, each material was used in solution-processed OLEDs. These devices exhibited deep blue-to-orange emission, all achieving luminance values suitable for display technology. Moreover, we were able to create a proof-of-concept WOLED for solid-state lighting application. While the brightness of this prototype did not meet the threshold for practical application, we successfully demonstrated a color temperature tuning motif which can be used for future materials. Future works include extensively optimizing the device architectures needed for each dopant to realize high-efficiency devices, spectroscopically identifying the radiative mechanism during operation, and studying degradation mechanisms that occur. This information will help inform synthetic efforts towards obtaining high-performance BBO dopants.Author contributions
David Wheeler: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, and visualization. Shambhavi Tannir: investigation, writing – revision writing – review & editing. Hakir Yadar: investigation and data curation. Dishi Or: investigation. Ori Gidron: conceptualization, methodology, investigation, resources, writing – original draft, writing – review & editing, visualization and funding acquisition. Malika Jeffries-EL: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, visualization supervision, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation (CHE-1808402) for financial support of this work. We also would like to thank Dr Paul Ralifo and Dr Norman Lee of the Boston University Chemistry Instrumentation Center for NMR and HR-MS access and measurements (respectively), Professor Allison Dennis for fluorimeter/integrating sphere access for quantum yield measurements, Dr Paul Mak for profilometry access. This research was also supported in part by the Israel Science Foundation (grant no. 3085/21).Notes and references
- R. M. Pankow and B. C. Thompson, Polymer, 2020, 207, 122874 CrossRef CAS
.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS PubMed
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed
- H. F. Haneef, A. M. Zeidell and O. D. Jurchescu, J. Mater. Chem. C, 2020, 8, 759–787 RSC
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Tang, B. Lin, K. Xian, B. Gao, C. An, P. Bi, W. Ma and J. Hou, Natl. Sci. Rev., 2020, 7, 1239–1246 CrossRef CAS PubMed
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed
- P. Cheng and Y. Yang, Acc. Chem. Res., 2020, 53, 1218–1228 CrossRef CAS PubMed
- S. Shi, H. Wang, M. A. Uddin, K. Yang, M. Su, L. Bianchi, P. Chen, X. Cheng, H. Guo, S. Zhang, H. Y. Woo and X. Guo, Chem. Mater., 2019, 31, 1808–1817 CrossRef CAS
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed
- D. T. Christiansen, S. Ohtani, Y. Chujo, A. L. Tomlinson and J. R. Reynolds, Chem. Mater., 2019, 31, 6841–6849 CrossRef CAS
- D. T. Christiansen, A. L. Tomlinson and J. R. Reynolds, J. Am. Chem. Soc., 2019, 141, 3859–3862 CrossRef CAS PubMed
- G. Yang, Y.-M. Zhang, Y. Cai, B. Yang, C. Gu and S. X.-A. Zhang, Chem. Soc. Rev., 2020, 49, 8687–8720 RSC
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 1605444, DOI:10.1002/adma.201605444
- J. Han and H.-J. Suk, J. Infect. Dis., 2019, 20, 31–38 CAS
- Z. Xu, B. Z. Tang, Y. Wang and D. Ma, J. Mater. Chem. C, 2020, 8, 2614–2642 RSC
- E. F. Kelley, Inf. Disp., 2013, 29, 6–11 Search PubMed
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed
- M.-T. Lee, C.-L. Wang, C.-S. Chan, C.-C. Fu, C.-Y. Shih, C.-C. Chen, K.-H. Lin, Y.-H. Chen, W.-J. Su, C.-H. Liu, C.-M. Ko, Z.-X. Weng, J.-H. Lin, Y.-C. Chin, C.-Y. Chen, Y.-C. Chang, A. T.-Y. Huang, H.-H. Lu and Y.-H. Lin, J. Soc. Inf. Disp., 2017, 25, 229–239 CrossRef
- R. Pode, Renewable Sustainable Energy Rev., 2020, 133, 110043 CrossRef
- R. Chavez, L. Diodati, D. L. Wheeler, J. Shaw, A. L. Tomlinson and M. Jeffries-El, J. Phys. Chem. A, 2019, 123, 1343–1352 CrossRef CAS PubMed
- R. Chavez III, M. Cai, B. Tlach, D. L. Wheeler, R. Kaudal, A. Tsyrenova, A. L. Tomlinson, R. Shinar, J. Shinar and M. Jeffries-El, J. Mater. Chem. C, 2016, 4, 3765–3773 RSC
- J. J. Intemann, E. S. Hellerich, M. D. Ewan, B. C. Tlach, E. D. Speetzen, R. Shinar, J. Shinar and M. Jeffries-El, J. Mater. Chem. C, 2017, 5, 12839–12847 RSC
- J. J. Intemann, E. S. Hellerich, B. C. Tlach, M. D. Ewan, C. A. Barnes, A. Bhuwalka, M. Cai, J. Shinar, R. Shinar and M. Jeffries-El, Macromolecules, 2012, 45, 6888–6897 CrossRef CAS
- B. C. Tlach, A. L. Tomlinson, A. G. Ryno, D. D. Knoble, D. L. Drochner, K. J. Krager and M. Jeffries-El, J. Org. Chem., 2013, 78, 6570–6581 CrossRef CAS PubMed
- D. L. Wheeler, A. V. Diodati, A. L. Tomlinson and M. Jeffries-El, ACS Omega, 2020, 5, 12374–12384 CrossRef CAS PubMed
- H. Ishikawa, Q. Chen, Y. Bin, K. Komatsu and M. Matsuo, J. Mater. Sci., 2007, 42, 7772–7779 CrossRef CAS
- Y.-H. So, S. J. Martin, B. Bell, C. D. Pfeiffer, R. M. Van Effen, B. L. Romain and S. M. Lefkowitz, Macromolecules, 2003, 36, 4699–4708 CrossRef CAS
- J. F. Wolfe and F. E. Arnold, Macromolecules, 1981, 14, 909–915 CrossRef CAS
- X. Yin, T. Zhang, Q. Peng, T. Zhou, W. Zeng, Z. Zhu, G. Xie, F. Li, D. Ma and C. Yang, J. Mater. Chem. C, 2015, 3, 7589–7596 RSC
- J. A. Osaheni and S. A. Jenekhe, Macromolecules, 1994, 27, 739–742 CrossRef CAS
- Q. Feng, K. Tan, X. Zheng, S. Xie, K. Xue, Y. Bo, H. Zhang, D. Lin, J. Rao, X. Xie, L. Xie, H. Cao, H. Zhang, Y. Wei and W. Huang, ChemPhotoChem, 2020, 4, 321–326 CrossRef CAS
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119–126 CrossRef CAS
- H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef
- W. Che, Y. Xie and Z. Li, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS
- L. Zhang, N. S. Colella, B. P. Cherniawski, S. C. B. Mannsfeld and A. L. Briseno, ACS Appl. Mater. Interfaces, 2014, 6, 5327–5343 CrossRef CAS PubMed
- M. Jeffries-El, B. M. Kobilka and B. J. Hale, Macromolecules, 2014, 47, 7253–7271 CrossRef CAS
- O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS PubMed
- O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov and D. F. Perepichka, Chem. Commun., 2011, 47, 1976–1978 RSC
- O. Gidron, Y. Diskin-Posner and M. Bendikov, J. Am. Chem. Soc., 2010, 132, 2148–2150 CrossRef CAS PubMed
- Y. Qiu, A. Fortney, C.-H. Tsai, M. A. Baker, R. R. Gil, T. Kowalewski and K. J. T. Noonan, ACS Macro Lett., 2016, 5, 332–336 CrossRef CAS PubMed
- Z. Zhao, H. Nie, C. Ge, Y. Cai, Y. Xiong, J. Qi, W. Wu, R. T. K. Kwok, X. Gao, A. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1700005 CrossRef PubMed
- J. Du, A. Fortney, K. E. Washington, M. C. Biewer, T. Kowalewski and M. C. Stefan, J. Mater. Chem. A, 2017, 5, 15591–15600 RSC
- P. Huang, J. Du, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2015, 3, 6244–6257 RSC
- Y. Gao, Z. Wang, J. Zhang, H. Zhang, K. Lu, F. Guo, Z. Wei, Y. Yang, L. Zhao and Y. Zhang, Macromolecules, 2018, 51, 2498–2505 CrossRef CAS
- B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Chem. Sci., 2016, 7, 2201–2206 RSC
- K. R. Idzik, P. J. Cywiński, W. Kuznik, J. Frydel, T. Licha and T. Ratajczyk, Phys. Chem. Chem. Phys., 2015, 17, 22758–22769 RSC
- D. L. Wheeler, L. Fisher, P. Friederich, C. Cunningham, A. K. Muthike, A. Aspuru-Guzik, T. Goodson and M. Jeffries-El, J. Mater. Chem. C, 2023, 11, 211–222 RSC
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC
- D. Wheeler, S. Tannir, E. Smith, A. Tomlinson and M. Jeffries-El, Mater. Adv., 2022, 3, 3842–3852 RSC
- J. Lim, T. A. Albright, B. R. Martin and O. Š. Miljanić, J. Org. Chem., 2011, 76, 10207–10219 CrossRef CAS PubMed
- A. E. Steen, T. L. Ellington, S. T. Nguyen, S. Balasubramaniam, I. Chandrasiri, J. H. Delcamp, G. S. Tschumper, N. I. Hammer and D. L. Watkins, J. Phys. Chem. C, 2019, 123, 15176–15185 CrossRef CAS
- S.-H. Yang and T.-L. Huang, Opt. Mater., 2021, 111, 110725 CrossRef CAS
- J. Zhao, Z. Yang, X. Chen, Z. Xie, T. Liu, Z. Chi, Z. Yang, Y. Zhang, M. P. Aldred and Z. Chi, J. Mater. Chem. C, 2018, 6, 4257–4264 RSC
- Z. Chen, X.-K. Liu, C.-J. Zheng, J. Ye, C.-L. Liu, F. Li, X.-M. Ou, C.-S. Lee and X.-H. Zhang, Chem. Mater., 2015, 27, 5206–5211 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01154b |
| This journal is © The Royal Society of Chemistry 2024 |