
Synthesis of functionalized γ-lactones via a three-component cascade reaction catalyzed by consecutive N-heterocyclic carbene systems†
Qian Zhao‡
a,
Bo Han‡b,
Biao Wanga,
Hai-Jun Lengb,
Cheng Peng*ab and
Wei Huang*a
aMinistry of Education Key Laboratory of Standardization of Chinese Medicine, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, P. R. China. E-mail: huangwei@cdutcm.edu.cn; Fax: +86 28 61800234
bState Key Laboratory Breeding Base of Systematic Research, Development and Utilization of Chinese Medicine Resources, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, P. R. China. E-mail: pengcheng@cdutcm.edu.cn; Fax: +86 28 61800231
First published on 6th March 2015
Abstract
Two consecutive N-heterocyclic carbene (NHC) catalytic systems were combined in a one-pot cascade reaction for the assembly of aromatic aldehydes and 2-haloenals into a structurally complex γ-lactone backbone. To our knowledge, this is the first report of NHC-catalyzed [3 + 2] annulation of α,β-unsaturated acylazoliums with 1,2-bisnucleophiles.
N-Heterocyclic carbene (NHC) catalysis, an elegant organocatalytic method for forming new bonds, is used broadly in organic synthesis.1 A number of NHC catalytic multicomponent cascade reactions have been developed, but most rely on a single catalyst, to perform continuous operations.2
Combining two catalytic bond-forming tactics into a single cascade reaction can offer unique synthetic opportunities, as it can allow unprecedented transformations that would be impossible in the presence of either catalyst on its own. NHC catalysis has been successfully integrated with transition metal catalysis,3 Lewis acid catalysis4 or aminocatalysis.5 These combinations have opened up synthetic possibilities, but they are severely limited by self-quenching between NHCs and transition metals/Lewis acids and by mutual interference between NHCs and the acidic additive in aminocatalytic systems. Over the past decade, NHC-mediated HOMO/LUMO activation of carbonyl compounds at the ipso-, α-, β- or γ-position has become well established; these carbonyl compounds include aldehydes, ketones and esters, as well as their α,β unsaturated variants.1 Despite these advances, few studies have explored the combination of two NHC-mediated activation modes in one multicomponent cascade reaction in a way that takes advantage of the inherent basicity and compatibility of both catalysts.
An efficient NHC-catalyzed activation mode that recently received immense attention is the generation of α,β-unsaturated acylazolium, which may be achieved from α,β-unsaturated acyl fluorides, enol esters, enals (under oxidative conditions), ynals, or 2-haloenals.6–14 This reactive intermediate may serve as an analogue of 1,3-biselectrophiles and form the basis for novel domino reactions. In fact, the groups of Lupton,6 Bode,7 Studer,8 Biju,9 You,10 Ye,11 Yao,12 Chi13 and Xiao14 have independently described NHC-catalyzed [3 + 3] annulation of α,β-unsaturated acylazoliums with 1,3-bisnucleophiles, leading to the formation of dihydropyranone or dihydropyridinone derivatives (Scheme 1, eqn (a)). To the best of our knowledge, the analogous [3 + 2] cycloaddition with 1,2-bisnucleophiles to synthesize five-membered heterocycles has not been reported (Scheme 1, eqn (b)).
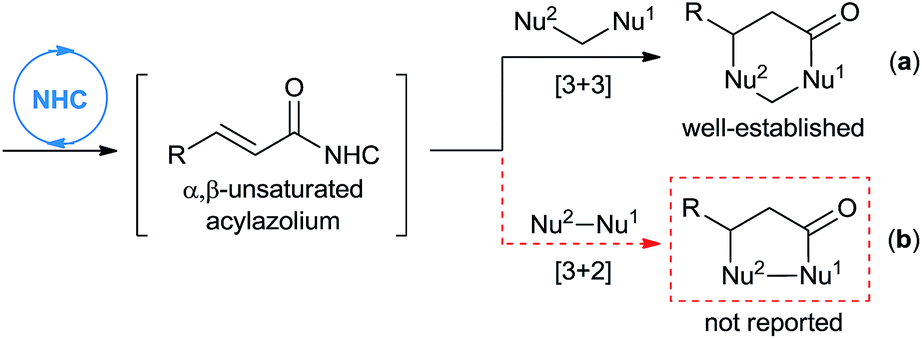 | ||
| Scheme 1 Annulation based on NHC-catalyzed generation of α,β-unsaturated acylazoliums. | ||
As part of our ongoing investigations aimed at assembling multiple substrates into synthetically important cyclic molecules,15 we envisaged a two-step organocatalytic relay cascade, beginning with NHC-catalyzed benzoin condensation,16 as a way to synthesize functionalized γ-lactones (Scheme 2). Condensation of aldehyde 1 generates an α-hydroxyketone intermediate 2, which then serves as the 1,2-bisnucleophile in the second NHC catalytic cycle. Subsequent [3 + 2] cyclization between the NHC-generated α,β-unsaturated acylazolium with α-hydroxyketone affords the desired γ-lactone 4. This approach, if successful, would provide an alternative method for preparing pharmacologically interesting γ-lactones or dihydrofuranones,17 and it would broaden the applications of sequential dual catalysis.18
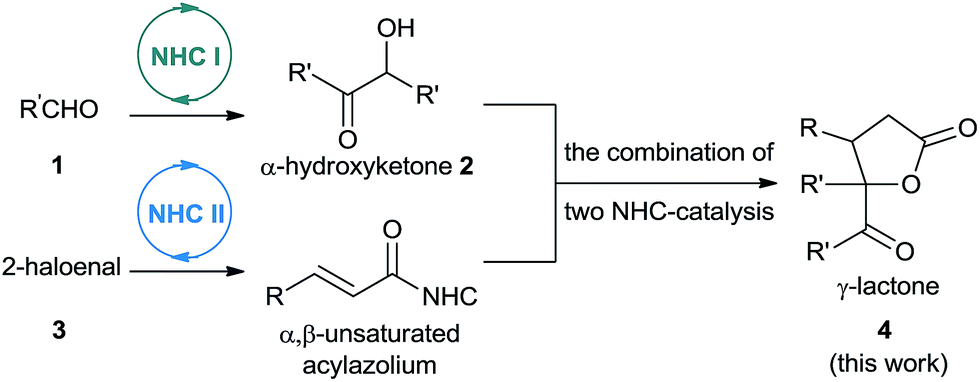 | ||
| Scheme 2 Combination of two NHC catalytic cycles to construct γ-lactone bearing a quaternary carbon center. | ||
At the very first beginning, we wondered whether NHC-catalyzed [3 + 2] annulation of α,β-unsaturated acylazoliums with 1,2-bisnucleophiles could work. So we investigated the reaction of benzoin 2a and α-bromo cinnamaldehyde 3a in the presence of NHC precursor with cesium carbonate. Fortunately, we were able to isolate the desired 4a product in moderate yield (Scheme 3). Since NHCs are known to catalyze benzoin condensation, we also wondered whether benzoin 2a could be generated in situ. If successful, two sequential NHC catalytic systems could be combined.
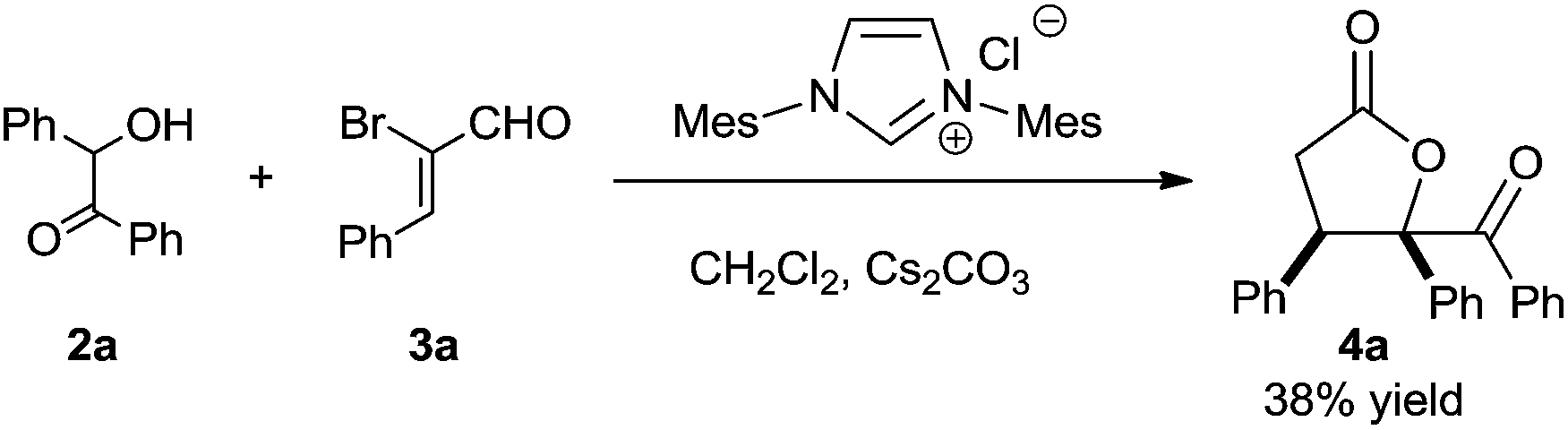 | ||
| Scheme 3 NHC-catalyzed [3 + 2] annulation of benzoin and 2-bromoenal. | ||
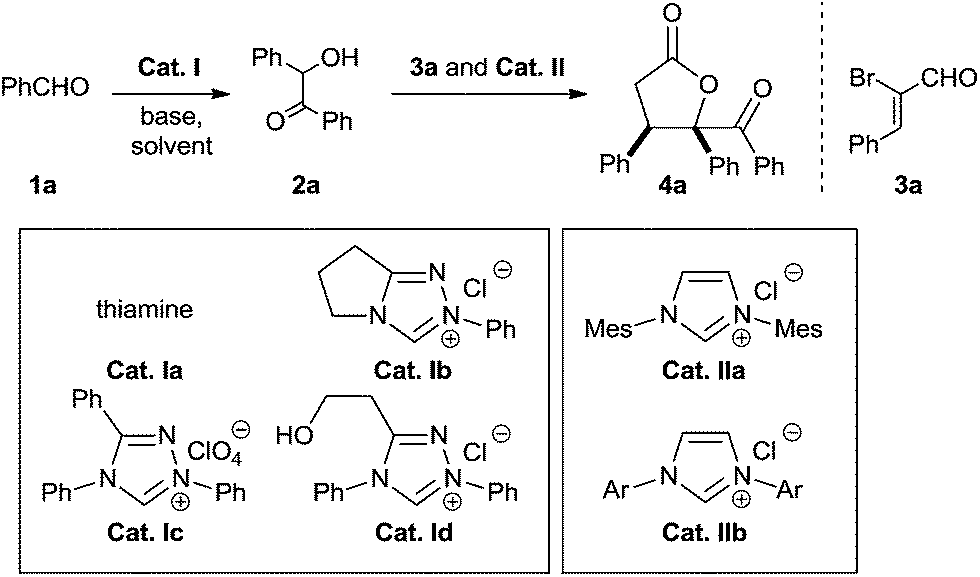
To probe the feasibility of the proposed two-step cascade process, the preliminary experiment was carried out with precatalyst Ia and benzaldehyde 1a to afford benzoin 2a in situ, after which precatalyst IIa and α-bromocinnamic aldehyde 3a were added in the reaction mixture. To our delight, by performing the reaction in dichloromethane with Cs2CO3, we were able to isolate the desired product with good diastereoselectivity, albeit in moderate yield (Table 1, entry 1). Screening various catalyst combinations (entries 1–5) showed Id and IIa to be the most promising catalyst pair for the reaction (entry 4). Different bases led to different yields, with DBU giving the best results (entries 6–10). Replacing dichloromethane with other solvents did not improve yield or diastereoselectivity (entries 10–13). We wondered whether one NHC catalyst could be used in both catalytic cycles. Performing the cascade reaction in the absence of precatalyst Id furnished only a trace amount of the desired product (entry 14), as did the reaction with only precatalyst Id (entry 15). At the same time, α-hydroxyketone 2a was detected by 1H NMR and HRMS of the crude reaction mixture (entry 15). These results suggest that different NHC catalysts are required in the two catalytic systems.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. I | Cat. II | Base | Solvent | Yieldb (%) | drc |
| a Unless otherwise noted, reactions were performed with precatalyst I (0.1 mmol), base (0.3 mmol) and 1a (1.6 mmol) in solvent (2 mL) at 50 °C for a specified reaction time until the sufficient benzoin 2a was generated (monitored by TLC), after which precatalyst II (0.1 mmol) and 3a (0.4 mmol) were added.b Isolated yield of pure diastereomer 4a.c Based on 1H NMR analysis of the crude reaction mixture. Mes = 2,4,6-(CH3)3C6H2; Ar = 2,6-(CH3CHCH3)2C6H3. | ||||||
| 1 | Ia | IIa | Cs2CO3 | CH2Cl2 | 41 | 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 :15 |
| 2 | Ib | IIa | Cs2CO3 | CH2Cl2 | 37 | 80:20 |
| 3 | Ic | IIa | Cs2CO3 | CH2Cl2 | 49 | 85:15 |
| 4 | Id | IIa | Cs2CO3 | CH2Cl2 | 59 | 88:12 |
| 5 | Id | IIb | Cs2CO3 | CH2Cl2 | 52 | 88:12 |
| 6 | Id | IIa | K2CO3 | CH2Cl2 | 59 | 86:14 |
| 7 | Id | IIa | tBuOK | CH2Cl2 | 54 | 78:22 |
| 8 | Id | IIa | TEA | CH2Cl2 | 39 | 85:15 |
| 9 | Id | IIa | DABCO | CH2Cl2 | 46 | 82:18 |
| 10 | Id | IIa | DBU | CH2Cl2 | 64 | 90:10 |
| 11 | Id | IIa | DBU | Toluene | 48 | 85:15 |
| 12 | Id | IIa | DBU | MeCN | 59 | 86:14 |
| 13 | Id | IIa | DBU | THF | 34 | 80:20 |
| 14 | IIa | IIa | DBU | CH2Cl2 | <10 | n.d. |
| 15 | Id | Id | DBU | CH2Cl2 | <10 | n.d. |
With the optimal reaction conditions in hand, we explored the substrate scope of the procedure (Table 2). Annulation of α-bromo cinnamaldehyde 3a proceeded smoothly with a variety of aldehydes 1 to give moderate to high yield with good diastereoselectivity (entries 1–10). Most substituted aromatic aldehydes 1 bearing electron-withdrawing groups led to higher yield than those possessing electron-donating groups, and dr values for products varied in the trend ortho > meta and para. This implies that the substitution pattern on the phenyl rings affects the reaction to some extent. Heteroaryl aldehydes participated efficiently, giving even higher yields than aryl aldehydes but with slightly lower dr values (entries 9 and 10). We also evaluated the use of aliphatic aldehydes (such as octanal and 3-phenylpropanal) in the optimal reaction conditions, but no [3 + 2] annulation products were obtained.19 To extend the scope of the reaction further, we explored other 2-bromoenals 3. The position and electronic properties of substituents on the aromatic ring of aryl-substituted 2-bromoenals did not significantly affect reaction efficiency (entries 11–19). The transformation was rather sluggish in the case of 2-bromo-3-furanylacrylaldehyde, and the corresponding γ-butyrolactone was afforded in only 35% yield after prolonged reaction time (entry 20). The relative configuration of 4a was determined by X-ray crystallographic analysis.20 Configurations of the other dihydrofuranones were tentatively assigned by analogy.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yieldb (%) | drc |
| a See entry 13 and footnote a in Table 1.b Isolated yield of pure diastereomer 4.c Based on 1H NMR analysis of the crude reaction mixture. | |||||
| 1 | Ph | Ph | 4a | 64 | 90:10 |
| 2 | 2-ClC6H4 | Ph | 4b | 65 | 95:5 |
| 3 | 4-ClC6H4 | Ph | 4c | 68 | 88:15 |
| 4 | 3-BrC6H4 | Ph | 4d | 65 | 92:8 |
| 5 | 4-BrC6H4 | Ph | 4e | 70 | 85:15 |
| 6 | 4-FC6H4 | Ph | 4f | 65 | 87:13 |
| 7 | 2,4-Cl2C6H3 | Ph | 4g | 70 | 92:8 |
| 8 | 4-i-PrC6H4 | Ph | 4h | 54 | 86:14 |
| 9 | 2-Furyl | Ph | 4i | 72 | 80:20 |
| 10 | 2-Thienyl | Ph | 4j | 71 | 75:25 |
| 11 | Ph | 2-ClC6H4 | 4k | 60 | 92:8 |
| 12 | Ph | 4-ClC6H4 | 4l | 61 | 86:14 |
| 13 | Ph | 4-BrC6H4 | 4m | 66 | 85:15 |
| 14 | Ph | 2-FC6H4 | 4n | 57 | 90:10 |
| 15 | Ph | 3-FC6H4 | 4o | 61 | 85:15 |
| 16 | Ph | 4-FC6H4 | 4p | 68 | 82:18 |
| 17 | Ph | 4-NO2C6H4 | 4q | 63 | 84:16 |
| 18 | Ph | 4-MeC6H4 | 4r | 54 | 80:20 |
| 19 | Ph | 2-OCH3C6H4 | 4s | 50 | 87:13 |
| 20 | Ph | 2-Furyl | 4t | 35 | 80:20 |
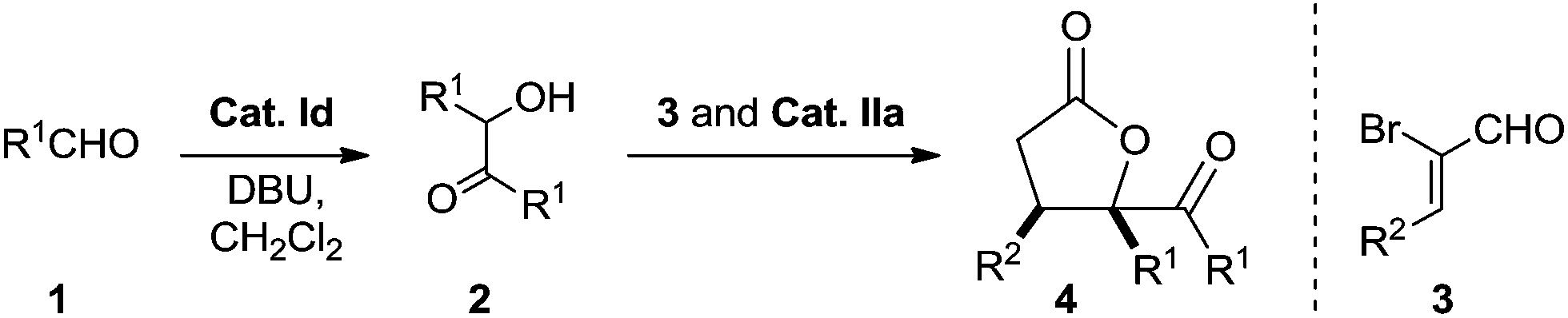
We also performed experiments on the asymmetric version of this [3 + 2] annulation (Scheme 4). We envisioned that treatment of benzoin 2a and 2-bromoenal 3a in the presence of chiral triazolium salts IIc and IId, which has proven effective in many asymmetric [3 + 3] cycloadditions involving activation of α,β-unsaturated acylazolium, might furnish the desired chiral product. Unfortunately, numerous attempts were unsuccessful, with only the α,β-unsaturated ester product 5a obtained in nearly quantitative yield. This linear product presumably formed via direct intermolecular O-acylation of the benzoin to the chiral α,β-unsaturated acylazolium intermediate (Scheme 4). Notably, the final product 5a was a racemic mixture, suggesting no kinetic or dynamic kinetic resolution during the conversion.
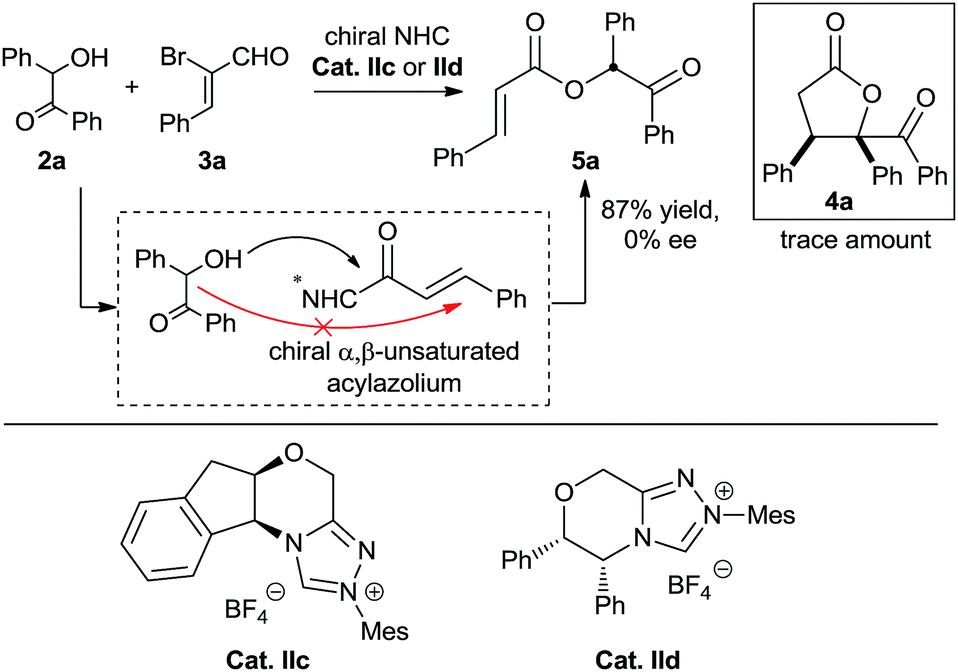 | ||
| Scheme 4 Studies on the enantioselectivity of the annulation process. | ||
Based on these results, we were puzzled by what the reaction mechanism might be. We reasoned that the γ-butyrolactone might form either by tandem 1,4-addition followed by intramolecular acylation (Scheme 5, path A) or by tandem intermolecular acylation followed by 2,3-wittig rearrangement-acylation (Scheme 4, path B). Importantly, the product 5a (generated in Scheme 4) did not undergo cyclization under various alkaline conditions,21 suggesting that pathway A is more reasonable to describe this α,β-unsaturated acylazolium-mediated [3 + 2] annulation.
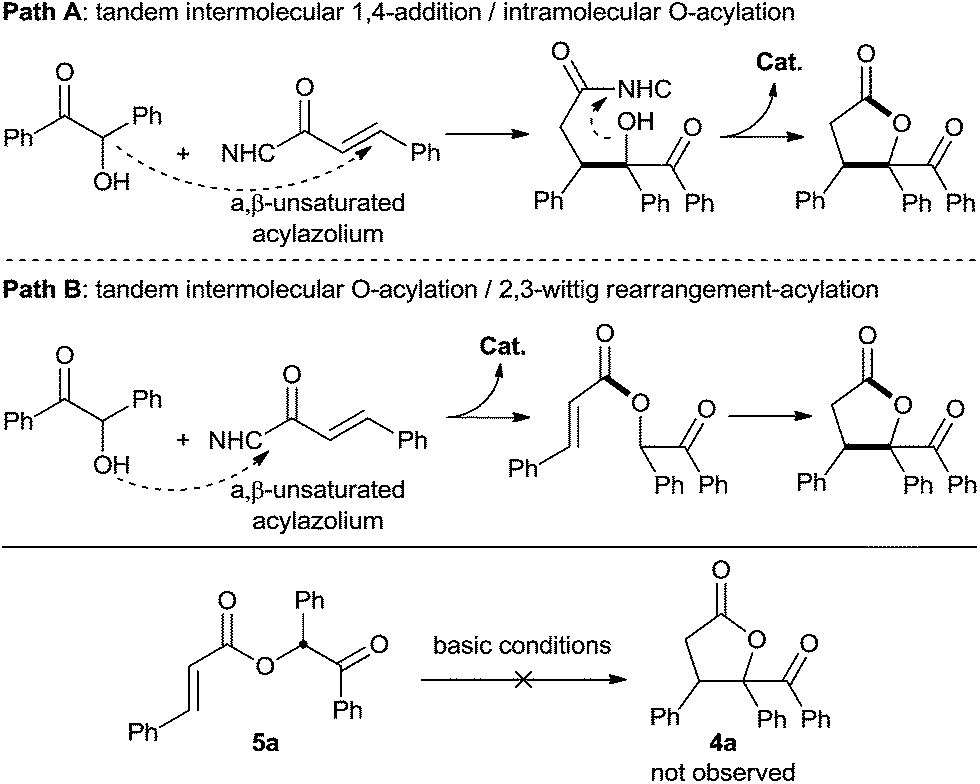 | ||
| Scheme 5 Two possible pathways for [3 + 2] annulation mediated by α,β-unsaturated acylazolium. | ||
Conclusions
In summary, we have developed a flexible and simple organocatalytic cascade reaction involving two sequential NHC catalytic cycles, and we have used it to assemble a functionalized γ-lactone (or dihydrofuranone) scaffold bearing a quaternary carbon center. Starting from aromatic aldehydes and 2-haloenals, we obtained the desired products in moderate to good yield with high diastereoselectivity. The present work is the first report of NHC-catalyzed [3 + 2] annulation of α,β-unsaturated acylazoliums with 1,2-bisnucleophiles. Studies are under way in our laboratory to clarify the detailed reaction mechanism and explore whether this method can be applied to asymmetric domino reactions.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21302016 and 81303208), the Science & Technology Department of Sichuan Province (2014JQ0020) and the Foundation for the Author of National Excellent Doctoral Dissertation of PR China.Notes and references
- For recent reviews on NHC catalysis, see: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC; (c) E. M. Phillips, A. Chan and K. A. Scheidt, Aldrichimica Acta, 2009, 42, 55 CAS; (d) J. L. Moore and T. Rovis, Top. Curr. Chem., 2010, 291, 77 CrossRef CAS; (e) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182 CrossRef CAS PubMed; (f) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336 RSC; (g) H. U. Vora and T. Rovis, Aldrichimica Acta, 2011, 44, 3 CAS; (h) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686 CrossRef CAS PubMed; (i) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617 CrossRef CAS PubMed; (j) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC; (k) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC; (l) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC; (m) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696 CrossRef CAS PubMed; (n) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- For a comprehensive review, see: A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS PubMed.
- (a) K. Hirano, I. Piel and F. Glorius, Chem. Lett., 2011, 40, 786 CrossRef CAS; (b) N. T. Patil, Angew. Chem., Int. Ed., 2011, 50, 1759 CrossRef CAS PubMed.
- D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53 RSC.
- (a) S. P. Lathrop and T. Rovis, J. Am. Chem. Soc., 2009, 131, 13628 CrossRef CAS PubMed; (b) B.-C. Hong, N. S. Dange, C.-S. Hsu and J.-H. Liao, Org. Lett., 2010, 12, 4812 CrossRef CAS PubMed; (c) B.-C. Hong, N. S. Dange, C.-S. Hsu, J.-H. Liao and G.-H. Lee, Org. Lett., 2011, 13, 1338 CrossRef CAS PubMed; (d) K. E. Ozboya and T. Rovis, Chem. Sci., 2011, 2, 1835 RSC; (e) C. B. Jacobsen, K. L. Jensen, J. Udmark and K. A. Jørgensen, Org. Lett., 2011, 13, 4790 CrossRef CAS PubMed; (f) D. Enders, A. Grossmann, H. Huang and G. Raabe, Eur. J. Org. Chem., 2011, 4298 CrossRef CAS; (g) Y.-Z. Liu, J. Zhang, P.-F. Xu and Y.-C. Luo, J. Org. Chem., 2011, 76, 7551 CrossRef CAS PubMed; (h) Y. Liu, M. Nappi, E. C. Escudero-Adán and P. Melchiorre, Org. Lett., 2012, 14, 1310 CrossRef CAS PubMed; (i) G. He, F. Wu, W. Huang, R. Zhou, L. Ouyang and B. Han, Adv. Synth. Catal., 2014, 356, 2311 CrossRef CAS.
- (a) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176 CrossRef CAS PubMed; (b) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2011, 133, 4694 CrossRef CAS PubMed; (c) L. Candisha and D. W. Lupton, J. Am. Chem. Soc., 2013, 135, 58 CrossRef PubMed; (d) L. Candish, C. M. Forsyth and D. W. Lupton, Angew. Chem., Int. Ed., 2013, 52, 9149 CrossRef CAS PubMed.
- (a) A. G. Kravina, J. Mahatthananchai and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9433 CrossRef CAS PubMed; (b) J. Mahatthananchai, J. Kaeobamrung and J. W. Bode, ACS Catal., 2012, 2, 494 CrossRef CAS PubMed; (c) E. Lyngvi, J. W. Bode and F. Schoenebeck, Chem. Sci., 2012, 3, 2346 RSC; (d) J. Mahatthananchai, P. Zheng and J. W. Bode, Angew. Chem., Int. Ed., 2011, 50, 1673 CrossRef CAS PubMed; (e) B. Wanner, J. Mahatthananchai and J. W. Bode, Org. Lett., 2011, 13, 5378 CrossRef CAS PubMed; (f) J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810 CrossRef CAS PubMed.
- (a) R. C. Samanta, S. De Sarkar, R. Frøhlich, S. Grimme and A. Studer, Chem. Sci., 2013, 4, 2177 RSC; (b) R. C. Samanta, B. Maji, S. De Sarkar, K. Bergander, R. Fröhlich, C. Mück-Lichtenfeld, H. Mayr and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 5234 CrossRef CAS PubMed; (c) A. Biswas, S. De Sarkar, R. Fröhlich and A. Studer, Org. Lett., 2011, 13, 4966 CrossRef CAS PubMed; (d) S. De Sarkar and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 9266 CrossRef CAS PubMed.
- (a) S. R. Yetra, T. Kaicharla, S. S. Kunte, R. G. Gonnade and A. T. Biju, Org. Lett., 2013, 15, 5202 CrossRef CAS PubMed; (b) S. R. Yetra, A. Bhunia, A. Patra, M. V. Mane, K. Vanka and A. T. Biju, Adv. Synth. Catal., 2013, 355, 1089 CrossRef CAS; (c) S. R. Yetra, T. Roy, A. Bhunia, D. Porwal and A. T. Biju, J. Org. Chem., 2014, 79, 4245 CrossRef CAS PubMed.
- Z.-Q. Rong, M.-Q. Jia and S.-L. You, Org. Lett., 2011, 13, 4080 CrossRef CAS PubMed.
- F.-G. Sun, L.-H. Sun and S. Ye, Adv. Synth. Catal., 2011, 353, 3134 CrossRef CAS.
- (a) C. Yao, W. Jiao, Z. Xiao, R. Liu, T. Li and C. Yu, Tetrahedron, 2013, 69, 1133 CrossRef CAS PubMed; (b) C. Yao, D. Wang, J. Lu, T. Li, W. Jiao and C. Yu, Chem.–Eur. J., 2012, 18, 1914 CrossRef CAS PubMed.
- J. Cheng, Z. Huang and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 8592 CrossRef CAS PubMed.
- (a) Z.-Q. Zhu, X.-L. Zheng, N.-F. Jiang, X. Wan and J.-C. Xiao, Chem. Commun., 2011, 47, 8670 RSC; (b) Z.-Q. Zhu and J.-C. Xiao, Adv. Synth. Catal., 2010, 352, 2455 CrossRef CAS.
- (a) X. Xie, C. Peng, G. He, H.-J. Leng, B. Wang, W. Huang and B. Han, Chem. Commun., 2012, 48, 10487 RSC; (b) X. Li, L. Yang, C. Peng, X. Xie, H.-J. Leng, B. Wang, Z.-W. Tang, G. He, L. Ouyang, W. Huang and B. Han, Chem. Commun., 2013, 49, 8692 RSC; (c) B. Han, X. Xie, W. Huang, X. Li, L. Yang and C. Peng, Adv. Synth. Catal., 2014, 356, 3676 CrossRef CAS.
- For selected examples, see: (a) Y. Shimakawa, T. Morikawa and S. Sakaguchi, Tetrahedron Lett., 2010, 51, 1786 CrossRef CAS PubMed; (b) D. Enders and A. Henseler, Adv. Synth. Catal., 2009, 351, 1749 CrossRef CAS.
- Several groups have reported the synthesis of the analogous γ-lactone derivatives via benzoin-triggered multicomponent tandem reactions, see: (a) O. Winkelmann, C. Näther and U. Lüning, Org. Biomol. Chem., 2009, 7, 553 RSC; (b) M. Yoshida, N. Terai and K. Shishido, Tetrahedron, 2010, 66, 8922 CrossRef CAS PubMed; (c) W. Ye, G. Cai, Z. Zhuang, X. Jia and H. Zhai, Org. Lett., 2005, 7, 3769 CrossRef CAS PubMed.
- For recent reviews on dual catalysis, see: (a) C. C. J. Loh and D. Enders, Chem.–Eur. J., 2012, 18, 10212 CrossRef CAS PubMed; (b) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337 RSC; (c) M. Rueping, R. M. Koenigs and I. Atodiresei, Chem.–Eur. J., 2010, 16, 9350 CrossRef CAS PubMed; (d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (e) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (f) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef PubMed; (g) J. Zhou, Chem.–Asian J., 2010, 5, 422 CrossRef CAS PubMed; (h) S. Piovesana, D. M. ScarpinoSchietroma and M. Bella, Angew. Chem., Int. Ed., 2011, 50, 6216 CrossRef CAS PubMed; (i) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (j) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211 RSC; (k) H. Pellissier, Tetrahedron, 2013, 69, 7171 CrossRef CAS PubMed.
- The mixture of the aliphatic α-hydroxyketone and the aldol condensation product could be detected.
- ESI.†.
- We screened Cs2CO3, K2CO3, tBuOK, TEA, DABCO and DBU.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for new compounds. CCDC 958885. For ESI and crystallographic data in CIF or other electronic formats see: DOI: 10.1039/c5ra01254f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |