
Efficient as-cast thick film small-molecule organic solar cell with less fluorination on the donor†
Kai
Wang‡
a,
Xin
Song‡
 b,
Xiao
Guo
a,
Yunhao
Wang
a,
Xue
Lai
ac,
Fei
Meng
ac,
Mengzhen
Du
a,
Dongyu
Fan
a,
Ren
Zhang
a,
Gongqiang
Li
*a,
Aung Ko Ko
Kyaw
*c,
Jianpu
Wang
a,
Wei
Huang
ad and
Derya
Baran
*b
b,
Xiao
Guo
a,
Yunhao
Wang
a,
Xue
Lai
ac,
Fei
Meng
ac,
Mengzhen
Du
a,
Dongyu
Fan
a,
Ren
Zhang
a,
Gongqiang
Li
*a,
Aung Ko Ko
Kyaw
*c,
Jianpu
Wang
a,
Wei
Huang
ad and
Derya
Baran
*b
aKey Laboratory of Flexible Electronic (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergistic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211816, P. R. China. E-mail: iamgqli@njtech.edu.cn
bKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Physical Sciences and Engineering Division (PSE), Thuwal, 23955-6900, Saudi Arabia. E-mail: derya.baran@kaust.edu.sa
cGuangdong University Key Laboratory for Advanced Quantum Dot Displays and Lighting, Shenzhen Key Laboratory for Advanced Quantum Dot Displays and Lighting, and Department of Electrical & Electronic Engineering, Southern University of Science and Technology, Shenzhen 518055, P. R. China. E-mail: aung@sustech.edu.cn
dShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), 127 West Youyi Road, Xi’an 710072, China
First published on 14th November 2019
Abstract
Herein, two donor materials based on fluorine substituted isatin units (DI3T-1F, DI3T-2F) were designed and synthesized for thick-film small molecule solar cells. The devices based on DI3T-1F demonstrate balanced charge transport and less trap-assisted recombination, leading to higher power conversion efficiency (PCE) of 8.33% with a thickness ca. 150 nm compared to DI3F-2F (PCE of 7.32% with thick film at ca. 160 nm) without additives, solvent or thermal annealing treatments. More interestingly, the device based on DI3T-1F small molecules demonstrates good tolerance to active layer thickness from 150 to 300 nm with device performance over 5%. Our results indicate that less fluorine atoms on the donor units can optimize the charge transport, and phase-segregated morphology in small molecule solar cells without the need for post-treatment.
Introduction
Organic solar cells (OSCs) have attracted significant attention owing to their advantages of light weight, mechanical flexibility and large-area solution processibility.1–4 In the past several years, the OSC field has witnessed much progress with high power conversion efficiencies (PCEs) of over 16%5 achieved in a single-junction configuration with a polymer donor on a laboratory scale. Yet, some of the drawbacks of polymer donors still limit the reproducible scale-up, such as batch-to-batch variation and material purification. Compared with their polymer counterparts, all small molecule solar cells (SMSCs) show more potential for industrialization because of their well-defined molecule structures, reproducible synthesis, and easy purification.6–10 With the continuous endeavors of molecule design and synthesis, the PCEs of SMSCs are reported to be over 12%,11,12 with the blend of fused-ring electron acceptors.Although significant progress has been achieved in SMSCs, blend morphology optimization methods such as thermal annealing,13–16 solvent annealing16–20 or trace amount of solvent additive21–24 are still essential to improve efficiencies, because of the control of aggregation. These tedious methods would induce extremely low device reproducibility as it is challenging to finely control the morphologies of the film within a few seconds. The thickness of the active layer of SMSCs typically lies below 100 nm25 (normally 60–80 nm), which is unfavorable for large-area fabrication technologies, such as roll-to-roll coating, and printing techniques.26 In contrast, thick-film polymer solar cells have been widely explored by several strategies. These include: (1) enhancing the molecular crystallinity by extending the conjugated backbone;27,28 (2) adding a third component;29 and (3) tuning the dipole monents and steric hindrance of polymers or non-fullerene acceptors.30–32 Inspired by these strategies to improve the performance of polymer-containing devices, we seek to employ such methods for all-small-molecule solar cells.
In 2018, our group developed a small molecule donor DI3T-2F with di-fluorine substituted isatin (1H-indole-2,3-dione) as a peripheral unit,33 and the as-cast devices based on the DI3T-2F:PC71BM blend obtained a champion performance of 7.8% with a thick active layer (∼150 nm). However, thicker DI3T-2F based devices deliver only a moderate PCE (lower than 4%) due to charge recombination issues in the thick films. Another problem with DI3T-2F is the too strong intermolecular interaction because of the formation of F⋯H, F⋯S, and F⋯O bonds, which results in its low miscibility with fullerene derivatives (PC71BM) and further coarse blend morphology in thick film devices. So decreasing the numbers of fluorine atoms on the peripheral isatin units can be adopted as a promising strategy to reduce the excessive donor–acceptor intermolecular interactions to optimize the morphology and thus give better performance.
Thus, we synthesized DI3T-1F (Scheme 1), and investigated the comparison to DI3T-2F based on DI3T-1F:PC71BM and DI3T-2F:PC71BM devices. As a result, the PCE of devices based on DI3T-1F was 8.33% without any pre or post device treatment, which represented a 14% improvement compared with that of the devices based on DI3T-2F (7.32%). More interestingly, after a systematic study, DI3T-1F featured more balanced charge transport and suppressed trap-assisted recombination behavior compared to that of DI3T-2F devices, and the DI3T-1F devices yielded a champion PCE of 7.8% with an active layer thickness of 200 nm, and even with a thickness of 300 nm, the PCE is still as high as 5.43%. These obvious differences manifest that our mono-fluorine strategy provides a fascinating approach for the construction of all-small-molecule solar cells with as-cast thick films.
 | ||
| Scheme 1 Structures of DI3T-1F and DI3T-2F. | ||
Results and discussion
Synthesis and material properties
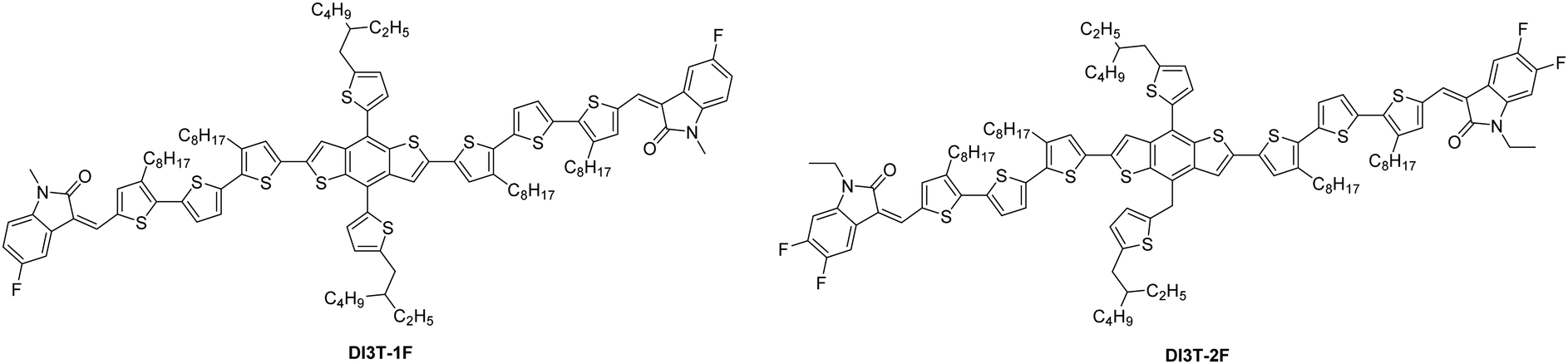
DI3T-1F and DI3T-2F were synthesized via Knoevenagel condensation from the dialdehyde intermediate 3TBDTT33 and the corresponding commercially available fluorination substituted oxindole derivatives in good yields as shown in Scheme S1 and the detailed synthesis procedure is described in the ESI.† Both structures were determined by NMR (Fig. S7 and S8, ESI†). As expected, DI3T-1F exhibits a much better solubility than DI3T-2F in common organic solvents (dichloromethane and chloroform) due to weaker intermolecule intereaction, which is beneficial for easy-processability, specifically in thick-film device fabrication.The optical properties of the two donors were investigated by UV-vis absorption spectra for solution in chloroform and film conditions (Fig. 1a). DI3T-1F and DI3T-2F can be classified as medium bandgap materials since the absorption onsets of the two donors are over 700 nm. Although the two molecules show very similar UV-vis absorption in solution, there are still some differences in their thin films. The absorption maximum peak featured a slightly larger red-shift of approximately 10 nm in DI3T-1F, indicating a better self-assembly property from DI3T-2F to DI3T-1F.34 Moreover, DI3T-1F displays an obvious vibronic shoulder peak, suggesting the stronger interchain aggregation in the film.35 Likewise, we determined the energy levels of DI3T-1F by cyclic voltammetry (CV) in DCM solution (Fig. S1, ESI†) and all detailed optical and electrochemical data are summarized in Table 1. According to the theoretical calculations, the highest occupied molecular orbital (HOMO) is mainly stationed in the electron-donating core, while the lowest unoccupied molecular orbital (LUMO) is delocalized across the entire molecule. In this study, the middle skeleton is kept and the slight end group modification (from 1F to 2F) would push the HOMO energetic level down, which is directly related to the Voc of the final OSC devices (which will be shown in the next part).
 | ||
| Fig. 1 (a) Absorption spectra of DI3T-F and DI3T-2F. (b) The J–V characteristics and (c) EQE spectra of the OSCs. (d) Photocurrent density (Jph) versus effective voltage (Veff) curves of the OSCs. | ||
| λ max(sol)a [nm] | λ max(film)b [nm] | λ onset(film)b [nm] | E optg [eV]c | E HOMO [eV] | E LUMO [eV] | |
|---|---|---|---|---|---|---|
| a Solution absorption spectra (1 × 10−5 M in chloroform). b Absorption spectra of small molecular films on quartz glass. c Optical bandgap derived from the absorption onset of small molecular films: Eoptg = 1240/λonset eV. d Electrochemically determined vs. Fc/Fc+, EHOMO = −(Eonsetox + 4.8), ELUMO = −(Eonsetred + 4.8). | ||||||
| DI3T-F | 496 | 572 | 714 | 1.74 | −4.99 | −3.46 |
| DI3T-2F 24 | 498 | 561 | 717 | 1.73 | −5.15 | −3.42 |
Differential scanning calorimetry (DSC) was carried out to verify the crystallinity of our DI3T-1F (Fig. S2, ESI†). DI3T-1F displayed a melting point (Tm) at 182 °C, whereas the DI3T-2F exhibits a Tm at 192 °C, indicating the strong fluorination effect and thus crystallinity of these two small molecules. Thermogravimetric analysis (TGA) shows that DI3T-1F possesses a 5% weight loss at 371 °C, exhibiting an excellent thermal stability for constructing the solar cell devices.
Device characterization and performance analysis of SM-OSCs
To investigate the effect of the terminal unit on the photovoltaic properties of the fullerene solar cells, two devices of DI3T-1F:PC71BM and DI3T-2F:PC71BM were fabricated with a conventional configuration of ITO/PEDOT:PSS/active layer/Phen-NaDPO/Al,36,37 where PEDOT:PSS used as a hole-transporting layer and PhenNa-DPO as an electron-transporting layer.Moreover, since DI3T-1F and DI3T-2F have a higher solubility in CF than other solvents, CF was selected as the spin-coating solvent for the two binary systems. In our previous report, the DI3T-2F:PC71BM system can obtain impressive performance with a D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A ratio of 2:1 without any pre and post-treatment. Herein, we cultivate the same device fabrication condition as a control device. DI3T-2F:PC71BM devices presented a champion performance of 7.32% with an open-circuit voltage of 835 mV, short-circuit current density (Jsc) of 12.8 mA cm−2, and fill factor (FF) of 68.5%, which is similar to our previous results. While fabricating with DI3T-1F:PC71BM, the corresponding devices exhibited better photovoltaic performance of 8.33% with Voc of 815 mV, Jsc of 13.6 mA cm−2, and FF of 75.1%, and such a performance enhancement originated from the increased Jsc and FF. To further understand the improvement, we focused on the improvement of Jsc and FF from dual-fluorine to mono-fluorine in the end group next.
:A ratio of 2:1 without any pre and post-treatment. Herein, we cultivate the same device fabrication condition as a control device. DI3T-2F:PC71BM devices presented a champion performance of 7.32% with an open-circuit voltage of 835 mV, short-circuit current density (Jsc) of 12.8 mA cm−2, and fill factor (FF) of 68.5%, which is similar to our previous results. While fabricating with DI3T-1F:PC71BM, the corresponding devices exhibited better photovoltaic performance of 8.33% with Voc of 815 mV, Jsc of 13.6 mA cm−2, and FF of 75.1%, and such a performance enhancement originated from the increased Jsc and FF. To further understand the improvement, we focused on the improvement of Jsc and FF from dual-fluorine to mono-fluorine in the end group next.
Firstly, we shone our light into the light-to-current conversion, which can be performed by external quantum efficiency (EQE) measurements. As shown in Fig. 1c, the DI3T-1F based devices show a slightly higher EQE value than those of DI3T-2F devices in all light response regions. In addition, the integrated Jsc value from the EQE spectrum for DI3T-1F:PC71BM (13.0 mA cm−2) is higher than that for DI3T-2F:PC71BM (12.4 mA cm−2). The calculated values showed around 5% mismatch compared with the Jsc values obtained from the J–V measurements. In addition, the charge carrier dynamics plays a critical role in the charge generation and collection. To investigate that, the photocurrent density (Jsc) as a function of the effective voltage (Veff)38 is plotted in Fig. 1d. Jph is defined as Jph = Jl − Jd, where Jl and Jd are the photocurrent densities under illumination and in the dark, respectively. Veff is defined as Veff = Vo − Va, where Vo is the voltage at which Jph = 0 and Va is the applied bias. When Veff arrived at ca. 0.5 V, the Jph value could reach the saturation condition (Jsat), which indicates that almost all mobile carriers are swept out by the high internal electric field and collected by the individual electrodes. The charge collection probability P(E,T) could be assessed by the formula of P(E,T) = Jph/Jsat. Under the short-circuited conditions, the P(E,T) for DI3T-1F devices is 96.2%, whereas the DI3T-2F shows a P(E,T) of 93.5%, suggesting a better charge collection probability in DI3T-1F devices.
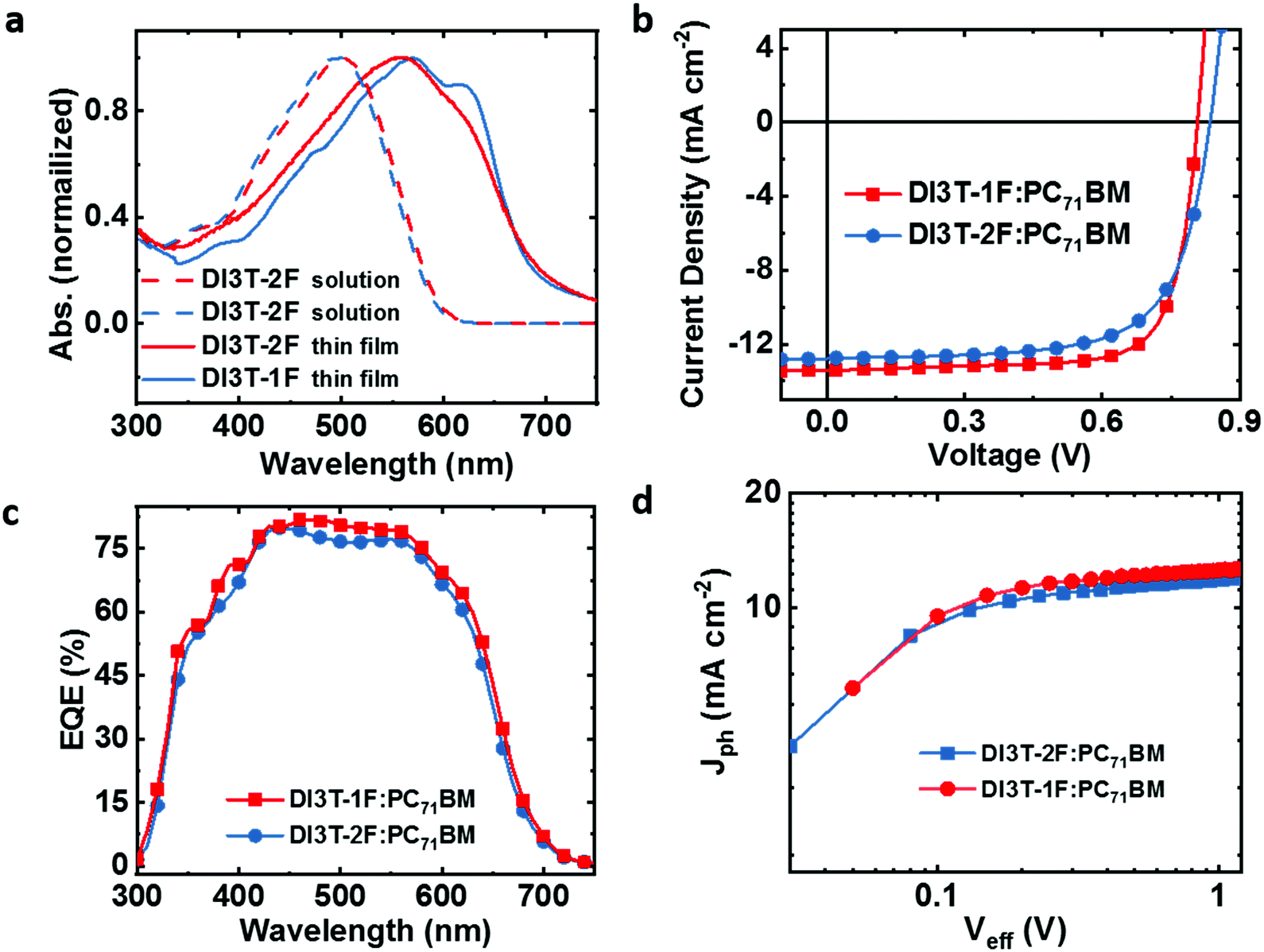
The charge carrier mobilities of two binary blend films were measured by the space-charge limited current method (SCLC) with device structures of ITO/PEDOT:PSS/active layer/MoOx/Ag for hole-only devices and ITO/ZnO/active layer/Ca/Al for electron-only devices (shown in Fig. 2a and b). The hole and electron mobilities of DI3T-2F:PC71BM were 2.4 × 10−4 and 1.03 × 10−4 cm2 V−1 s−1 with μh/μe ratio of 2.36, while in a DI3T-1F system, higher hole and electron mobilities of 4.04 × 10−4 and 3.67 × 10−4 cm2 V−1 s−1 were achieved with a much better balanced μh/μe ratio of 1.11.
 | ||
| Fig. 2 (a and b) The dark current density versus voltage curves of the OSCs. (c) The plot of Jscvs. light intensity of the OSCs. (d) The plot of Vocvs. light intensity of the OSCs. | ||
Such higher and more balanced charge carrier mobilities in the DI3T-1F system could contribute to the much higher Jsc and FF, which is consistent with a previous report.38b On the other hand, the bimolecular recombination process was investigated by measuring the dependence of Jsc values on light intensity (Plight), which can be expressed as a power-law formula Jsc ∝ (Plight)S, where S close to 1 implies that the bimolecular recombination in negligible. As shown in Fig. 2c, both S values of DI3T-1F and DI3T-2F are 0.99, indicating that the bimolecular recombination is not the main factor to contribute the improvement in performance. Furthermore, we investigate the Voc as a function of light intensity to obtain the initial information on the trap-assisted recombination. In general, a slope of kT/q is expected for solely bimolecular recombination, where k is the Boltzmann constant, q is elementary charge and T is temperature, in the plot of Vocversus the natural logarithm of the light intensity. In contrast, a signature of trap-assisted recombination is pronounced by an enhanced slope of Vocvs. light intensity (2kT/q). As shown in Fig. 2d, the DI3T-2F:PC71BM device delivers a slope of 1.31kT/q, suggesting the big potential of the recombination originated from traps. The trap-assisted recombination is drastically reduced in a DI3T-1F:PC71BM system (slope of 1.15kT/q), in agreement with the higher FF and better performance.
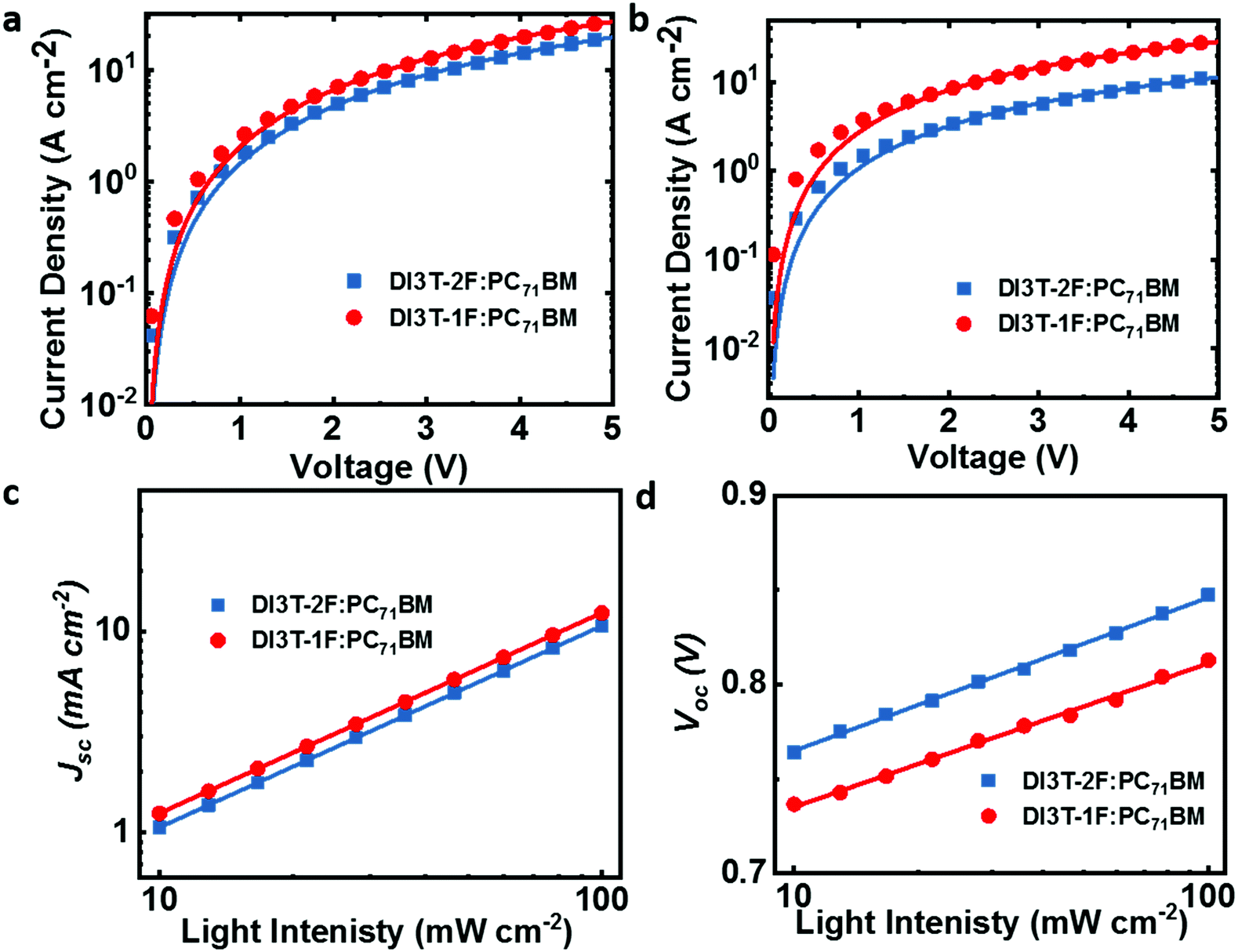
The bulk blend morphology of the two optimal blend films was examined by transmission electron microscopy (TEM). Normally, for the fullerene-based blend morphology, the dark regions are regarded as fullerene-rich domains, whereas the bright regions are believed to be small-molecule donor-rich domains. As shown in Fig. 3a and b, both DI3T-1F:PC71BM and DI3T-2F:PC71BM exhibit a homogeneous and intermixed blend morphology. However, comparing with the DI3T-2F system, the DI3T-1F:PC71BM blend shows a smaller domain size, indicating a better miscibility of DI3T-1F and fullerene acceptors (which will be discussed below).
 | ||
| Fig. 3 The TEM image of the blend film of (a) DI3T-1F:PC71BM and (b) DI3T-2F:PC71BM. The image of GIWAXS for the pristine films of (c) DI3T-1F and (d) DI3T-2F. The image of GIWAXS for the blend film of (e) DI3T-1F:PC71BM and (f) DI3T-2F:PC71BM. | ||
Grazing incidence wide-angle X-ray scattering (GIWAXS) measurements were performed to further understand the molecule crystallinity and orientation of the neat and blend films, which always generate a consequent impact on the device performance.39,40 In detail, DI3T-1F and DI3T-2F featured a similar scattering pattern, with a distinct π–π diffraction peak (010) in the in-plane (IP) direction and first-order (100), second-order (200), and third-order (300) lamellar diffraction peaks in the out-of-plane (OOP) direction41 (shown in Fig. 3c and d). Further calculations from the IP and OOP profiles (Fig. S4, ESI†) showed that the DI3T-2F shows a closer π–π stacking distance (3.55 Å) than that of DI3T-1F (3.62 Å), indicating the higher ordered structure and preferential edge-on orientation, which is due to the stronger fluorination effect as there are more fluorine atoms in DI3T-2F. After blending with PC71BM, the original edge-on orientation has been weakened in both DI3T-1F and DI3T-2F small molecules. And more interestingly, there is a signal of (010) peak in the out-of-plane direction of the DI3T-1F:PC71BM blend film, showing an inclination to face-on orientation, which is important for a vertical transport device such as a solar cell. As reported in a previous report,42 the mixed face-on and edge-on orientation would enable high efficiency because of the establishment of 3D charge carrier transport pathways, so we believe that 3D pathways in DI3T-1F:PC71BM films implement efficient carrier transport, even in the film with thickness over 300 nm.43,44
To investigate the dependence of performance on the thickness of the photoactive layer from 150 nm to 300 nm, devices with different thicknesses of active layers are fabricated, and Fig. 4a shows the variation of the solar cell performance as a function of the thickness of the photoacitive layer. For the DI3T-2F-based devices, the PCE drops dramatically from 7.32% to 3.62% with the thickness increasing from ∼150 nm to ∼300 nm, which is mainly caused by the decreased Jsc (from 12.8 mA cm−2 to 8.4 mA cm−2) and FF (from 68.5% to 51.6%) (Table S1, ESI†). Meanwhile, the PCE drops from 8.33% to 5.43% in the DI3T-1F system, with a very slight decrease of Jsc (from 13.6 mA cm−2 to 10.6 mA cm−2) (Table S2, ESI†). These results show that the performance, especially Jsc, in DI3T-1F is not as sensitive to the thickness of the active layers as for DI3T-2F.
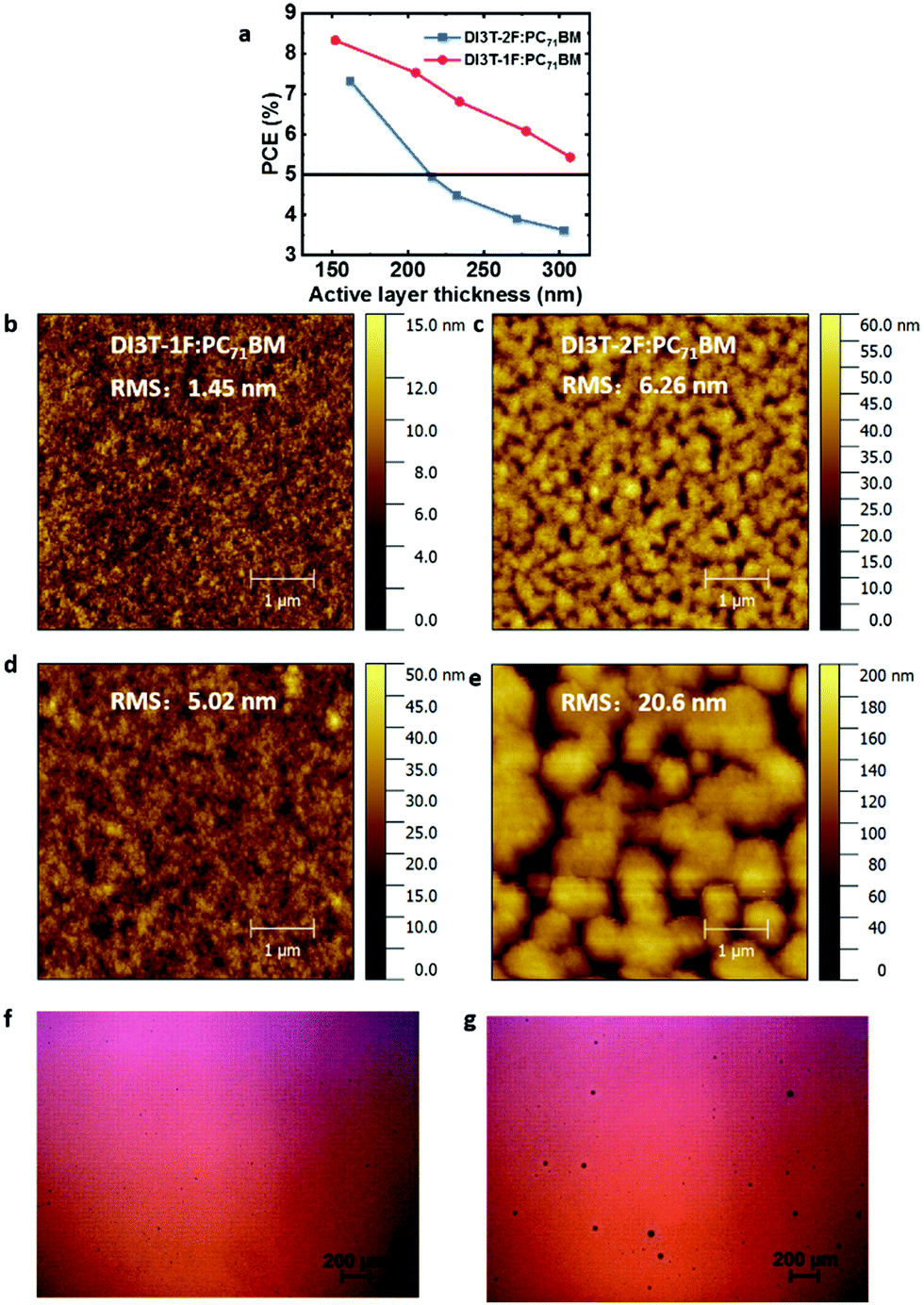 | ||
| Fig. 4 (a) The performance of the DI3T-1F:PC71BM and DI3T-2F:PC71BM in different film thicknesses. The AFM topography of (b) DI3T-1F:PC71BM and (c) DI3T-2F:PC71BM with film thickness of 150 nm. The AFM topography of (d) DI3T-1F:PC71BM and (e) DI3T-2F:PC71BM with film thickness of 300 nm. The microscopy image of (f) DI3T-1F:PC71BM and (g) DI3T-2F:PC71BM with film thickness of 300 nm. | ||
As reported before in the OSCs, the difference in surface energies of the donors and acceptors plays a vital role in the device performance with increasing thickness of the active layers.
Therefore, we calculated the surface tensions of DI3T-1F, DI3T-2F and PC71BM by the contact angles of water (Fig. S6, ESI†), ethylene glycol and benzylic alcohol on top of the corresponding film. The contact angle of the small molecules increases from DI3T-1F to DI3T-2F, indicating their enhanced hydrophobicity along with the increased amount of fluorine atoms in the end groups. Based on the contact angles, the surface tensions (γ) of them were obtained according to Wu's model, which are summarized in Table S3 (ESI†). To afford an accurate comparison of the miscibility, the Flory–Huggins interaction parameter,45,46χ of the two blends was further evaluated based on the equation:
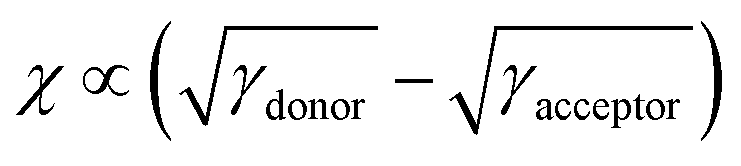
Conclusions
In conclusion, we synthesized DI3T-1F with less fluorine atoms on the peripheral isatin unit and compared it to DI3T-2F as donors in a blend with PC71BM in thick organic solar cells. Both devices based on DI3T-1F and DI3T-2F demonstrate a very good tolerence for a thick active layer from 150 nm to 300 nm. However, the devices based on DI3T-1F show a better balance of charge transport, morphology separation and less trap-assisted recombination, leading to a higher champion PCE of 8.33%, which is 14% higher than that based on DI3T-2F (7.32%) even with almost the same thickness of the active layer (ca. 150 nm). More interestingly, devices based on DI3T-1F with less fluorine show a better tolerence of thick active layers than that of DI3T-2F, and even with a thickness of ∼300 nm, the PCE of the devices based on DI3T-1F is 5.43%, which is about 50% higher than that of the devices based on DI3T-2F (3.62%). The thick active layer is still one of the challenges for the whole organic photovoltaics community, so herein we believe it can be an efficient way to develop new materials for thick-layer organic solar cells by tuning the number of fluorines on the tails of the donors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. G. Li acknowledges the general supported by the National Key R&D Program of China (2017YFA0204704), the National Science Foundation of China (51773091, 61604069, and 61761136013), and the Natural Science Foundation of Jiangsu Province (BK20171465);Prof A. K. Kyaw acknowledges general support by Thousand Talents Program of China and Shenzhen Municipal Science and Technology Innovation Committee and Shenzhen Peacock Team Project (No. KQTD2016030111203005).
Prof. D. Baran acknowledges the general support by KAUST Solar Center Competitive Fund (CCF) for financial support.
Notes and references
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- J. Zhou, X. Wan, Y. Liu, Y. Zuo, Z. Li, G. He, G. Long, W. Ni, C. Li, X. Su and Y. Chen, J. Am. Chem. Soc., 2012, 134, 16345–16351 CrossRef CAS.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS.
- (a) S. D. Collins, N. A. Ran, M. C. Heiber and T.-Q. Nguyen, Adv. Energy Mater., 2017, 7, 1602242 CrossRef; (b) D. Baran, S. E. Ela, A. Kratzer, T. Ameri, C. J. Brabec and A. Hirsch, RSC Adv., 2015, 5, 64724–64730 RSC.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, R. A. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282–7285 CrossRef CAS.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS.
- K. Sun, Z. Xiao, S. Lu, W. Zajaczkowski, W. Pisula, E. Hanssen, J. M. White, R. M. Williamson, J. Subbiah, J. Ouyang, A. B. Holmes, W. W. Wong and D. J. Jones, Nat. Commun., 2015, 6, 6013 CrossRef CAS.
- L. Yuan, K. Lu, B. Xia, J. Zhang, Z. Wang, Z. Wang, D. Deng, J. Fang, L. Zhu and Z. Wei, Adv. Mater., 2016, 28, 5980–5985 CrossRef CAS.
- M. Li, K. Gao, X. Wan, Q. Zhang, B. Kan, R. Xia, F. Liu, X. Yang, H. Feng, W. Ni, Y. Wang, J. Peng, H. Zhang, Z. Liang, H.-L. Yip, X. Peng, Y. Cao and Y. Chen, Nat. Photonics, 2016, 11, 85–90 CrossRef.
- B. Kan, H. Feng, H. Yao, M. Chang, X. Wan, C. Li, J. Hou and Y. Chen, Sci. China: Chem., 2018, 61, 1307–1313 CrossRef CAS.
- J. A. Love, C. M. Proctor, J. Liu, C. J. Takacs, A. Sharenko, T. S. van der Poll, A. J. Heeger, G. C. Bazan and T.-Q. Nguyen, Adv. Funct. Mater., 2013, 23, 5019–5026 CrossRef CAS.
- J. Miao, H. Chen, F. Liu, B. Zhao, L. Hu, Z. He and H. Wu, Appl. Phys. Lett., 2015, 106, 183302 CrossRef.
- J. D. Zimmerman, X. Xiao, C. K. Renshaw, S. Wang, V. V. Diev, M. E. Thompson and S. R. Forrest, Nano Lett., 2012, 12, 4366–4371 CrossRef CAS.
- S. Chen, L. Yan, L. Xiao, K. Gao, W. Tang, C. Wang, C. Zhu, X. Wang, F. Liu, X. Peng, W.-K. Wong and X. Zhu, J. Mater. Chem. A, 2017, 5, 25460–25468 RSC.
- J.-L. Wang, Z. Wu, J.-S. Miao, K.-K. Liu, Z.-F. Chang, R.-B. Zhang, H.-B. Wu and Y. Cao, Chem. Mater., 2015, 27, 4338–4348 CrossRef CAS.
- J. Min, Y. N. Luponosov, N. Gasparini, L. Xue, F. V. Drozdov, S. M. Peregudova, P. V. Dmitryakov, K. L. Gerasimov, D. V. Anokhin, Z.-G. Zhang, T. Ameri, S. N. Chvalun, D. A. Ivanov, Y. Li, S. A. Ponomarenko and C. J. Brabec, J. Mater. Chem. A, 2015, 3, 22695–22707 RSC.
- V. Piradi, X. Xu, Z. Wang, J. Ali, Q. Peng, F. Liu and X. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 6283–6291 CrossRef CAS.
- C. D. Wessendorf, A. Perez-Rodriguez, J. Hanisch, A. P. Arndt, I. Ata, G. L. Schulz, A. Quintilla, P. Bäuerle, U. Lemmer, P. Wochner, E. Ahlswede and E. Barrena, J. Mater. Chem. A, 2016, 4, 2571–2580 RSC.
- Y. Huang, W. Wen, S. Mukherjee, H. Ade, E. J. Kramer and G. C. Bazan, Adv. Mater., 2014, 26, 4168–4172 CrossRef CAS.
- Y. Liu, C. C. Chen, Z. Hong, J. Gao, Y. M. Yang, H. Zhou, L. Dou, G. Li and Y. Yang, Sci. Rep., 2013, 3, 3356 CrossRef.
- S. Chen, L. Xiao, X. Zhu, X. Peng, W.-K. Wong and W.-Y. Wong, Chem. Commun., 2015, 51, 14439 RSC.
- S. Mukherjee, C. M. Proctor, G. C. Bazan, T.-Q. Nguyen and H. Ade, Adv. Energy Mater., 2015, 5, 1500877 CrossRef.
- (a) X. Song, N. Gasparini, M. M. Nahid, H. Chen, S. M. Macphee, W. Zhang, V. Norman, C. Zhu, D. Bryant, H. Ade, I. McCulloch and D. Baran, A Highly Crystalline Fused-Ring n-Type Small Molecule for Non-Fullerene Acceptor Based Organic Solar Cells and Field-Effect Transistors, Adv. Funct. Mater., 2018, 28, 1802895 CrossRef; (b) C. Cui, X. Guo, J. Min, B. Guo, X. Cheng, M. Zhang, C. J. Brabec and Y. Li, Adv. Mater., 2015, 27, 7469–7475 CrossRef CAS.
- D. Corzo, K. Almasabi, E. Bihar, S. Macphee, D. Rosas-Villalva, N. Gasparini, S. Inal and D. Baran, Adv. Mater. Technol., 2019, 8, 1900040 CrossRef.
- G. Zhang, K. Zhang, Q. Yin, X.-F. Jiang, J. Xin, W. Ma, H. Yan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2017, 139, 2387–2395 CrossRef CAS.
- J. Lee, D. H. Sin, B. Moon, J. Shin, H. G. Kim, M. Kim and K. Cho, Energy Environ. Sci., 2017, 10, 247–257 RSC.
- J. Huang, J. H. Carpenter, C.-Z. Li, J.-S. Yu, A. Ade and A. K.-Y. Jen, Adv. Mater., 2016, 28, 967–974 CrossRef CAS.
- S. Feng, C. Zhang, Y. Liu, Z. Bi, Z. Zhang, X. Xu, W. Ma and Z. Bo, Adv. Mater., 2017, 29, 1703527 CrossRef.
- Y. Liu, M. Li, J. Yang, W. Xue, S. Feng, J. Song, Z. Tang, W. Ma and Z. Bo, Adv. Energy Mater., 2019, 9, 1901280 CrossRef.
- M. Li, Y. Zhou, J. Zhang, J. Song and Z. Bo, J. Mater. Chem. A, 2019, 7, 8889–8896 RSC.
- Y. Yang, K. Wang, G. Li, X. Ran, X. Song, N. Gasparini, Q. Q. Zhang, X. Lai, X. Guo, F. Meng, M. Du, W. Huang and D. Baran, Small, 2018, 14, e1801542 CrossRef.
- C. J. Takacs, S. D. Collins, J. A. Love, A. A. Mikhailovsky, D. Wynands, G. C. Bazan, T. Q. Nguyen and A. J. Heeger, ACS Nano, 2014, 8, 8141–8151 CrossRef CAS.
- D. Xie, T. Liu, W. Gao, C. Zhong, L. Huo, Z. Luo, K. Wu, W. Xiong, F. Liu, Y. Sun and C. Yang, Sol. RRL, 2017, 1, 1700044 CrossRef.
- X. Song, N. Gasparini, M. M. Nahid, S. H. K. Paleti, J.-L. Wang, H. Ade and D. Baran, Joule, 2019, 3, 846–857 CrossRef CAS.
- W.-Y. Tan, R. Wang, M. Li, G. Liu, P. Chen, X.-C. Li, S.-M. Lu, H. L. Zhu, Q.-M. Peng, X.-H. Zhu, W. Chen, W. C. H. Choy, F. Li, J. Peng and Y. Cao, Adv. Funct. Mater., 2014, 24, 6540–6547 CrossRef CAS.
- (a) S. Li, L. Ye, W. Zhao, X. Liu, J. Zhu, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1704051 CrossRef; (b) A. K. K. Kyaw, D. H. Wang, C. Luo, Y. Cao, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Adv. Energy Mater., 2014, 4, 1301469 CrossRef.
- H. Yao, L. Ye, J. Hou, B. Jang, G. Han, Y. Cui, G. M. Su, C. Wang, B. Gao, R. Yu, H. Zhang, Y. Yi, H. Y. Woo, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1700254 CrossRef.
- S. Dai, F. Zhao, Q. Zhang, T. K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS.
- H. Li, Y. Zhao, J. Fang, X. Zhu, B. Xia, K. Lu, Z. Wang, J. Zhang, X. Guo and Z. Wei, Adv. Energy Mater., 2018, 8, 1702377 CrossRef.
- Y. Chang, Y. Chang, X. Zhu, X. Zhou, C. Yang, J. Zhang, K. Lu, X. Sun and Z. Wei, Adv. Energy Mater., 2019, 9, 1900190 CrossRef.
- Z. Zhou, S. Xu, J. Song, Y. Jin, Q. Yue, Y. Qian, F. Liu, F. Zhang and X. Zhu, Nat. Energy, 2018, 3, 952–959 CrossRef CAS.
- B. Guo, X. Guo, W. Li, X. Meng, W. Ma, M. Zhang and Y. Li, J. Mater. Chem. A, 2016, 4, 13251–13258 RSC.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2013, 135, 12057–12067 CrossRef CAS.
- J. J. van Franeker, D. Hermida-Merino, C. Gommes, K. Arapov, J. J. Michels, R. A. J. Janssen and G. Portale, Adv. Funct. Mater., 2017, 27, 1702516 CrossRef.
- G. Zhang, K. Zhang, Q. Yin, X. F. Jiang, Z. Wang, J. Xin, W. Ma, H. Yan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2017, 139, 2387–2395 CrossRef CAS.
- T. Wang, X.-K. Chen, A. Ashokan, Z. Zheng, M. K. Ravva and J.-L. Brédas, Adv. Funct. Mater., 2018, 28, 1705868 CrossRef.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS.
- C. R. McNeill, H. Frohne, J. L. Holdsworth, J. E. Furst, B. V. King and P. C. Dastoor, Nano Lett., 2004, 4, 219–223 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00605b |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2020 |