
Targeted removal of blood cancer cells from mixed cell populations by cell recognition with matching particle imprints†
Perrine
Remaud
a,
Jevan
Medlock
 a,
Anupam A. K.
Das
a,
David J.
Allsup
b,
Leigh A.
Madden
c,
Dieter
Nees
d,
Paul J.
Weldrick
a and
Vesselin N.
Paunov
*a
a,
Anupam A. K.
Das
a,
David J.
Allsup
b,
Leigh A.
Madden
c,
Dieter
Nees
d,
Paul J.
Weldrick
a and
Vesselin N.
Paunov
*a
aDepartment of Chemistry and Biochemistry, University of Hull, Hull, HU67RX, UK. E-mail: V.N.Paunov@hull.ac.uk; Tel: +44 1482 465660
bHull York Medical School, University of Hull, University of Hull, Hull, HU67RX, UK
cDepartment of Biomedical Sciences, University of Hull, Hull, HU67RX, UK
dJoanneum Research FmbH, Leonhardstrasse 59, 8010 Graz, Austria
First published on 21st November 2019
Abstract
We report a new approach for separation of blood cancer cells from healthy white blood cells based on cell recognition by surface functionalised particle imprints. We prepared polymeric particle imprints from a layer of suspension of monodisperse PMMA microbeads which closely match the size of in vitro cultured human leukaemia cells (HL60). The imprints were replicated on a large scale with UV curable polyurethane resin using nanoimprinting lithography and surface functionalized with a cationic polymer, a branched polyethylene imine (bPEI), and a Pluronic surfactant, Poloxamer 407, to engineer a weak attraction towards the cells. The latter is amplified several orders of magnitude when a cell of a closely matching size and shape fits into the imprint cavity which multiplies the contact area between the cell surface and the imprint. The particle imprints were optimised for their specificity toward blood cancer cells by treatment with oxygen plasma and then subsequent coatings with bPEI and Poloxamer 407 with various functionalisation concentrations. We tested the surface functionalised imprints for their specificity in retaining in vitro cultured human leukaemic cells (HL60) over healthy human peripheral blood mononuclear cells (PBMCs) in a flow through chamber. The effect of the flushing flow rate of the mixed cell suspension over the particle imprint and the imprint length were also investigated. At each step the selectivity towards HL60 was assessed. Selective isolation of an increased amount of HL60 tumour cells over PBMC was ultimately achieved as a function of the cell seeding ratio on the particle imprint. The effect is attributed to the substantial size difference between the HL60 cell and the PBMCs. The data presented show that relatively inexpensive PMMA microbeads imprints can be utilised as a cell separation technique which could ultimately lead to novel therapies for removal of neoplastic cells from the peripheral blood of acute myeloid leukaemia patients.
Introduction
Bioimprints are polymeric replicas of cells or microorganisms which produce surface cavities of the same shape and size as the original cells.1,2 These polymer-based cavities can then be optimised to allow specific recognition of target cells of a matching size and shape. The bioimprint acts by the same principle as that of the Lock and Key model which enables the shape and size based recognition of specific cells.1,3–5 The first report of bioimprints is credited to Dickert et al.6 who created a highly selective yeast imprint from a sol–gel matrix. Further whole cell imprints have been made from a range of micro-organisms and human cells. As utilised here, Hayden et al.7 functionalised red blood cell polyurethane imprints capable of distinguishing cells via blood group antigen recognition. Subsequently they were able to separate cells from blood groups A1 and A2.8 Molecular imprinted polymers have also been utilised for biological applications such as bacteria, virus and chemical sensing.9–11 The advantages of the imprinting technologies lie in their ease of manufacture, stability, reusability and importantly, specificity for biological targets.12Acute myeloid leukaemia (AML) presents as uncontrolled proliferation of neoplastic cells (myeloblasts) causing an imbalance in haematopoiesis leading to bone marrow failure and resulting in the death of the patient if untreated.13–15 In the UK, the average overall 5-year survival is 16% from the point of diagnosis. The prognosis is significantly worse in the majority of patients due to disease presentation in the later years of life.13,16–19 Current AML treatment is myeloablative chemotherapy followed by an allogeneic stem cell transplant.20,21 AML patients spend a long time hospitalised and suffer considerable morbidity related to anaemia, sepsis and bleeding. Outcomes for AML patients have only improved in relation to supportive care pathways rather than any new drug regimens.22 There is an urgent need to seek alternative therapies for these patients. Selective leukophoresis can potentially be used more in the extraction of myeloblasts from peripheral blood which is critical in stabilizing AML patients with leukostasis associated with hyperleuocytosis.23 By reducing the number of circulating tumour cells, the likelihood of early relapse is also diminished.24
AML blood cancer cells present an ideal target for cell shape recognition by prefabricated bioimprinted surfaces where a very strong specific adhesion can occur between the imprinted cell surface pattern and cells of a matching shape and size. The separation between the myeloblasts and healthy white blood cells on a polymeric imprint should occur due to the distinct cell size and morphological differences between the myeloblasts and normal blood cells (Fig. 1). The cell surfaces are normally negatively charged due to the presence of integral membrane proteins and carbohydrates with negatively charged COO− groups on the phospholipid bilayer.25 Hence, the imprints can be first treated with a biocompatible cationic polyelectrolyte to promote attractive electrostatic interactions between the target cells and the imprint surface. The polymeric imprints can be potentially used for various applications in different fields due to their low production cost, unique mechanical, thermal and chemical properties, their stability and the ease of their preparation.26 Molecular imprinting technology has been used in many different areas such as separation methods, biosensors, organic synthesis and drug development.27 This imprinting technology also has great prospects in the field of biomedicine, hence, this study is aimed at the development of matching imprints for medical applications such as a supplementary therapy for blood cancer that can be used alongside conventional chemotherapy. There are two types of methods to create imprinted polymers:28 cell-membrane-molecular imprinting and the whole-cell-imprinting strategy. The former uses only part of the cell surface while the second method replicates the cells as whole entities or their constituent bio-macromolecules. We have recently utilised a bioimprinting approach to remove tumour cells from healthy cells.29 This approach, whilst very successful would be time consuming to replicate for individual patients where a bioimprint must be made from the individuals tumour cells, which would first have to be isolated from patient whole blood. A simple particle imprint of matching target cell size could simplify this approach and negate the requirement for individualised bioimprints. In addition, casting cells with polymers opens a range of challenges as the cells can potentially shrink, flatten and change their shape during the replication process due to drying or upon contact with the replicating resins. In this project, a novel imprinting strategy using monodisperse poly(methyl methacrylate) (PMMA) microbeads of a similar size to target blood cancer cells (∼14 μm) was used to mimic whole cell bioimprinting. The strategy is reminiscent of the method used to fabricate micro-lens arrays by dual templating of monodisperse particle monolayers with PDMS and a UV-curable optical adhesive.40 Its application produced a positive imprint of a microbead layer imprint which could then be upscaled using the Roll-to Roll-nanolithography method (R2R NIL).30 In order to increase the selectivity of the particle imprint towards blood cancer cells, the particle imprint was first treated with the cationic polyelectrolyte bPEI in order to promote attractive electrostatic interaction between the target cells and the particle imprint surface. In order to weaken the attraction and make the interaction cell-imprint more specific with respect to the cell shape and size, a secondary treatment in the form of POL407 was used to passivate the surface of the positively charged particle imprint in order to make healthy white blood cells less likely to non-specifically attach to the imprint. Hence the interaction needs to be weak enough to rely on the amplification of the contact area between the cells and the imprint cavity surface. The function of the POL407 is to offset the approaching cells from the imprint surface which weakens the attraction. This is needed to reduce the non-specific adhesion of the PBMCs with the imprint upon point contact, while amplifying the interaction between HL60 cells and the size-matched imprint cavities. The particle imprint used for this specific study was made using 15 μm PMMA microbeads (Spheromers™ CA15, Microbeads) (Fig. 2) which have a matching size to the targeted HL60 cells of an average diameter of 14 μm (see Fig. S1, ESI†).
 | ||
| Fig. 1 The principle of particle imprint-based AML cell depletion from a mixture with PBMCs. The substantially larger (AML) cells are preferentially attracted and retained on the imprint due to the large contact area whereas smaller, healthy PBMCs are flushed through with little interaction. | ||
 | ||
| Fig. 2 The process of imprint production from the PMMA microbeads layers. CA15 PMMA particles were formulated as an aqueous suspension containing glucose and spread evenly over a glass substrate. The produced microbeads layer was levelled off with a bespoke tool and dried up in air. The microbeads layer was cast with curable silicone (PDMS). The latter was peeled off after 24 h of curing and cleaned up to make a negative imprint. This imprint was cast a second time with UV-curable polyurethane (PU) resin to produce a positive master imprint (shim) which was used in replication of the original patter by Roll-to-Roll Nanoimprinting (see Fig. S2, ESI†). | ||
Materials and methods
Preparation of the CA15 particle imprints
Spheromers® C15 microbeads were sourced from Microbeads AS, Norway (http://www.micro-beads.com). Glass substrates (70 cm × 40 cm) were cleaned with acetone and alcoholic KOH (10 wt%) for 1 h, washed with deionized water and treated with 20 wt% poly(diallyldimethylammonium chloride) (PDAC) aqueous solution for 30 min, followed by further cleaning with deionized water and drying under air stream. A sample of 6.0 g of Spheromers® CA15 PMMA microbeads and 2.5 g of glucose was mixed together in 30 mL of 0.1% (w/v)% xanthan gum solution. Spreading of this CA15 suspension on the glass substrate was done using a glass tool comprising a square frame of four glass strips, one of which was offset by 100 μm to create a gap. The CA15 suspension was added to the frame cavity and the tool was moved over the glass substrate (with a lead) in a steady motion in the direction opposite to the higher side. This allowed an aqueous film of CA15 microbeads suspension (40 cm × 70 cm) of uniform thickness (∼100 μm) to be deposited, followed by evaporation to a semi-dry state at room temperature in a laminar flow cabinet. The curable Sylgard 184 elastomer (polydimethylsiloxane, PDMS, from Dow Corning) was mixed at a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of elastomer-to-accelerator and degassed by 10 min centrifugation at 4000g. A metal frame of interior space 65 × 30 × 4 cm was placed on top of the produced CA15 microbeads layer and the PDMS elastomer mixture (900 mL) was poured evenly inside.
:1 ratio of elastomer-to-accelerator and degassed by 10 min centrifugation at 4000g. A metal frame of interior space 65 × 30 × 4 cm was placed on top of the produced CA15 microbeads layer and the PDMS elastomer mixture (900 mL) was poured evenly inside.
For structural support, a polyester fabric sheet (Boyes UK, dimensions 65 × 30 × 0.1 cm) was added on top of the PDMS layer and the composite was allowed to cure at 25 °C for 48 h. The cured PDMS cast of the CA15 microbeads layer was peeled off the glass surface and washed using warm aqueous solutions of detergent, ethanol and deionized water in sequence, followed by drying under an air stream. The negative PDMS imprint (Fig. 3A and B) was then replicated further into a photo-curable urethane acrylate resin layer (supplied from Joanneum Research FmbH, Graz, Austria) pre-deposited onto a polyethylene terephthalate (PET) foil for the manufacturing of a positive roller imprint shim (20 cm × 62 cm).
This positive imprint shim was than used in a continuous Roll-to-Roll UV-imprint process creating the final negative imprint into a photo-curable acrylate resin layer on a PET film (see Fig. S2, ESI†). We were able to Roll-to-Roll print hundreds of meters of negative polymeric imprints on PET foil from a single shim which was used for further cell recognition experiments.
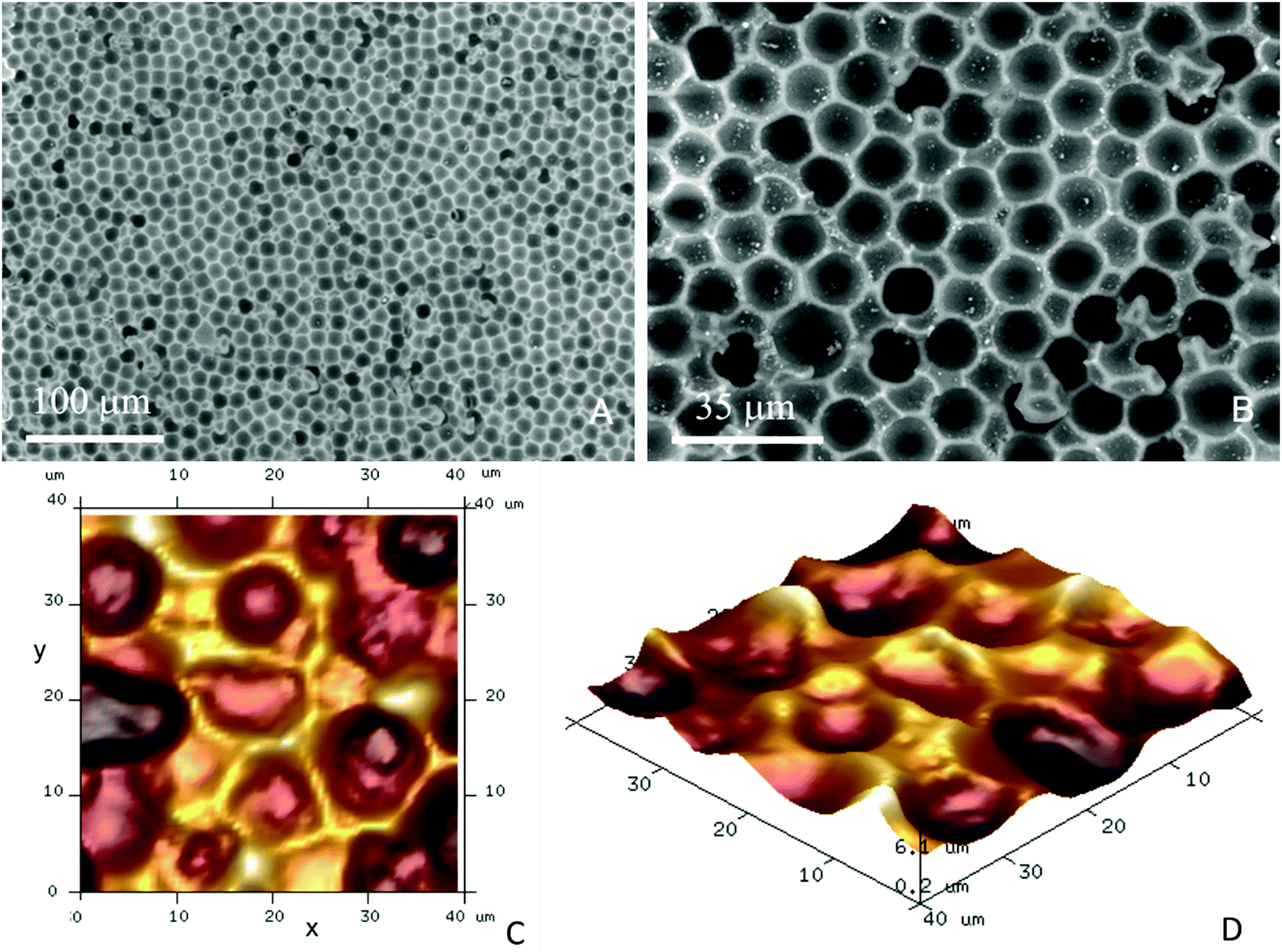 | ||
| Fig. 3 SEM images of typical negative PDMS imprints of a layer of 15 μm PMMA microbeads (Spheromers® CA15) deposited on glass slides as a suspension. The scale bars represent (A) 100 μm and (B) 35 μm, respectively. (C and D) Atomic force microscope images of the CA15 particle imprints with different perspectives. | ||
Surface modification of CA15 particle imprints
The negative CA15 microbeads imprints on PET foil were cut to different lengths (2, 4, 8 and 12 cm) × 8 mm and were further surface functionalised to incur a weak attraction between the target HL60 cells and the imprint. The particle imprint samples were first treated with oxygen plasma (Harrick Plasma PDC 32G) at 147 Pa, using an RF power of 16 W for 4 min. The oxygen plasma treated CA15 imprint samples were further incubated for 15 minutes with aqueous solutions of the cationic polyelectrolyte branched polyethylene imine (bPEI, Polysciences Inc.) with different concentrations (0.01%, 0.015%, 0.02%, 0.025%, 0.03%, 1% and 2% (w/w)). Further, they were rinsed with deionised water and air-dried before being integrated in the PDMS-based flow-through chip (Fig. 4A and B) in order to test the CA15 microbeads imprint selectivity towards HL60 cells in a mixture with PBMCs and HL60 cells. Additional treatment of the imprint and the whole chamber and tubing with solutions of Poloxamer 407 (Sigma-Aldrich) was applied, with a range of POL407 concentrations of 0.25%, 0.5%, 1%, 2% or 3% (w/w) in deionised water which were loaded in the PDMS-based chip after the bPEI-treated particle imprint was integrated in the chip. The imprint was incubated with the POL407 solution for 30 min and finally washed by flushing with 10 mL of phosphate buffered saline (PBS, Corning) at a flowrate of 219 mL h−1 using a syringe pump (World Precision Instruments SP100i Syringe Pump). Tapping mode atomic force microscopy was carried out using a Dimension Edge (Bruker) with TESPA-V2 probes (Bruker). A scan rate of 0.1 Hz was used with 1024 or 512 lines for a 50 μm scan range (Fig. 3C and D). | ||
| Fig. 4 (A) Schematic and (B) top side view of the actual particle imprint (of length 4 cm and width 6 mm) attached on a microscope slide with PDMS channelled casing with input and output channels. (C) Photographs showing the setup used for the static experiment using the syringe pump, the cell loaded chip and the collected effluent. | ||
Flow-through imprint device
A phosphate buffer solution at 5 mM was prepared by adding 0.78 g of sodium dihydrogen orthophosphate dihydrate (Fisher Chemicals) in 1 L of deionised water. The pH was adjusted to 3 using orthophosphoric acid (Fisher Chemicals). The solution was heated at 50 °C and 1 g of hydroxypropyl methylcellulose (HPMC) (sourced from Dow Chemical Company) was added slowly to obtain a solution of HPMC at 0.1% (w/v). Sylgard 184 elastomer (10:1 elastomer:accelerator, Dow Corning) was mixed thoroughly and degassed by centrifugation (Sorvall Biofuge Primo, Thermofisher Scientific) at 5000g for 10 min. 3 mL of the HPMC solution was put on the glass substrates and was incubated for 30 min.
It was then rinsed with water and air dried. To form the PDMS chamber of the device, 100 micrometres thick stripes of plastic foil were cut with width: 0.5 cm and lengths, 4 cm, 8 cm and 12 cm, respectively. 2 layers of the plastic foil were layered with 2 layers of double-sided adhesive tape to form a template for a channel depth of 250 micrometres. This stripe was adhered on the middle of the glass substrate. 25 mL of PDMS-hardener 10:1 mixture was poured over the glass substrates (5 × 50 × 1 mm) with the plastic stripe (channel template), immobilised on the glass surface, and left to cure at 40 °C for 24 h. The PDMS was removed from the glass surface in order to yield identical channels on the PDMS substrates. Another microscope slide (ThermoScientific) and PDMS channel was treated with Oxygen Plasma (1100 mTorr, 32 W) for 2 min. The PDMS channel was punched with at each end with a 2 mm biopsy puncher to form an inlet and an outlet for the circulating fluid (see Fig. 4B). The prepared microbeads imprint was placed over the channel aperture and the glass slide was used to enclose the system (Fig. 4). The device was clamped together and put in a drying oven at 40 °C for 30 min. Inlets and outlets were added by puncturing the PDMS and feeding with PTFE tubing (∼0.5 mm diameter).
Cell retention tests, staining and counting
HL60 cells (sourced from Public Health England) were cultured aseptically in a mixture of 80 mL of Roswell Park Memorial Institute Medium (RPMI) 1640 (Gibco), mixed with 10 mL foetal bovine serum (Gibco), 5 mL penicillin and 5 mL streptomycin solutions (Lonza) at 37 °C with 5% CO2.PBMCs were obtained from anonymous apparently healthy donors via the NHS blood transfusion service (under IRAS 214660 with REC ethical approval 16/LO/1948) and stored in liquid nitrogen prior to use. PBMCs are composed of monocyte and lymphocyte populations and the shape and size are generally accepted to be the same to a large degree. Flow cytometry can highlight the cell populations without the need for additional staining to differentiate between monocytes and lymphocytes. The cells were slowly defrosted and washed 3 times in PBS. Removal of platelet contamination was achieved by triple centrifugation at 120g for 10 min and resuspension in PBS. HL60 cells surfaces were fluorescently tagged by dropwise addition of 100 μL of 0.025 wt% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine N(carboxy-fluorescein) in ethanol to 2 mL of the stock HL60 cell suspension (1 × 106 cells per mL). PBMCs were treated via a similar method using 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N (Lissamine rhodamine B sulphonyl) (ammonium salt). The cells were washed twice separately using centrifugation (VWR, Mega Star 3.0R) at 400g for 5 min. The supernatant was removed and the cells were redispersed in deionised water. The cells were counted using an Improved Neubauer haemocytometer observed under the microscope. An Olympus BX51 fluorescence microscope coupled with a mercury lamp excitation source and DP70 camera (Olympus) with ImageProPlus software was used to acquire images of the captured cell populations retained on the CA15 imprint.
Mixed cell sample preparation
The desired amount of cells was taken from the master stock suspensions of PBMC and HL60 cells and transferred into two tubes (Eppendorf) in order to have a (HL60/PBMC) cell ratio of 25%/75% or 10%/90%, respectively, measured by the number of cells.The ratios were chosen to mimic in vivo blast loads in AML where diagnosis is made at a blast proportion of 20%.31 The same ratio of (HL60/PBMC) cells was loaded into the fluidic chips of different imprint lengths. The total number of cells was calculated to be enough in order to saturate the smallest imprint (6.54 × 105 cells for the 2 cm chip and 1.31 × 106 cells for the 4 cm chip). PBS buffer was used to top up the total volume of the cell suspension to 1 mL. The suspensions were centrifuged at 400g for 6 min (with Minispin Plus Eppendorf centrifuge). The supernatant was then removed leaving the separated cells in the tube. PBS buffer was added to the PBMC tube (2 cm: 50 μL, 4 cm: 100 μL, 8 cm: 200 μL and 12 cm: 300 μL), mixed with the cells and then with the HL60 cell suspension at the desired ratios (see below). The centrifuge tubes were sonicated using an ultrasonic bath for 5 min to prevent any cell aggregation. The prepared mixed cell suspension of the desired cell ratio was injected into the fluidic imprint chip and allowed to settle on the imprint for 30 min. 10 mL of PBS was eluted over the particle imprint at different flowrates (50 mL h−1, 126 mL h−1 or 219 mL h−1) using a syringe pump (see Fig. 4C). The CA15 particle imprint samples with the residual cells were analysed by bright field and fluorescence microscopy by taking images before and after flushing of the chip. The HL60 and PBMC cells on the imprint were counted using the ImageJ software with the macro script described in ESI.† In the case of multiple seeding of the imprint with the HL60/PBMC cell mixture, the cell solution was added to the chip after the flushing step with PBS without removing the retained cells from the previous run. Cell viability was not determined throughout the tests as the latter are time consuming and it is very challenging to keep every batch of cells with the same viability. It was not possible to test viability by this method as the cells were already fixed and stained for recognition purposes with dyes that would be detected with the same fluorescence emission. However, in principle it could be tested using an Annexin V staining procedure prior to flow cytometry analysis. Retention of HL60 cells was assessed by taking images (n = 10) with bright field and fluorescence microscopy at various sites across the bioimprint. Cells were enumerated via the automatic method using images collected using FITC and TRITC in order to separately assess each cell type collected at each site. Each experiments was reproduced up to 3 times and the standard deviation was calculated from these results.
Results and discussion
The EDX results presented in Fig. S8 (ESI†) clearly show that there is nitrogen present on the surface of the CA15 negative particle imprints after the treatment with 0.015 wt% bPEI and 0.03 wt% bPEI. In comparison, the non-treated CA15 particle imprint sample shown has no nitrogen. Note that the nitrogen content on the surface increases with the increase of the bPEI concentration. We characterised the average diameter of the CA15 imprint cavities which was determined to be 12.9 ± 0.9 μm (Fig. S3 and Table S1, ESI†). Note that this value does not correspond to the equatorial diameter of the templated CA15 particles which is 14.5 ± 0.4 μm. The reason being that the glucose layer partially filled the voids of the CA15 layer and obstructed the replicating resin to avoid formation of “necks” in the imprinted particles which would make it impossible for a cell of the same radius of curvature to freely fit in. The average diameters of the HL60 cells and the PBMCs are 13.1 μm and 9.2 μm, respectively. This substantial difference between the radii of the curvature of the malignant and the healthy white blood cells and the close match of the former to the CA15 microbeads imprint makes it possible to selectively retain them on the imprint.HL60 selectivity optimisation
To assess the CA15 imprint selectivity towards HL60 cells, different parameters such as flowrate, the pre-treatment of the imprint with oxygen plasma, the bPEI and the POL407 solution concentration were investigated. The aim was to improve the selectivity of the CA15 microbeads imprint towards the HL60 blood cancer cells while allowing the PBMCs to pass through the imprint. Oxygen plasma treatment was firstly found to positively impact cell selection with 38% selectivity observed after such treatment but only 25% selectivity with a non-treated imprint (ESI,† Fig. S4A). This is explained with the formation of additional negatively charged carboxyl groups (–COO−) on the imprint surface after the oxygen plasma treatment which serve as better anchoring points of the cationic polyelectrolyte (bPEI).Therefore, all further experiments were carried out with oxygen plasma treated imprints. The length of the chip was then investigated and it was observed that imprints over 4 cm in length captured more cells proportionately, however at the expense of lower selectivity towards HL60 (Fig. S4B, ESI†). As it may be expected, the higher flowrate was associated with a lower retention of cells on the imprint. We found that the selectivity towards HL60 cells was optimal with a flowrate of 219 mL h−1 at this treatment of the imprint. The particle imprint had an average selectivity of 43% in HL60 at 219 mL h−1 after flushing but only 30% at a flowrate of 50 mL h−1. These results enabled us to select the flowrate of 219 mL h−1 as the optimal for losing fewer HL60 and more PBMC from the imprint (Fig. 5A and B). Different concentrations of bPEI (between 0.01 wt% and 0.03 wt%) were tested as a pre-treatment of the imprint in order to explore the effect of this cationic polymer on the HL60 cell selectivity of the imprint. This positively charged polymer adheres to the particle imprint and as a result increases the electrostatic attraction between the cells and the particle imprint surface. This effect is shown in terms of percentage of decrease of HL60 in Fig. 5C: at 0.015 wt% bPEI treatment, 90% of the PBMCs were released from the imprint whereas at 0.03 wt% bPEI, only 40% were lost because they show higher affinity for binding with the positively charged imprint surface. In terms of HL60 selectivity, 0.015 wt% bPEI treatment turned out to be the optimal concentration as is evident from Fig. 5D. For the other concentrations, the HL60 selectivity remains the same before and after flushing. For optimisation of the concentration of the POL407 treatment of the imprint, different concentrations (between 0% and 3 wt%) of POL407 were used to evaluate the effect on the particle imprint in terms of cell capture and selectivity for HL60 (Fig. 5E and F).
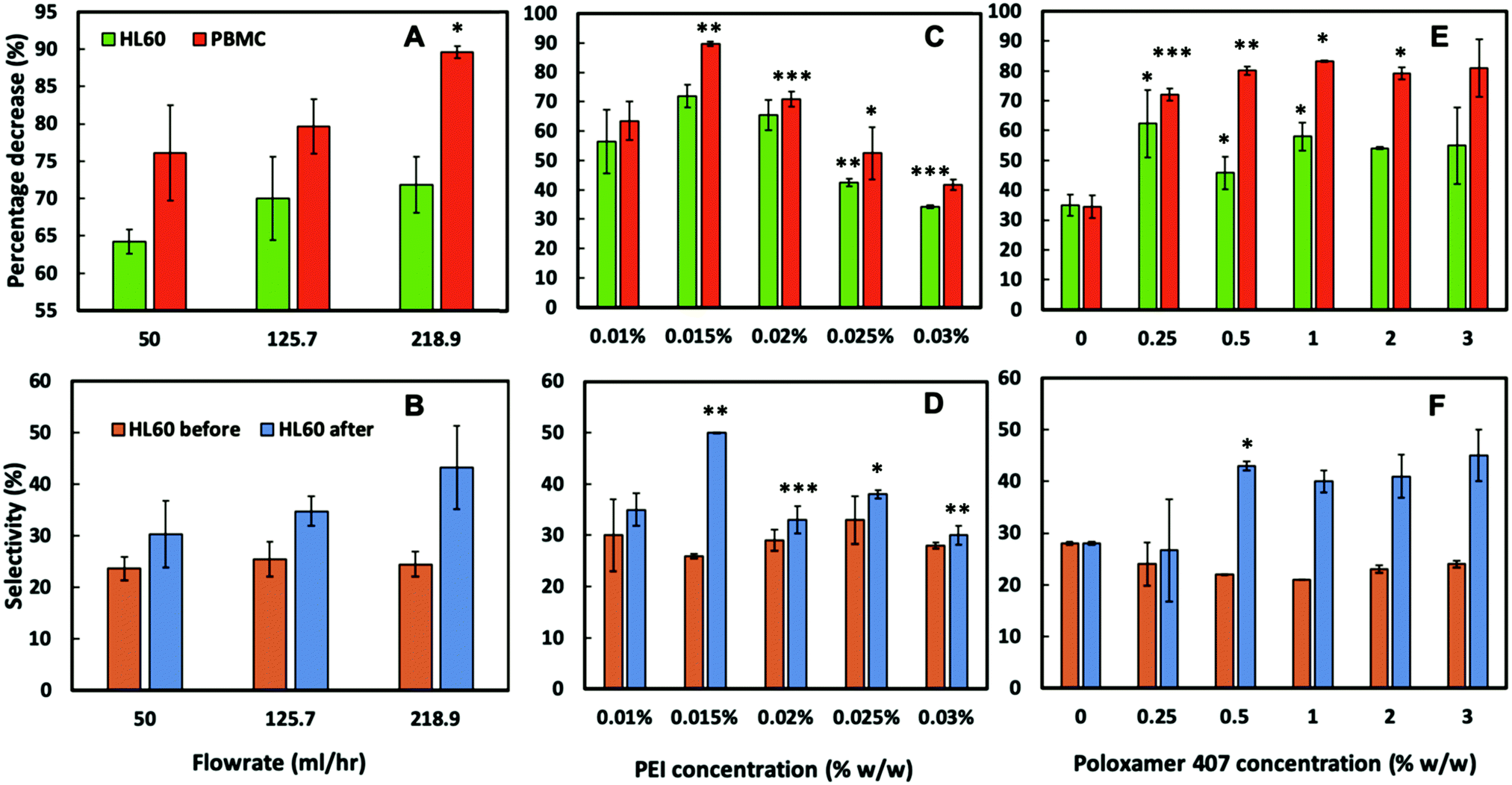 | ||
| Fig. 5 The influence on cell percentage decrease (due to cell capture) and cell selectivity of (A and B) flow rate, (C and D) bPEI concentration on the imprint, and (E and F) POL407 concentration on the imprint after being passed through a oxygen plasma treated 4 cm CA15 particle imprint treated with 0.015 wt% bPEI, at a flow rate of 218.9 mL h−1. Data were expressed as average values ± standard deviations of the mean. P-Values of less than 0.05 were considered significant. *P < 0.05, **P < 0.01, ***P < 0.001. All unpaired two-tailed t-tests were performed in GraphPad v7.0.4. Numeric data are given in Tables S2–S7 (ESI†). | ||
Selectivity for HL60 cells was achieved at POL407 concentrations above 0.5 wt%. The POL407 coating prevents very strong adhesions between the cells and the imprint and therefore improves the HL60 cell selectivity. Thus, predominantly cells that fit tightly into the imprint cavities will be retained as the interaction force of the cell with the imprint is proportional to the contact surface area. For PBMCs, which are much smaller than the imprint cavities, the contact area is very small (point contact) which makes them loosely bound and easy to flush out, which allows HL60 selectivity with respect to the PBMCs. At 0 wt% POL407 (no passivation), the particle imprint showed no selectivity between HL60 cells and the PBMCs: the percentage decrease is the same for both type of cells (35%) and the ratio between HL60 cells and PBMCs did not change (28%/72%) as evident from Fig. 5E and F. This is explained with the indiscriminate strong attraction between the bPEI-treated imprint and the negatively charged PBMCs and HL60 cells. Above 0.25 wt% POL407 treatment, the particle imprint became selective for the HL60 cells. Our results show that above 0.5 wt% POL407, the particle imprint had been saturated with POL407 as the percentage decrease for the two types of cells and the selectivity were similar between 0.5 wt% and 3 wt%.
The particle imprint surface appeared to have become saturated with the highest cell concentration when pre-treated with 1% bPEI and 3% POL 407 (Fig. 5). The cell retention was shown to be proportional to both the seeding cell ratio and the bPEI concentration. Higher bPEI concentrations, whilst retaining more HL60 to the imprint (Fig. 6A) had the effect of loss of selectivity (Fig. 5D) and therefore also increased the PBMC retention. The optimal bPEI concentration was determined to be 0.015% (Fig. 5C and D).
 | ||
| Fig. 6 Cell retention on imprint as a factor of the cell seeding density and bPEI concentration (A). Imprint used was 4 cm in length, coated with 3% POL407 and (B) preferential capture of HL60 against human PBMC on 4 cm PMMA microbeads imprint, oxygen plasma treated and coated with 0.015 wt% bPEI, 3 wt% POL407. The flushing flow rate was of 218.9 mL h−1. | ||
Finally, the cell retention of PBMCs and HL60 was determined (Fig. 6B) with the optimised parameters. The proportion of retained cells was increased with increasing of the cell seeding concentrations for both cell types, however, this effect was far more pronounced for HL60 suggesting a specificity for HL60 over PBMCs was achieved (Fig. 6B). We also conducted experiments with multiple seeding of the imprint up to four times on the chip to check the retention of HL60 cells on the imprint and its selectivity towards HL60 from the HL60/PBMC mixture. According to Fig. S7A (ESI†), the percentage decrease of PBMC and HL60 follow a trend. These percentages decrease with the number of seeding up to a certain value. At this moment, the imprint becomes mostly saturated with HL60 cells.
The percentage decrease at the 3rd and the 4th seeding are similar. This result can also be observed in Fig. S7B (ESI†), where the selectivity of the imprint for the HL60 cells increased until the 3rd seeding and then stagnated. The HL60 cell selection parameters were optimised to achieve blood tumour cell (HL60) specificity over healthy PBMCs. The experiments showed that the higher the flushing flowrate, the fewer the number of cells retained on the imprint. However, it also indicated that the imprint becomes more selective with respect to HL60 cells when the flowrate is increased. The positively charged polyelectrolyte bPEI increased the electrostatic interaction between the cells and the imprint, hence the higher the concentration of the bPEI, the greater the number of cells remaining on the imprint. The passivating polymer, POL407 was used to weaken the cell adhesion and reduce non-specific binding of PBMCs therefore improving selectivity. Selectivity increased with increasing POL407 concentration up to a threshold of 0.5 wt%. Due to the smooth, spherical nature of the imprinted particles, relatively little asperity is observed with the CA15 particle imprint cavities as seen with the cell imprints (see ref. 29). Interactions dependent on the extracellular features are not seen with particle imprints and retention is dependent only on size matching between the targeted cells and the imprint. The optimal parameters for HL60 selective capture from the CA15 PMMA microbeads imprint were the following: flowrate: 219 mL h−1 and imprint treatment with oxygen plasma, 0.015 wt% bPEI and 3 wt% POL407 from a ratio of 25%/75% between the HL60 cells and the PBMCs.
Particle imprints are cheaper and easier to mass produce than cell bioimprints which we have previously demonstrated to also be selective for circulating tumour cells.29 The ease of production and functionalisation of particle imprints, as described here, offer an advantage over bioimprinting, however selectivity and retention rates were lower here, suggesting a compromise between possible universal usage of a particle imprint versus increased selectivity and retention of a personalised bioimprint.
Particle imprints, however, have fewer surface targets compared to whole cell or molecular imprints where relatively small macromolecules such as polysaccharides, enzymes, aptamers, DNA sequences, antibodies as well as whole mammalian cells can also be targeted.32–38 The methodology described here could be adapted for a range of purposes such as described here for capture but also for cell interrogation, for example response to drug treatments on an individualised basis. Depletion of blood cancer cells from healthy cells could result in improved outcomes for patients.39
Conclusions
Here we developed an alternative and inexpensive way to remove blood cancer cells from a mixture with peripheral blood mononuclear cells by using particle imprints. We identified microbeads of closely matching size distribution to the blood cancer cells. A layer of monodisperse CA15 PMMA microbeads of an average diameter of ∼15 μm was produced and replicated on a large scale by PDMS and UV curable PU resin followed by copying of the produced master positive replica with Roll-to-Roll Nanoimprinting lithography to fabricate negative polymeric replicas of the original layer of microbeads. After suitable surface functionalisation of the imprints, we carried out experiments with mixtures of HL60 (blood cancer cells) and healthy PBMCs to explore the selectivity of the imprints towards blood cancer cells. The imprint was functionalised with oxygen plasma treatment, cationic polyelectrolyte and passivation with the Poloxamer 407 polymer. Our method worked very well based on the large size difference between the HL60 and the PBMCs. The HL60 cells closely fitted in the imprint cavities and maximised their contact area with the imprint, while the PBMC have only a point contact and can be easily flushed out, leaving the HL60 cell trapped on the imprint.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PR acknowledges Ecole Nationale Supérieure de Chimie de Rennes for the opportunity to complete this work and the support from the ERASMUS+ mobility grant. AAKD, DJA, LAM and VNP thank Cancer Research UK for the Pioneer Award funding and Higher Education Innovation Fund (UK) for supporting this work. JM also thanks University of Hull for supporting his PhD scholarship. We also thank Help for Health charity (Hull, UK) for supporting this research. The authors thank Mr Tony Sinclair for the SEM images of the CA15 microbeads imprints.References
- J. Medlock, A. A. K. Das, L. A. Madden, D. J. Allsup and V. N. Paunov, Chem. Soc. Rev., 2017, 46, 5110–5127 RSC.
- K. Ren and R. N. Zare, ACS Nano, 2012, 6, 4314–4318 CrossRef CAS.
- A. L. Bole and P. Manesiotis, Adv. Mater., 2016, 28, 5349–5366 CrossRef CAS.
- Y. Ge and A. P. Turner, Trends Biotechnol., 2008, 26, 218–224 CrossRef CAS.
- G. Vasapollo, R. Del Sole, L. Mergola, M. R. Lazzoi, A. Scardino, S. Scorrano and G. Mele, Int. J. Mol. Sci., 2011, 12, 5908–5945 CrossRef CAS PubMed.
- F. L. Dickert and O. Hayden, Anal. Chem., 2002, 74, 1302–1306 CrossRef CAS PubMed.
- O. Hayden, K. J. Mann, S. Krassnig and F. L. Dickert, Angew. Chem., Int. Ed., 2006, 45, 2626–2629 CrossRef CAS PubMed.
- A. Mujahid and F. L. Dickert, Sensors, 2015, 16, 51 CrossRef PubMed.
- Z. Zhang, J. Li., L. Fu, D. Liu and L. Chen, J. Mater. Chem. A, 2015, 3, 7437–7444 RSC.
- M. Jia, Z. Zhang, J. Li, X. Ma, L. Chen and X. Yang, Trends Anal. Chem., 2018, 106, 190–201 CrossRef CAS.
- Z. Altinas, M. Gittens, A. Guerreio, K.-A. Thompson, J. Walker, S. Piletsky and I. E. Tothill, Anal. Chem., 2015, 87, 6801–6807 CrossRef.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- C. O'Brien, E. S. Prideaux and T. Chevassut, Adv. Hematol., 2014, 103175 Search PubMed.
- E. Estey and H. Döhner, Lancet, 2006, 368, 1894–1907 CrossRef.
- J. L. Shipley and J. N. Butera, Exp. Hematol., 2009, 37, 649–658 CrossRef CAS.
- Y. Ofran, M. S. Tallman and J. M. Rowe, Blood, 2016, 128, 488–496 CrossRef CAS.
- D. A. Pollyea, H. E. Kohrt and B. C. Medeiros, Br. J. Haematol., 2011, 152, 524–542 CrossRef.
- J. E. Kolitz, Br. J. Haematol., 2006, 134, 555–572 CrossRef CAS.
- J. M. Bennett, D. Catovsky, M.-T. Daniel, G. Flandrin, D. A. G. Galton, H. R. Gralnick and C. Sultan, Br. J. Haematol., 1976, 33, 451–458 CrossRef CAS.
- A. K. Burnett, Hematol. Amer. Soc. Hematol. Educ. Program, 2012, 1–6 Search PubMed.
- C. S. Viele, Semin. Oncol. Nurs., 2003, 19, 98–108 CrossRef.
- H. Dombret and C. Gardin, Blood, 2016, 127, 53–61 CrossRef CAS.
- C. S. Hourigan and J. E. Karp, Nat. Rev. Clin. Oncol., 2013, 10, 460–471 CrossRef CAS PubMed.
- F. Ravandi, R. B. Walter and S. D. Freeman, Blood Adv., 2018, 2, 1356–1366 CrossRef CAS.
- R. Fakhrullin, I. Choi and Y. Lvov, Cell Surface Engineering: Fabrication of Functional Nanoshells, Royal Society of Chemistry, Cambridge, 2014, p. 164 Search PubMed.
- J. Pan, W. Chen, Y. Mab and G. Pan, Chem. Soc. Rev., 2018, 47, 5574–5587 RSC.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- E. Estey and H. Döhner, Lancet, 2006, 368, 1894–1907 CrossRef.
- A. A. K. Das, J. Medlock, H. Liang, D. Nees, D. Allsup, L. A. Madden and V. N. Paunov, J. Mater. Chem. B, 2019, 7, 3497–3504 RSC.
- M. Leitgeb, D. Nees, S. Ruttloff, U. Palfinger, J. Götz, R. Liska, M. R. Belegratis and B. Stadlober, ACS Nano, 2016, 10, 4926–4941 CrossRef CAS.
- H. Döhner, E. H. Estey, S. Amadori, F. R. Appelbaum, T. Büchner, A. K. Burnett, H. Dombret, P. Fenaux, D. Grimwade, R. A. Larson, F. LoCoco, T. Naoe, D. Niederwieser, G. J. Ossenkoppele, M. A. Sanz, J. Sierra, M. S. Tallman, B. Löwenberg and C. D. Bloomfield, Blood, 2010, 115, 453–474 CrossRef.
- A. L. Bole and P. Manesiotis, Adv. Mater., 2016, 28, 5349–5366 CrossRef CAS.
- T. S. Bedwell and M. J. Whitcombe, Anal. Bioanal. Chem., 2016, 408, 1735–1751 CrossRef CAS.
- S. Sacanna, W. T. M. Irvine, P. M. Chaikin and D. J. Pine, Nature, 2010, 464, 575–578 CrossRef CAS.
- J. Borovicka, S. D. Stoyanov and V. N. Paunov, Nanoscale, 2013, 57, 8560–8568 RSC.
- J. Borovicka, W. J. Metheringham, L. A. Madden, C. D. Walton, S. D. Stoyanov and V. N. Paunov, J. Am. Chem. Soc., 2013, 135, 5282–5285 CrossRef CAS.
- J. Borovicka, S. D. Stoyanov and V. N. Paunov, MRS Proc., 2013, 1498, 127–132 CrossRef.
- J. Borovicka, S. D. Stoyanov and V. N. Paunov, Phys. Rev. E, 2015, 92, 032730 CrossRef.
- C. S. Hourigan and J. E. Karp, Nat. Rev. Clin. Oncol., 2013, 10, 460–471 CrossRef CAS.
- O. J. Cayre and V. N. Paunov, J. Mater. Chem., 2004, 14, 3300–3302 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00531e |
| This journal is © the Partner Organisations 2020 |