
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselectivity of concerted proton–electron transfer at the surface of a polyoxovanadate cluster†
Eric
Schreiber
 ,
William W.
Brennessel
and
Ellen M.
Matson
*
,
William W.
Brennessel
and
Ellen M.
Matson
*
Department of Chemistry, University of Rochester, Rochester, NY 14611, USA
First published on 20th December 2022
Abstract
Proton-coupled electron transfer (PCET) is an important process in the activation and reactivity of metal oxide surfaces. In this work, we study the electronic structure of a reduced polyoxovanadate-alkoxide cluster bearing a single bridging oxide moiety. The structural and electronic implications of the incorporation of bridging oxide sites are revealed, most notably resulting in the quenching of cluster-wide electron delocalization in the most reduced state of the molecule. We correlate this attribute to a change in regioselectivity of PCET to the cluster surface (e.g. reactivity at terminal vs. bridging oxide groups). Reactivity localized at the bridging oxide site enables reversible storage of a single H-atom equivalent, changing the stoichiometry of PCET from a 2e−/2H+ process. Kinetic investigations indicate that the change in site of reactivity translates to an accelerated rate of e−/H+ transfer to the cluster surface. Our work summarizes the role which electronic occupancy and ligand density play in the uptake of e−/H+ pairs at metal oxide surfaces, providing design criteria for functional materials for energy storage and conversion processes.
Introduction
The net uptake and transfer of hydrogen-atom (H-atom) equivalents is an important function of metal oxide materials, allowing for applications in small molecule activation and hydrogen storage, as well as optoelectronic devices.1–11 Understanding and controlling thermochemical parameters of surface-bound H-atoms is important for tailoring the reactivity of a system (e.g. site-selectivity, reversible H-atom binding, O-atom vacancy formation). To date, the precise coordination modes of surface-doped H-atoms in reducible metal oxides remain unclear, as the heterogeneity of surface structures of these systems makes direct determination of binding sites challenging.1,12–17To gain atomistic understanding of interfacial proton and electron transfer at metal oxide surfaces, we turn to a class of molecular analogues of these materials known as polyoxometalates (POMs). These multimetallic structures comprise three or more MOx units featuring terminal and edge-sharing oxide ligands. The resultant surface composition of these molecular metal oxide assemblies mimics those of their heterogenous counterparts. While most POMs are derived from high-valent, d0, early transition metal ions (e.g. WVI, MoVI, VV), the reduced analogues have been demonstrated to possess diffuse electronic structures upon reduction, resembling those of extended metal oxides.18–21 The similarities in physicochemical properties between POMs and heterogenous oxides render these clusters superb candidates for modelling the interfacial reactivity of extended structures, providing an approach for probing the mechanisms of charge transfer and compensation at material surfaces.
Net H-atom transfer, or electron/proton transfer, to POM surfaces can be localized either at bridging (Ob) or terminal (Ot) oxide positions. Typically, preferential binding of protons and H-atom equivalents to bridging oxide moieties has been observed as a consequence of their heightened basicity.22–27 However, in a few examples, reduced POMs have been shown to generate terminal oxygen-atom (O-atom) defects following electron/proton co-doping of the assembly.28–31 Of most relevance this work, our group has reported this mode of reactivity in a series of Lindqvist-type polyoxovanadate-alkoxide (POV-alkoxide) clusters, [V6O7(OMe)12]n (n = 1−, 0, 1+). The operative form of net H-atom addition to these organofunctionalized assemblies involves activation of a terminal V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety, resulting in the formation of an unstable hydroxide which abstracts an additional H-atom equivalent to generate a labile aquo ligand at the surface of the cluster.32,33 We hypothesize that the observed localization of net H-atom uptake at Ot groups is a consequence of organic saturation of the bridging oxide positions at the surface of the assembly (i.e. μ2-OMe1− as opposed to μ2-O2−).
O moiety, resulting in the formation of an unstable hydroxide which abstracts an additional H-atom equivalent to generate a labile aquo ligand at the surface of the cluster.32,33 We hypothesize that the observed localization of net H-atom uptake at Ot groups is a consequence of organic saturation of the bridging oxide positions at the surface of the assembly (i.e. μ2-OMe1− as opposed to μ2-O2−).
To further understand site-selectivity in proton coupled electron transfer (PCET) at metal oxide surfaces, we turned to a complex with asymmetrical surface structure. First reported by Hartl in 2009, [V6O8(OMe)11]0 (1-V6O80) features a Lindqvist core with eleven bridging methoxide groups and a single bridging oxide (Fig. 1).34 The isolated cluster is neutral in charge, with a VIV3VV3 oxidation state distribution. Notably, like its dodecamethoxide congener, [V6O7(OMe)12]n, 1-V6O80 exhibits rich redox activity, with four additional charge states accessible as suggested by cyclic voltammetry (CV; Fig. 1). With relevance to our interest in the selectivity of net H-atom installation to cluster surfaces, substitution of a single bridging methoxide functionality with a μ2-O2− provides a platform by which the removal of organic groups from M-O-M linkages can result in switching of reactivity from terminal to bridging oxides in reduced POMs. Additionally, the generation of a single μ2-O2− moiety provides the opportunity to manipulate the stoichiometry of PCET to a single e−/H+ process.
 | ||
| Fig. 1 CV of 1 mM 1-V6O80 (green, top) and V6O7(OMe)12 (black, bottom) in MeCN with 100 mM [nBu4N][PF6] supporting electrolyte (scan rate = 100 mV s−1). Labelled on the curves are the structures, oxidation state distributions for each charge state of the cluster, and the open circuit voltages indicated with a black arrow. | ||
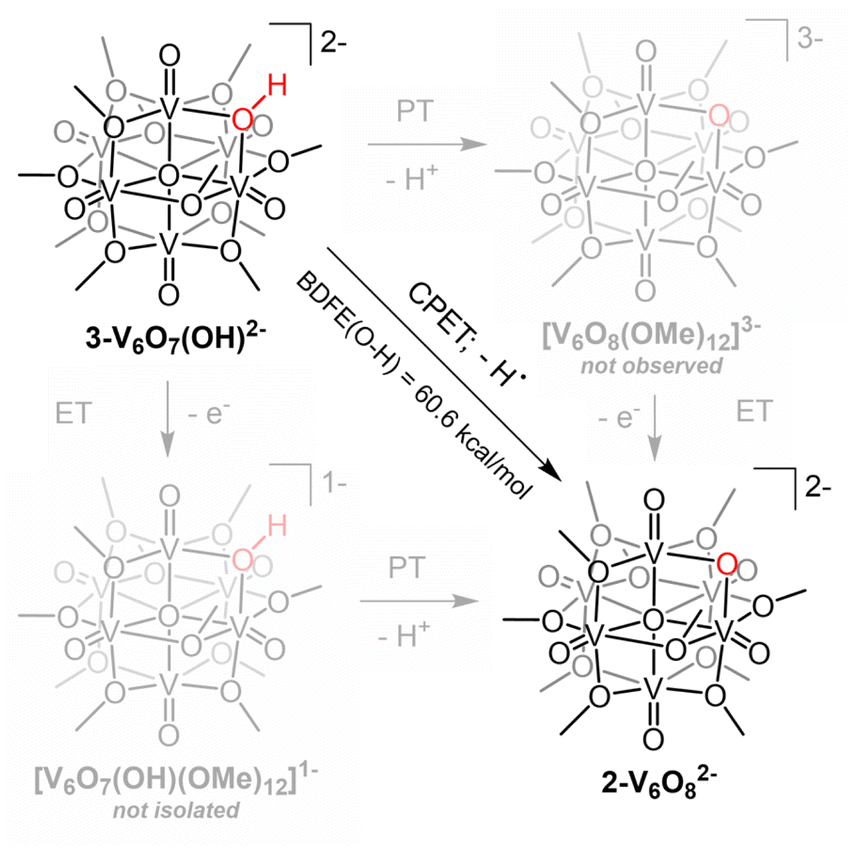
Here, we report concerted proton-electron transfer to 1-V6O80 and its di-reduced redoxomer, [V6O8(OMe)11]2− (2-V6O82−), revealing several features specific to PCET at the surface of this architecture:
(1) E−/H+ transfer to 1-V6O80 does not occur at a particular site or with well-defined stoichiometry, whereas regioselective, 1e−/1H+ transfer occurs exclusively to the bridging oxide in the reduced assembly, 2-V6O82−.
(2) Reduction of 2-V6O82− to a VIV6 state is only possible via PCET (i.e. by addition of a reductant capable of simultaneously delivering a proton along with its electron).
(3) The rate of PCET in 2-V6O82− (e− distrib. = VIV5VV) is accelerated in comparison to its isovalent, organofunctionalized congener, [V6O7(OMe)12]1− (e− distrib. = VIV5VV), suggesting a kinetic preference for net H-atom transfer to bridging oxide moieties at the surface of metal oxides.
In whole, these results outline structure–function relationships that manipulate the reactivity of metal oxide assemblies with H-atom equivalents, providing design considerations for optimizing PCET in ligand-capped metal oxide nanomaterials.
Results and discussion
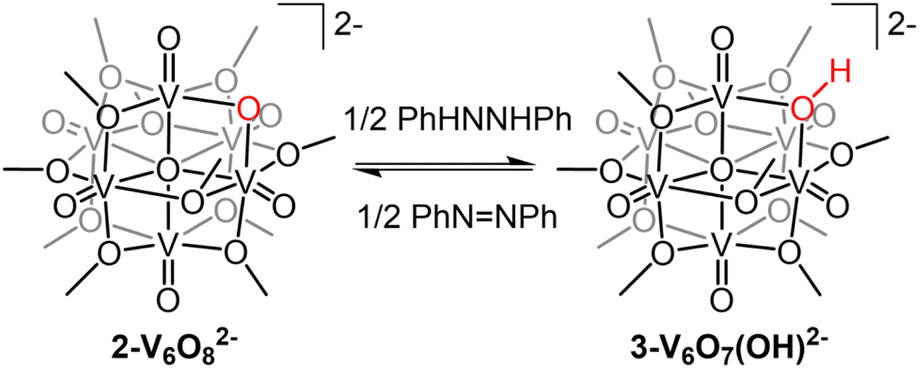
Investigation of e−/H+ transfer to 1-V6O80 was performed through addition of half an equivalent of the reductant 5,10-dihydrophenazine (H2Phen) in acetonitrile (MeCN). The 1H NMR spectrum of the crude reaction mixture revealed a series of paramagnetically shifted resonances ranging from 30 to −20 ppm, alongside the four signals of the starting material (Fig. S1†). The complicated spectrum, featuring a total number of resonances that exceeds the lowest-possible symmetry the cluster can adopt, suggests that addition of H2Phen to 1-V6O80 results in the formation of a mixture of products. In addition, the presence of resonances up-field of 0 ppm suggests that a terminal oxygen defect is formed in at least one reaction product, as similar features have been previously observed on O-atom deficient clusters.35–37 Attempts to separate vanadium-containing product(s) from the reaction mixture were unsuccessful.The lack of regioselectivity of PCET to 1-V6O80 is surprising, as typically the basicity of bridging oxide moieties at POM surfaces localizes proton interactions to these sites.23,38,39 We can justify the disparate reactivity observed in this case through discussion of the oxidation state distribution of vanadium centres in 1-V6O80 (VIV3VV3). The formal assignment of three vanadium(V) ions contained within the cluster core necessitates at least one VVO moiety occupy a position where the metal ion is flanked by four bridging methoxide ligands. Previous work from our group has demonstrated that PCET to these electron deficient vanadium-oxo moieties results in the irreversible formation of an O-atom vacancy.32,33 We thus hypothesized that chemical reduction would provide the opportunity to improve the selectivity of this process; reduction of the number of organosaturated VVO centres available for O-atom defect formation would translate to localization of net H-atom installation to the more basic, bridging oxide site.
The electrochemical profile of 1-V6O80 indicates that two reduced forms of the cluster should be chemically accessible (Fig. 1), following sequential 1e− reductions of the assembly to its mono- or di-anionic form. We opted to investigate the fully reduced form of the assembly, 2-V6O82−, as the oxidation state distribution of this cluster features a lone VV centre (VIV5VV), expected to reside adjacent to the μ2-O2− moiety. This cluster provides an opportunity to study PCET to a single vanadate unit, minimizing the potential for side reactions (e.g. O-atom defect formation). Reduction of 1-V6O80 was achieved by exposure of the assembly to 2 equiv of tetrabutylammonium borohydride ([nBu4N][BH4]) in MeCN at 90 °C (Scheme 1). Following work-up, the product was isolated as a dark blue powder in good yield (70%, see experimental section for additional information). Electrospray ionization mass spectrometry (ESI-MS) and CV of the product suggests that successful reduction of the cluster to the divalent anion was achieved under these reaction conditions (Fig. S2 and S3†).
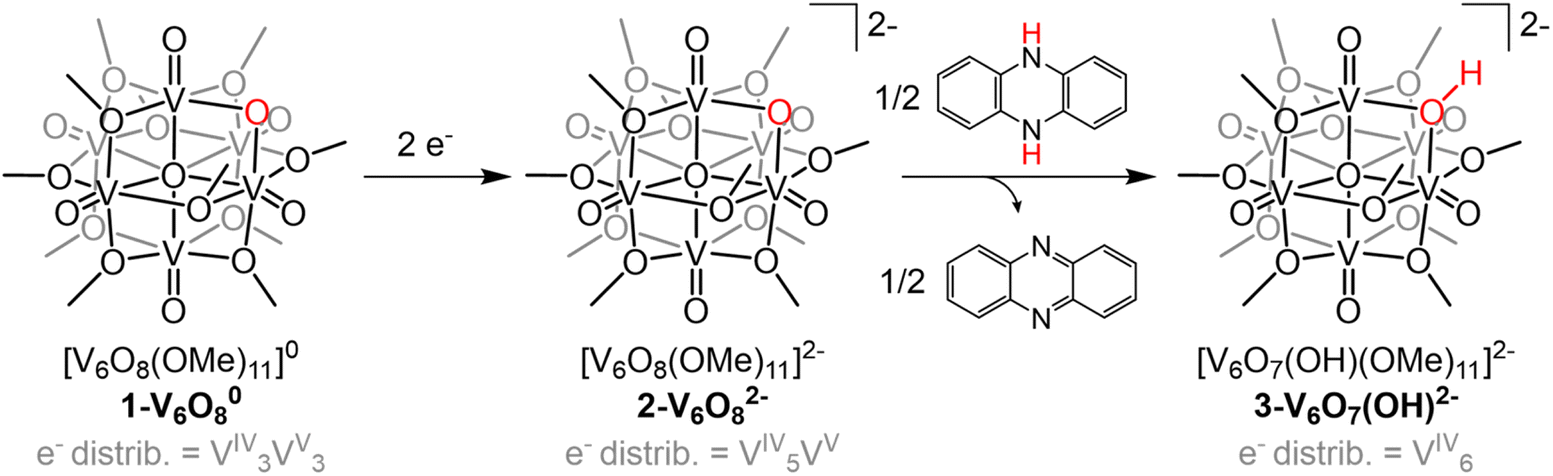 | ||
| Scheme 1 Synthesis of 2-V6O82− and 3-V6O7(OH)2−. | ||
Characterization of 2-V6O82− by 1H NMR spectroscopy provides a facile means of differentiating between charge states of the cluster. The 1H NMR spectrum of 1-V6O80 features four paramagnetically shifted and broadened resonances corresponding to the protons of the bridging methoxide ligands (δ: 11.2, 12.3, 19.3, and 20.2 ppm in MeCN-d3). The number of resonances is consistent with the C2v symmetry of the cluster. A similar pattern of resonances was observed for 2-V6O82− (δ: 20.7, 22.6, 23.5, 26.7 ppm), with significant deshielding of protons as a result of the reduction of the vanadium oxide core (Fig. S5†).
Single crystals of 2-V6O82− suitable for single crystal X-ray diffraction (SCXRD) were grown from vapor diffusion of pentane into a solution of cluster in tetrahydrofuran. To obtain crystals suitable for analysis, the bis-cobaltocenium counter ion was used; the non-coordinating nature of this cation does not alter the cluster structure. Solution of the structure confirmed the expected Lindqvist-type POV-alkoxide bearing 11 bridging methoxides and a single bridging oxide, with two organometallic cations (Fig. 2). Overall, the V–O bond metrics of this complex are consistent with other mixed-valent POV-alkoxides, with VOt and V–OMeb lengths of 1.599(5)–1.617(5) Å and 1.986(5)–2.020(5) Å, respectively (Table 1).
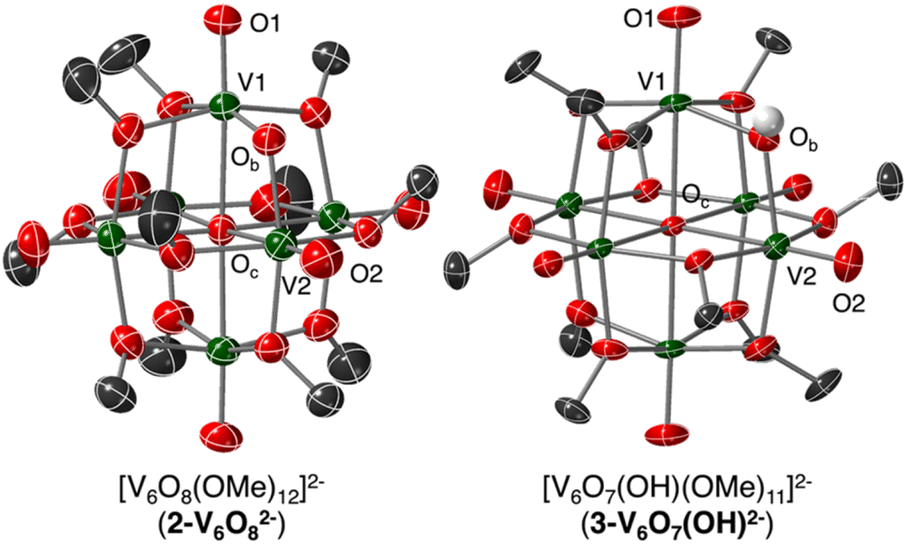 | ||
| Fig. 2 Molecular structures of 2-V6O82− and 3-V6O7(OH)2− shown with 50% probability ellipsoids. Counter ions, solvent molecules, and selected hydrogen atoms contained within the unit cell have been removed for clarity. Key: dark green ellipsoids, V; grey ellipsoids, C; red ellipsoids, O; white spheres, H. | ||
| Bond | 2-V6O82− | 3-V6O7(OH)2− |
|---|---|---|
| a sat = Sites where all V ions are bridged by four oxides saturated by either organic groups or a proton. | ||
| VOavg.t |
1.609(6) Å | 1.611(2) Å |
| V–OMe/Hb avg. | 1.989(50) Å | 2.002(7) Å |
| V1–Ob | 1.772(5) Å | — |
| V2–Ob | 1.952(4) Å | — |
| Vn–Oc (n = 1, 2) | 2.340(4), 2.337(4) Å | — |
| V(sat)–Oc avg.a | 2.280(21) Å | 2.312(7) Å |
The bond lengths between the bridging oxide ligand and its neighbouring V ions indicate a polarized bridging unit (V–Ob = 1.772(5), 1.952(4) Å; ΔV–Ob = 0.180 Å) in the solid state at reduced temperature (223 K). The significant differences in V–O bond lengths suggest charge localization across the V–O–V regime under the conditions of the SCXRD experiment. Such asymmetry in the solid state is consistent with previously reported VIV/V mixed-valent vanadium-oxo dimers which feature similarly differentiated V–Ob bond lengths on either side of the μ2-O2− ligand.40,41 Indeed, these works describe V2O3 subunits supported by multidentate ligands, and have been shown to feature ΔV–Ob values as high as 0.2 Å resulting from localization of VIV and VV in the solid phase at low temperature.40 In these examples, the high-valent, d0 vanadium ion is bound more tightly to the oxide ligand, as indicated by a truncated V–Ob bond distance.
The degree of charge localization in 2-V6O82− in the solid state is a distinct feature of this multimetallic complex. Other mixed-valent POV-alkoxides bearing bridging oxide ligands reported by Zubieta ([V6O9(OH)4(OCH2)3C2H3]2−, ΔV–Ob: 0.105 Å) and Matson ([V6O9(OH)4(OCH2)3CNO2]2−, ΔV–Ob: 0.1004 and 0.0729 Å), feature less polarized V–O–V moieties as a consequence of more diffuse electronic structures under similar SCXRD experimental conditions.19,42 This is likely due to the presence of four analogous OtV(μ2-O)(μ2-OH)(μ2-OR)2 units throughout the equatorial plane of these complexes with alternating VIV/VV oxidation states. The lack of additional equivalent vanadate units in 2-V6O82− further limits electron shuttling beyond the two μ2-oxide-bound V centres, rendering its electronic structure more similar to the ligand-supported oxide-bridged V dimers described above.40,41
To further understand the origins of the observed V–O–V bond polarization, bond valence sum (BVS) calculations were performed. This technique correlates bond metrics about an atom to its oxidation state. In the previous report on 1-V6O80, the single crystal X-ray structure was reported, but the solution found only a partial molecule in the asymmetric unit. Fortunately, in the case of 2-V6O82−, all vanadium centres are found in general positions. BVS calculations on the V ions of 2-V6O82− revealed the expected VIV5VV oxidation state distribution in the di-anionic state (Table S2†). The only VV centre was located adjacent to the μ2-O2-, assigned to the vanadium atom bound through the shorter (1.772(5) Å) bond. Importantly, this degree of charge localization is likely amplified in the solid state; subsequent spectroscopic observations suggest that some amount of charge delocalization can occur about the V–O–V region in solution at elevated temperatures (vide infra).
Analysis of 2-V6O82−via electronic absorption spectroscopy (UV-vis/NIR) provides insight into the effect that the introduction of a superexchange-mediating ligand (i.e. μ2-O2−) has on the electronic structure of the reduced assembly. The absorption profile of the fully reduced species 2-V6O82− features at least five transitions between 800 and 1300 nm with a λmax at 1015 nm (εmax = 548 M−1 cm−1) (Fig. 3). This is broadly indicative of intervalence charge transfer (IVCT) between d1 (VIV) and d0 (VV) ions. In comparing the electron absorption spectrum of 2-V6O82− to that reported for the isoelectronic, dodecamethoxide species [V6O7(OMe)12]1− (e− distrib. = VIV5VV; εIVCT: 944 M−1 cm−1), the IVCT band of 2-V6O82− is significantly less intense. An analogous comparison has been made between 1-V6O80 (e− distrib. = VIV3VV3; εIVCT: 903 M−1 cm−1) and its isoelectronic, organic-saturated congener [V6O7(OMe)12]SbCl6 (εIVCT: 1160 M−1 cm−1).34 Hartl and coworkers note that significant quenching of the IVCT band in 1-V6O80 is attributed to suppression of electronic communication across the cluster core by electronic isolation of the V ions bound to the bridging oxide. We anticipate that this phenomenon holds true across the accessible charge states of the undecamethoxide complex. Additional evidence for localization of electron density across the [V2O3]3+ regime in this framework can be obtained by comparing the UV-vis/NIR spectrum of 2-V6O82− to other [V2O3]3+-containing complexes in the literature.40,41 Mixed-valent dimeric species feature a broad absorption band beginning at 750 nm and stretching beyond 1000 nm in analogy the observed spectrum for 2-V6O82−.
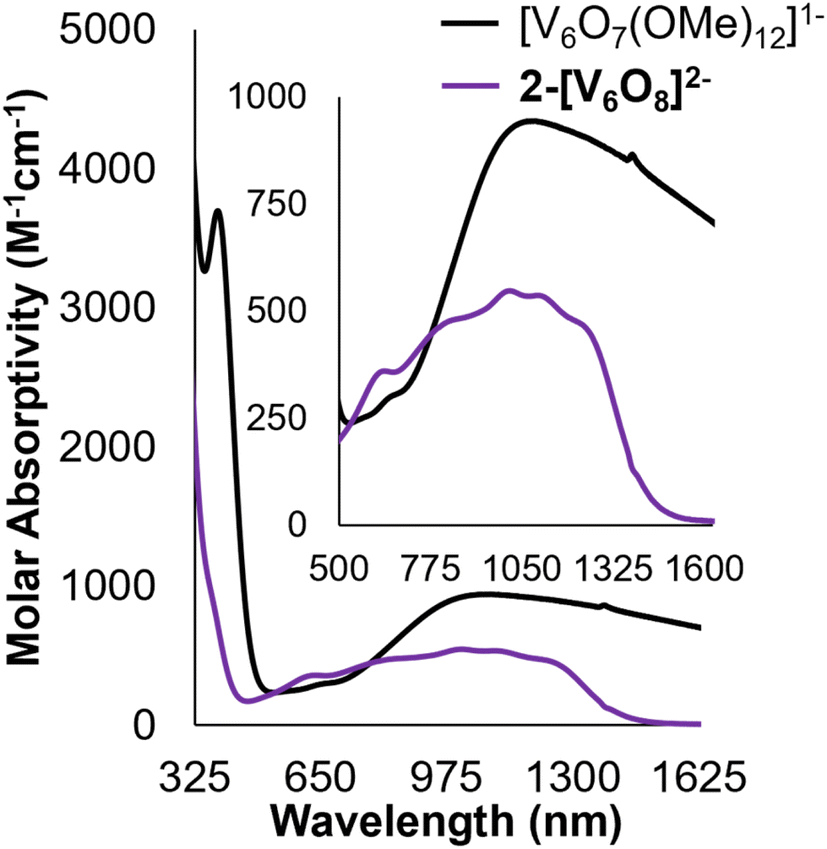 | ||
| Fig. 3 Electronic absorption spectra of 2-V6O82− and [V6O7(OMe)12]1− collected in MeCN at 21 °C. Inset shows magnification of the IVCT and d–d bands located in the NIR region of the spectra. | ||
The remaining features in the electronic absorption spectrum of 2-V6O82− can be assigned to a series of d–d transitions within individual VIV centres in the cluster. The molar absorptivities of these absorption features are more intense than expected, likely due to spectral overlap with the IVCT band.19,34 The presence of at least four resolved d–d transitions is consistent with a reduction in symmetry of the cluster core, with charge localization imparting multiple electronic environments for the remaining VIV centres.
With the electronic structure of 2-V6O82− established, we turned our attention to the reactivity of the reduced undecamethoxide complex with an H-atom equivalent (Scheme 1). Exposure of 2-V6O82− to half an equivalent of H2Phen results in an immediate colour change of the reaction solution from dark blue to teal. Analysis of the reaction mixture by 1H NMR spectroscopy reveals coalescence of the surface methoxide resonances to a single, asymmetric feature at 24.4 ppm (Fig. S5†). No resonances are observed at chemical shifts below 0 ppm, indicating that terminal O-atom vacancy formation did not occur under these reaction conditions. This data suggests that HAT to 2-V6O82− results in the selective conversion of the μ2-O2− ligand to a bridging hydroxide. ESI-MS of the reaction mixture possesses a peak with an observed mass approximately 0.5 m/z higher than 2-V6O82− (387.9 vs. 387.5 m/z; Fig. S6†), consistent with formation of [V6O7(OH)(OMe)11]2− (3-V6O7(OH)2−).
Analysis of the electronic absorption spectrum of 3-V6O7(OH)2− provides strong evidence for the formation of a reduced POV-alkoxide core bearing six VIV ions with a bridging hydroxide on the cluster surface (Fig. 4). The overall line shape and absorption intensity of 3-V6O7(OH)2− is very similar to that of the fully reduced, dodecamethoxide compound [V6O7(OMe)12]2− (ox. state distrib.: VIV6). However, the d–d transition in the spectrum of complex 3-V6O7(OH)2− (λ = 1024 nm) is blue shifted by 24 nm with respect to [V6O7(OMe)12]2−; we attribute this to decreased electron donation by the μ2-O2−-bound H-atom versus a methoxide substituent.
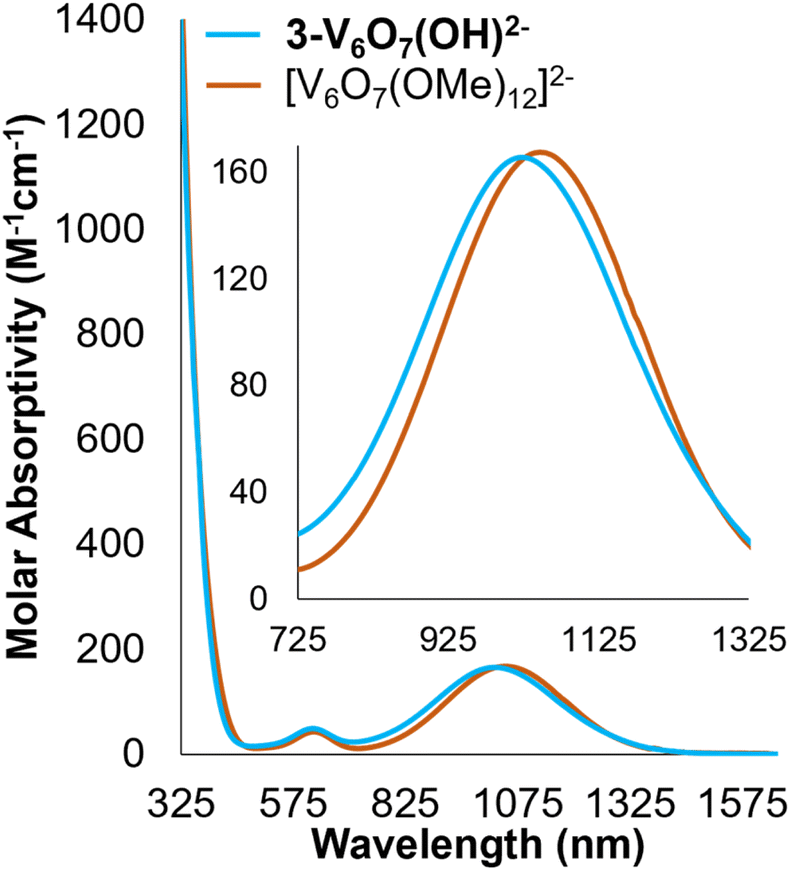 | ||
| Fig. 4 Electronic absorption spectra of 3-V6O7(OH)2− (light blue) and [V6O7(OMe)12]2− (orange) collected in MeCN at 21 °C. The inset spectrum shows a magnification of the NIR region of the spectra for clarity. | ||
To unambiguously determine formation of 3-V6O7(OH)2−, crystals suitable for SCXRD were grown by vapor diffusion of diethyl ether into a solution of cluster in MeCN. The asymmetric unit constitutes one half of a cluster with a fully-occupied [nBu4N]+ counterion, supporting the purported dianionic charge state (Fig. S7†). Subsequent analysis and refinement of the structural data confirmed the expected structure to be a Lindqvist hexavanadate cluster featuring 11 bridging methoxides and a single bridging oxygen featuring V–O–V bond lengths and angles which support the presence of a hydroxide functionality (see Experimental section for more details).
The overall di-anionic charge of 3-V6O7(OH)2−, confirmed through X-ray crystallography, provides additional support for the proposed for the formation of an isovalent, VIV6 assembly upon uptake of a net H-atom equivalent. This result is intriguing, as additional reduction of the parent Lindqvist ion to this state is not possible by electron transfer alone (Fig. S8†). The simultaneous addition of a proton is necessary to facilitate reduction of the Lindqvist ion.
The observed regioselectivity in PCET to 2-V6O82− supports our hypothesis that reduction-induced charge localization directs reactivity to the bridging oxide of the cluster surface. This system represents only the second example of tuneable regioselectivity for net H-atom uptake at the surface of POMs; the first having been reported by Launay and Pope. In their work, 6e−/6H+ reduction of Keggin-type polyoxotungstates produces a complex with three terminal WIV-aqua moieties on a single cluster face.28,29 Despite subsequent mechanistic studies by Christian, no proposals have been put forward to rationalize the change in site-selectivity of e−/H+ uptake.43 In our system, by analysing the electronic structure of the V6O8n platform, we have identified the underlying basis behind the change in PCET regioselectivity as the extent of electronic delocalization throughout the cluster core.
Thermochemical and kinetic analysis of PCET to 2-V6O82−
Interested in understanding how the localized electronic structure and e−/H+ stoichiometry of 3-V6O8(OH)2− might influence the thermodynamics of the μ2-O–H bond, we performed studies aimed at abstracting the H-atom equivalent. Reversibility of the PCET process was confirmed through the reactivity of 3-V6O7(OH)2− with (2,2,6,6,-tetramethylpiperidin-1-yl)oxyl (TEMPO; Fig. S5†). Addition of one equivalent of the substrate to the reduced cluster results in an immediate colour change to dark blue, consistent with conversion of 3-V6O7(OH)2− to 2-V6O82−. Analysis of the 1H NMR spectrum of the crude reaction mixture indicated that the nitroxyl radical was converted to the corresponding N-hydroxyl moiety of TEMPO-H (Fig. S5†). In addition, four paramagnetically shifted and broadened resonances were observed between 18–28 ppm, supporting formation of 2-V6O82−via abstraction of a H-atom from the surface of the cluster.To quantify the BDFE(O–H) of complex 3-V6O7(OH)2−, we next monitored the extent of reaction between the 2e−/2H+ donor hydrazobenzene (Hydz, BDFE(N–H)avg = 60.9 kcal mol−1) and 2-V6O82− in MeCN-d3 (Scheme 2).44 The 1H NMR spectrum revealed partial conversion of the cluster to 3-V6O7(OH)2−, as evidenced by the growth of a signal at ∼24 ppm (Fig. S10†). Further evidence for successful e−/H+ transfer is noted in the observation of resonances corresponding to azobenzene (Azo), the oxidation product of Hydz. Signals of the starting materials, Hydz and 2-V6O82−, were also observed, suggesting that the PCET process had reached equilibrium (Fig. S11–14†). By employing a modified version of the Nernst Equation, the adjusted BDFE of a reagent under equilibrium conditions can be determined.1 We use the BDFE(N–H)avg of Hydz, number of H-atom equivalents transferred per molecule (n = 2) and the ratio of [Hydz] to [Azo] at equilibrium (values determined by integration of the 1H NMR spectrum after 14 days of reaction) to calculate the BDFE(O–H) of 3-V6O7(OH)2− (60.6 ± 0.1 kcal mol; Table S3†).
 | ||
| Scheme 2 H-atom exchange equilibrium between 2-V6O82− and Hydz. | ||
The BDFE(O–H) of 3-V6O7(OH)2− is in a similar range to values reported previously for reduced vanadium oxide assemblies by our research group. Specifically, a hexavanadate cluster bearing bridging hydroxide moieties and a VIV6 core, [V6O7(OH)6(TRIOLNO2)2]2−, was found to have a BDFE(O–H)avg of 61.6 kcal mol−1.19 It is important to note that this thermochemical value describes the average BDFE for the loss of two H-atoms ([V6O7(OH)6(TRIOLNO2)2]2− ⇌ [V6O9(OH)4(TRIOLNO2)2]2− + 2 H˙), rendering direct comparison between these two systems challenging. However, the overall similarity of observed BDFE(O–H)s indicates that despite differences in ligand identity and degree of net H-atom functionalization between both complexes, the oxidation state distribution of vanadium ions across the cluster core plays an important role in dictating the BDFE(O–H) of surface hydroxide moieties.
PCET processes typically proceed by one of three major pathways: electron-proton transfer (ET–PT), proton–electron transfer (PT–ET), or concerted proton-electron transfer (CPET), described in a thermochemical square scheme (Scheme 3). Electrochemical characterization of 2-V6O82− indicates that this cluster cannot be further reduced, eliminating ET–PT as a potential pathway for this reaction (Fig. S8†). To probe the relevance of PT–ET as a possible mechanism, we reacted organic acids with 2-V6O82−; exposure of the reduced assembly to protons generates a mixture of species, some of which contain up-field signals corresponding to the formation of an O-atom defect(s) (Fig. S9†). The lack of cluster stability in the presence of weak organic acids renders the PT–ET pathway similarly non-viable. These preliminary experiments suggest that CPET is the most likely pathway of net H-atom uptake at the cluster surface.
 | ||
| Scheme 3 Thermochemical square scheme for PCET reactivity at the surface of 2-V6O82−, outlining the basic mechanisms for e−/H+ uptake at μ2-O2− ligand. | ||
To confirm this mechanism, we performed a series of kinetic analyses. Electronic absorption spectroscopy was used to monitor the reduction of 2-V6O82− by H2Phen under pseudo-first order reaction conditions at −25 °C (Fig. 5). The absorbance at 1024 nm was tracked over the course of the reaction, as complex 2-V6O82− features significantly higher absorbance than 3-V6O7(OH)2− at this wavelength. Fitting the data produced a series of pseudo-first order rate constants (kobs) which, when related to the concentration of reductant, yields a linear plot. The experimental rate constant, kexp (0.53 ± 0.01 M−1 s−1), can be found from the slope of this line (Fig. 6). Overall, the rate law is determined to be first order in H2Phen and second order overall, indicating a bimolecular transformation in the rate determining step of the PCET reaction.
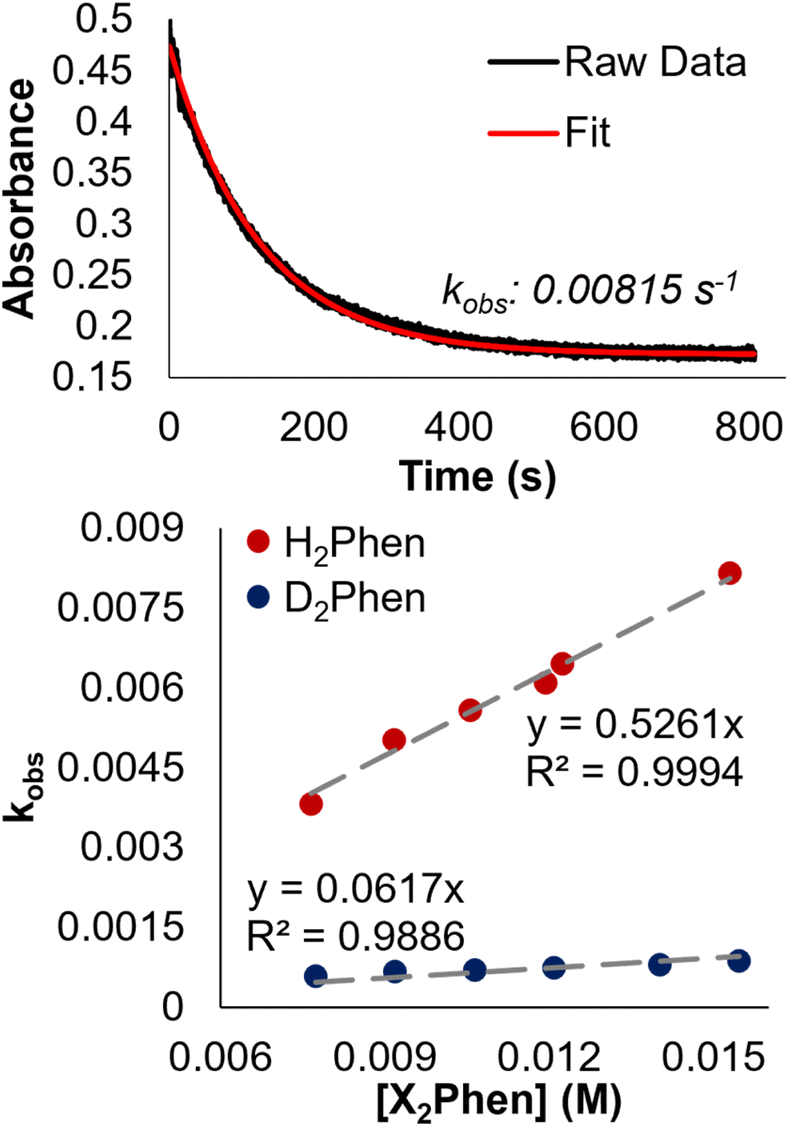 | ||
| Fig. 5 Determination of the observed pseudo first-order rate constant, kobs, for the reduction of 2-V6O82− (1.45 mM) by H2Phen (15.3 mM) in MeCN at −25 °C by monitoring absorbance at 1024 nm over the reaction coordinate (top). Plot of kobs as a function of [X2Phen], where X = H, D. Concentration of 2-V6O82− was held constant at 1.45 mM, and reductant concentration was varied between 7.65 and 15.3 mM. The slope of the resultant lines provides the experimentally derived second-order rate constants, kexp. Comparison of kexp for H2Phen and D2Phen yields a KIE of 8.5. | ||
 | ||
| Fig. 6 Eyring plot for the reaction of 2-V6O82− (1.45 mM) with H2Phen (10.55 mM) in MeCN between −40 and 10 °C. Y-axis values were determined by dividing kobs by the concentration of H2Phen, providing the rate constant, kexp. | ||
Further evidence for CPET was derived from kinetic isotope effect (KIE) experiments using the deuterium-labelled reductant: D2Phen. Pseudo first-order reactions describing the rate of formation of [V6O7(OD)(OMe)11]2− using D2Phen yielded a sharp decrease in kexp (0.062 ± 0.003 M−1 s−1), translating to a KIE of 8.5.
Comparison of kexp for the reduction of 2-V6O82− at −25 °C with that determined for the isoelectronic species [V6O7(OMe)12]1− (0.033 ± 0.002 M−1 s−1) at the same temperature reveals that the generation of the bridging hydroxide proceeds ∼10 times faster than HAT to the terminal oxo (Table 2).32 Both clusters undergo bimolecular CPET reactions with H2Phen; however, this disparity in reaction rate and regioselectivity, despite fewer accessible reactive sites in 2-V6O82−, indicates that changes in the electronic structure of the cluster by the introduction of a single bridging oxide impart significant changes to the PCET process.
Interested in understanding the differences in the rates of PCET between 2-V6O82− and [V6O7(OMe)12]1−, we performed Eyring analysis for the reaction of 2-V6O82− with H2Phen. This involves variable temperature kinetic measurements, allowing us to assess activation parameters. The Eyring plot was obtained by tracking reactions at −40 to 10 °C, yielding the activation parameters listed in Table 2 (Fig. 6). Analogous experiments have been reported for the organosaturated species, allowing for direct comparison of transition state thermodynamics in both systems.32
We note a large, negative activation entropy (ΔS‡ = −34 cal K−1 mol−1) for this reaction, indicating a well-ordered transition state involving an H-bond between the μ2-oxide of 2-V6O82− and an N–H moiety of the reductant. A similar H-bonding transition state motif is invoked in the reduction of terminal V=Ot bonds by PCET reagents, and the Gibbs free energy of activation for reduction of 2-V6O82− is nearly identical to that of [V6O7(OMe)12]1− (Table 2, Fig. S18 and S19†). We hypothesize that, despite both systems featuring analogous activation parameters, differences in basicity of the relevant surface oxide moieties (i.e. terminal versus bridging) are principally the source of the disparate reaction rates. Indeed, tailoring basicity in PCET reactions has been shown to have a profound effect on reaction rates.45–49 By introducing a strongly basic region on the cluster face, the H-bond which forms prior to CPET may be stronger, enabling kinetically-controlled regioselectivity of PCET to the μ2-oxide-containing POV-alkoxide. The role of surface basicity in the rate of CPET is the subject of ongoing investigations in our group.
Conclusions
Control over site-selectivity in PCET processes at ligand-capped nanoparticles and molecular metal oxides is of significant importance for governing the reactivity of these systems. Here, we present the PCET reactivity of the Lindqvist framework V6O8(OMe)11 in different charge states. The as-synthesized, neutral-charged species features complex PCET reactivity, with terminal oxo activation being favoured over net H-atom installation at the bridging oxide. This is due to the presence of reactive VVO moieties throughout the entire molecule, which feature competitive reactivity with the bridging oxide.
Subsequent alterations to the electronic structure of the metal oxide core via reduction provides a method for controlling the regiochemistry of CPET at the surface of cluster. By injecting electrons into the complex to access its most reduced form, we induce charge localization and force the only reducible VV centre to bind to the μ2-O2− in 2-V6O82−. The high relative basicity of the bridging ligand asserts kinetic dominance for PCET over the terminal oxo, giving rise to the selective formation of a stable, bridging hydroxide on the cluster face. This reversal in regioselectivity provides only the second example to date of charge state-induced site-switching of PCET and a rare example of single e−/H+ reactivity at a POM surface. The kinetic advantage imparted by this basic moiety is highlighted by the acceleration of PCET rate at 2-V6O82− by almost an order of magnitude over the isovalent, dodacamethoxide congener [V6O7(OMe)12]1−, which undergoes irreversible 2e−/2H+ transfer to form an oxygen vacancy. Thermochemical analyses found the strength of the O–H bond (BDFE(O–H) = 60.6 kcal mol−1) to be comparable to those formed upon net H-atom installation at the bridging (BDFE(O–H) = 61.6 kcal mol−1) and terminal (BDFE(O–H) = ∼61 kcal mol−1) oxygens of other, isoelectronic POV clusters, suggesting that electronic occupancy plays a key role in dictating the O–H bond strengths in POVs despite differences in surface ligand identities, reactive sites, and e−/H+ stoichiometry.19,32
In sum, these results provide atomistic insight into site selectivity of PCET at metal oxide surfaces. Our findings indicate that both ligand density and electronic occupancy of metal oxides play an important role in the ability of these materials to perform desired PCET reactions; limiting the number of reactive sites may necessitate charge localization in order to facilitate reversible hydroxide formation on metal oxide surfaces. Future work will seek to study a continuum of complexes bearing diverse ligand densities to tune the physicochemical properties of POV-alkoxides and probe how these alterations affect PCET regioselectivity, stoichiometry, and thermodynamics.
Experimental
General considerations
All manipulations were carried out in the absence of water and oxygen using standard Schlenk techniques or in a UniLab MBraun inert atmosphere drybox under a dinitrogen atmosphere. All glassware was oven-dried for a minimum of 4 h and cooled in an evacuated antechamber prior to use in the drybox. Solvents were dried and deoxygenated on a glass contour system (Pure Process Technology, LLC) and stored over 3 Å molecular sieves purchased from Fisher Scientific and activated prior to use. 5,10-phenazine (Phen), hydrazobenzene (Hydz) (2,2,6,6,-tetramethylpiperidin-1-yl)oxyl (TEMPO), and 1.6 M n-Butyllithium in hexanes were purchased from Sigma-Aldrich and used as received. D2O was purchased from Cambridge Isotope Laboratories and used as received. V6O8(OMe)110 (1-V6O80),34 [V6O7(OMe)12]1−,50 [V6O7(OMe)12]2−,30 5,10-dihydrophenazine (H2Phen),51 and its deuterated analogue (D2Phen)30 were generated following literature precedent.1H NMR spectra were recorded at 500 MHz on a Bruker DPX-500 spectrometer locked on the signal of deuterated solvents. All chemical shifts were reported relative to the peak of the residual H signal in deuterated solvents. CD3CN was purchased from Cambridge Isotope Laboratories, degassed by three freeze–pump–thaw cycles, and stored over fully activated 3 Å molecular sieves. UV-vis-NIR spectroscopy was collected using an Agilent Cary 6000i spectrophotometer at room temperature. Samples were prepared in the drybox in MeCN and added to air-free cuvettes and sealed prior to removing from the drybox. All molar absorptivity values were determined by averaging spectra collected in triplicate at different concentrations. Kinetic experiments were carried out on an Agilient Cary 60 UV-vis spectrophotometer held at desired temperatures using an Unisoku CoolSpek UV cryostat. Mass spectrometry analyses were performed on an Advion ExpressionL Compact mass spectrometer equipped with an electrospray probe and an ion-trap mass analyser (instrument error: ±0.1 amu). Direct injection analysis was employed in all cases with a sample solution in MeCN. Infrared (FT-IR, ATR) spectra of compounds were recorded on a Shimadzu IRAffinity-1 Fourier Transform Infrared Spectrophotometer and are reported in wavenumbers (cm−1).
Cyclic voltammetry (CV) was performed using a BioLogic SP 150 potentiostat/galvanostat and the EC-Lab software suite. Glassy carbon discs (3 mm, CH Instruments, USA) were used as working electrodes. Working electrodes were polished using a micro cloth pad and 0.05 μM alumina powder. Potentials recorded during CV were measured relative to a nonaqueous Ag/Ag+ reference electrode with 1 mM AgNO3 and 100 mM [nBu4N][PF6] in MeCN (BASi) and ultimately referenced against the Fc+/0 couple using an internal reference. A platinum wire served as the counter electrode. All experiments were carried out at room temperature inside a nitrogen-filled glove box (MBraun, USA). All CV measurements were iR compensated at 85% with impedance taken at 100 kHz using the ZIR tool included with the EC-Lab software. CV experiments were conducted at 100 mV s−1 on solutions of 1 mM analyte and 100 mM [nBu4N][PF6] supporting electrolyte in MeCN.
Single crystals of 2-V6O82− and 3-V6O7(OH)2− were mounted on the tip of a thin glass optical fiber (goniometer head) and analysed on an XtaLab Synergy-S Dualflex diffractometer equipped with a HyPix-6000HE HPC area detector for data collection at 223.00(10) and 100.00(10) K, respectively. The structures were solved using SHELXT-2018/2 and refined using SHELXL-2018/3.52,53 Elemental analyses were performed on a PerkinElmer 2400 Series II Analyzer, at the CENTC Elemental Analysis Facility, University of Rochester.
Synthesis of 2-V6O82−
Synthesis of 3-V6O7(OH)2−
Two 20 mL scintillation vials were charged with 2-V6O82− (0.100 m, 0.079 mmol) with a stir bar and H2Phen (0.009 mg, 0.047 mmol, 0.6 equiv.), respectively. The cluster and H2Phen were dissolved in 6 mL and 4 mL MeCN, respectively. The reductant was added dropwise into the cluster solution with vigorous stirring. The solution was stirred for 1 h, during which time the solution rapidly turned from dark blue to pale teal. After the reaction was complete, the solution was concentrated to one fourth its original volume and 10 mL of diethyl ether was added to precipitate the resultant cluster as a pale teal powder. The solid was washed thoroughly with diethyl ether (3 × 5 mL) to remove the phenazine byproduct and unreacted H2Phen. The product was extracted in MeCN and evaporated to dryness to yield 3-V6O7(OH)2− (0.085 g, 0.067 mmol, 85%) 1H NMR (500 MHz, CD3CN, 21 °C): δ = 24.38, 3.08, 1.58, 1.35, 0.96 ppm. IR (ATR): C–H = 2962, 2935, 2907, 2877, 2800 cm−1; V–OCH3 = 1059; V=Ot: 937 cm−1. UV-vis/NIR (21 °C, CH3CN): λ (ε) = 618 nm (49 M−1 cm−1), 1024 nm (165 M−1 cm−1). Elemental analysis for C43H105N2V6O19 (MW: 1261.0 g mol−1) Calc'd (%): C, 40.96; H, 8.47; N, 2.22. Found (%): C, 40.85; H, 8.87; N, 2.19.Crystals suitable for analysis by single crystal X-ray diffraction were grown by vapor diffusion of diethyl ether into a concentrated solution of 3-V6O7(OH)2− in MeCN at −30 °C. For refinement of the solid-state structure of 3-V6O7(OH)2−, it was noted that five of the six methyl substituents found in the asymmetric unit featured occupancies less than 1, suggesting that the hydroxide-functionalized molecule crystallized in multiple orientations (Fig. S7†). A similar phenomenon was observed by Hartl and coworkers when reporting the crystallographic data for 1-V6O80; deviations in V-Ob bond distances and C occupancies suggested that a bridging oxide was occupationally disordered with a methoxide group.34 In the case of complex 3-V6O7(OH)2−, we found that of the six bridging groups in the unit cell, methoxide occupancies only add to 5.5, leaving the final 0.5 to be accounted for by a hydroxide moiety (Fig. S7†). Upon expansion of the asymmetric unit to the full formula unit, these occupancy values become 11 methoxides and 1 hydroxide, accounting for all 12 bridging substituents.
Procedure for thermochemical analysis of the BDFE(O–H) of 3-V6O7(OH)2−
Determination of the BDFE(O–H) of 3-V6O7(OH)2− was performed using reactions between 2-V6O82− and half an equivalent of Hydz (one molar equiv of H-atoms) in quadruplicate. 300 μL of cluster stock solution (6.03 mM) and 120 μL of reductant (15.09 mM) in CD3CN were combined in a J. Young tube and sealed prior to removal from the glovebox for analysis. Reactions were allowed to equilibrate over 14 days at room temperature, tracking progress by 1H NMR (Fig. S11–14†). Upon equilibration, the relative concentrations of azobenzene (Azo) to Hydz were determined by using the integrations of resonances corresponding with each compound and normalizing for the number of protons each signal represents (Table S3†). Upon determination of [Hydz]/[Azo], the adjusted BDFE of the reductant was determined for each reaction using the following equation:where BDFEHydz is the average BDFE(N–H) of the N–H bonds in Hydz (60.9 kcal mol−1), n is the number of H-atom equivalents transferred (n = 2), and BDFEadj is the adjusted BDFE under the reaction conditions.1 Averaging the observed BDFEadj values provides the equilibrium BDFEadj for the reductant, which is equivalent to that of the O–H bond of 3-V6O7(OH)2−. This parameter was found to be 60.6 ± 0.1 kcal mol−1 (Table S3†).
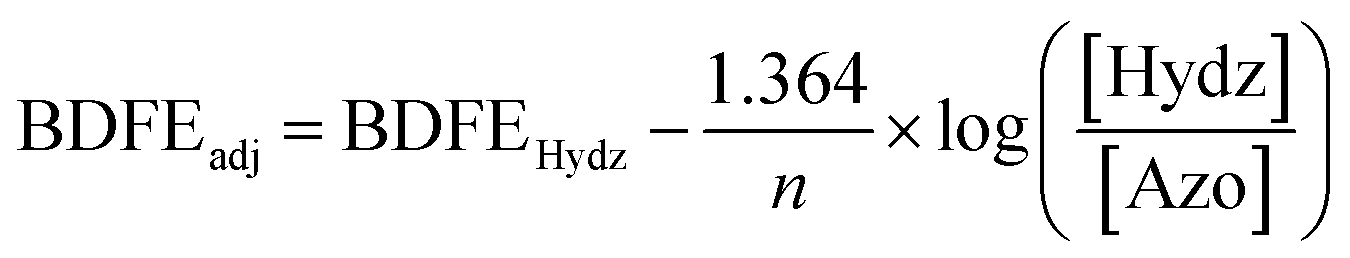
General procedure for pseudo-first order kinetic experiments between 2-V6O82− and H2Phen
Pseudo-first order reaction conditions were used to establish the rate constant of the PCET reaction between 2-V6O82− and H2Phen at −25 °C in MeCN. Using a UV-vis-NIR spectrometer with an N2-cooled cryostat set to −25 °C, reactions between 2-V6O82− and excess H2Phen (5.3–10.6 equivalents) were tracked by monitoring the absorbance at 1024 nm. Final reductant concentrations were varied from 15.30 to 7.65 mM, with a constant concentration of cluster of 1.45 mM. A 3 mL sample of H2Phen was loaded in a quartz cuvette, sealed with a septum, and was allowed to equilibrate to −25 °C, at which time the spectrometer began collecting absorbance data. After ∼20 seconds, 400 μL of cluster stock solution (12.32 mM) was forcefully injected to ensure homogeneity in the sample. The absorbance initially increased to that of 2-V6O82−, then, as the PCET reaction progressed, the absorbance decayed until the reaction reached completion, levelling to the absorbance for 3-V6O7(OH)2−. The plots of absorbance over time were fit to the following equation by least squares fitting (Fig. S15†):where At is the calculated absorbance at time, t, in seconds, Af is the absorbance value at the end of the experiment, Ai is the initial absorbance after injection of cluster to the cuvette, and kobs is the pseudo-first order rate constant. The excellent fit found for reaction curves indicated that the order of reductant in the rate expression was 1. Plotting kobs as a function of reductant concentration generated a linear plot, meaning that the reaction rate expression is second order overall, such that:
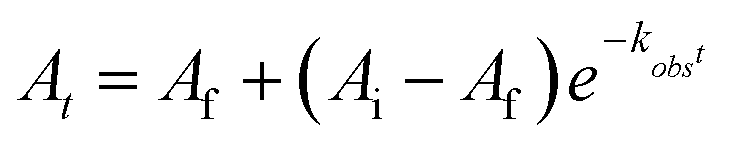
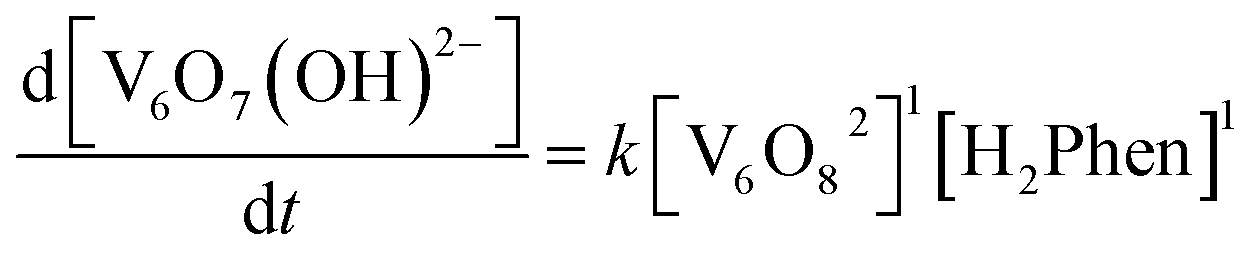
The slope of the line, when held to a y-intercept of 0, of 0.5261 (R2 = 0.9994), provides the experimentally determined rate constant, kexp at −25 °C of 0.53 ± 0.01 M−1 s−1.
To determine the kinetic isotope effect (KIE), analogous reactions were carried out at −25 °C using 3 mL samples of D2Phen solution between 7.73 and 15.46 mM, and 0.4 mL of cluster stock solution (13.23 mM) for a 1.45 mM final concentration (Fig. S16†). Similar treatment of the data produced a kexp for the formation of deuterium-labelled species of 0.062 ± 0.003 M−1 s−1.
Procedure for determination of activation parameters
Eyring analysis was performed by collecting absorbance vs. time data at temperatures between −42.5 and 10 °C. Reactions were assembled in an analogous fashion to previously run experiments, with constant reductant and cluster concentrations of 11.96 and 1.45 mM for reactions with 2-V6O82− (Fig. S17†) and 16.44 and 1.44 mM for reactions with [nBu4N][V6O7(OCH3)12] (Fig. S18†), respectively. Conversion of kobs to kexp was done by dividing kobs by the reductant concentration. Plotting ln(kexp/T) as a function of 1/T (temperature converted to K), the linear plot was used to solve for activation parameters using the below equations where R is the gas constant in units of cal (mol−1 K−1), kBoltz is Boltzmann's constant, and hPlanck is Planck's constant. The activation parameters for the activation of 2-V6O82− and [nBu4N][V6O7(OMe)12] are tabulated in Table 2, as well as Fig. 6 and S19.†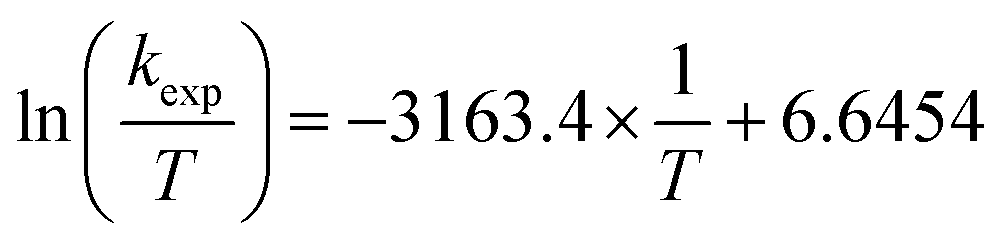
| ΔH‡ = −3163.4 × R |
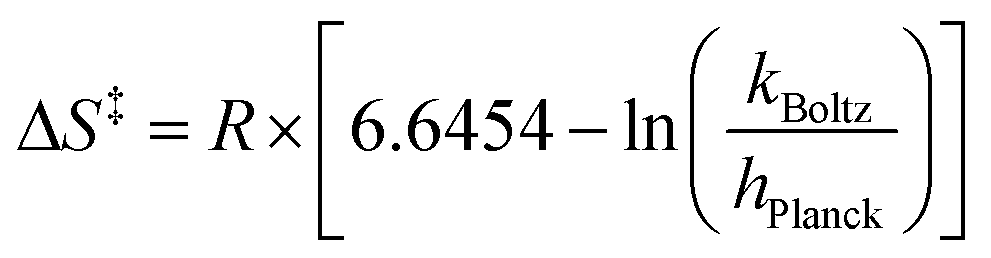
| ΔG‡ = ΔH‡ − TΔS‡ |
Data availability
All data for this project has been deposited in the main text or ESI,† as well as in the CCDC.Author contributions
E. S. and E. M. M. conceived and planned the experiments. E. S. performed the synthesis of all compounds and collected experimental data, except those involving X-ray crystallography. W. W. B. collected single crystal X-ray diffraction data and solved the crystal structures. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Department of Energy, Office of Basic Energy Sciences, under award DE-SC0023465. E. S. is the recipient of a Messersmith Fellowship from the University of Rochester that has also provided financial support for this work.Notes and references
- R. G. Agarwal, H.-J. Kim and J. M. Mayer, J. Am. Chem. Soc., 2021, 143, 2896–2907 CrossRef CAS PubMed.
- V. Aroutiounian, Int. J. Hydrogen Energy, 2007, 32, 1145–1158 CrossRef CAS.
- B. Chen, Z. Liu, C. Li, Y. Zhu, L. Fu, Y. Wu and T. van Ree, in Metal Oxides in Energy Technologies, ed. Y. Wu, Elsevier, Amsterdam, 1st edn, 2018, ch. 9, pp. 251–274 Search PubMed.
- Y. Chen, Z. Wang, S. Chen, H. Ren, L. Wang, G. Zhang, Y. Lu, J. Jiang, C. Zou and Y. Luo, Nat. Commun., 2018, 9, 818 CrossRef PubMed.
- V. Fung, G. Hu, Z. Wu and D.-e. Jiang, J. Phys. Chem. Lett., 2020, 11, 7049–7057 CrossRef CAS PubMed.
- S. M. Laga, T. M. Townsend, A. R. O'Connor and J. M. Mayer, Inorg. Chem. Front., 2020, 7, 1386–1393 RSC.
- B. Li, L. Xie, Z. Wang, S. Chen, H. Ren, Y. Chen, C. Wang, G. Zhang, J. Jiang and C. Zou, Angew. Chem., Int. Ed., 2019, 58, 13711–13716 CrossRef CAS PubMed.
- J. L. Peper, N. E. Gentry, B. Boudy and J. M. Mayer, Inorg. Chem., 2022, 61, 767–777 CrossRef CAS PubMed.
- C. F. Wise and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 14971–14975 CrossRef CAS PubMed.
- L. Xie, Q. Zhu, G. Zhang, K. Ye, C. Zou, O. V. Prezhdo, Z. Wang, Y. Luo and J. Jiang, J. Am. Chem. Soc., 2020, 142, 4136–4140 CrossRef CAS PubMed.
- Q. Zhu, S. Jiang, K. Ye, W. Hu, J. Zhang, X. Niu, Y. Lin, S. Chen, L. Song, Q. Zhang, J. Jiang and Y. Luo, Adv. Mater., 2020, 32, 2004059 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- K. Hermann, M. Witko and R. Druzinic, Faraday Discuss., 1999, 114, 53–66 RSC.
- K. Hermann, M. Witko, R. Druzinic and R. Tokarz, Top. Catal., 2000, 11, 67–75 CrossRef.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- L. Zhuang, Y. Jia, T. He, A. Du, X. Yan, L. Ge, Z. Zhu and X. Yao, Nano Res., 2018, 11, 3509–3518 CrossRef CAS.
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2005, 127, 13978–13987 CrossRef CAS PubMed.
- A. A. Fertig, W. W. Brennessel, J. R. McKone and E. M. Matson, J. Am. Chem. Soc., 2021, 143, 15756–15768 CrossRef CAS PubMed.
- M. T. Pope, in Mixed-Valence Compounds: Theory and Applications in Chemistry, Physics, Geology, and Biology, ed. D. B. Brown, Springer Netherlands, Dordrecht, 1st edn, 1980, ch. 4, pp. 365–386 Search PubMed.
- J. Spandl, C. Daniel, I. Brüdgam and H. Hartl, Angew. Chem., Int. Ed., 2003, 42, 1163–1166 CrossRef CAS PubMed.
- J. A. Fernández, X. López and J. M. Poblet, J. Mol. Catal. A: Chem., 2007, 262, 236–242 CrossRef.
- X. López, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2002, 124, 12574–12582 CrossRef PubMed.
- K. Nakajima, K. Eda and S. Himeno, Inorg. Chem., 2010, 49, 5212–5215 CrossRef CAS PubMed.
- K. Eda and T. Osakai, Inorg. Chem., 2015, 54, 2793–2801 CrossRef CAS PubMed.
- T. Ueda, ChemElectroChem, 2018, 5, 823–838 CrossRef CAS.
- A. A. Fertig and E. M. Matson, Inorg. Chem., 2022 DOI:10.1021/acs.inorgchem.2c02541.
- J. P. Launay, J. Inorg. Nucl. Chem., 1976, 38, 807–816 CrossRef CAS.
- K. Piepgrass and M. T. Pope, J. Am. Chem. Soc., 1987, 109, 1586–1587 CrossRef CAS.
- E. Schreiber, W. W. Brennessel and E. M. Matson, Inorg. Chem., 2022, 61, 4789–4800 CrossRef CAS PubMed.
- E. Schreiber, B. E. Petel and E. M. Matson, J. Am. Chem. Soc., 2020, 142, 9915–9919 CrossRef CAS PubMed.
- E. Schreiber, A. A. Fertig, W. W. Brennessel and E. M. Matson, J. Am. Chem. Soc., 2022, 144, 5029–5041 CrossRef CAS PubMed.
- S. E. Cooney, A. A. Fertig, M. R. Buisch, W. W. Brennessel and E. M. Matson, Chem. Sci., 2022, 13, 12726–12737 RSC.
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2009, 131, 5101–5114 CrossRef CAS PubMed.
- B. E. Petel, W. W. Brennessel and E. M. Matson, J. Am. Chem. Soc., 2018, 140, 8424–8428 CrossRef CAS PubMed.
- B. E. Petel, A. A. Fertig, M. L. Maiola, W. W. Brennessel and E. M. Matson, Inorg. Chem., 2019, 58, 10462–10471 CrossRef CAS PubMed.
- B. E. Petel, R. L. Meyer, M. L. Maiola, W. W. Brennessel, A. M. Müller and E. M. Matson, J. Am. Chem. Soc., 2020, 142, 1049–1056 CrossRef CAS PubMed.
- V. W. Day, W. G. Klemperer and D. J. Maltbie, J. Am. Chem. Soc., 1987, 109, 2991–3002 CrossRef CAS.
- J. M. Maestre, J. P. Sarasa, C. Bo and J. M. Poblet, Inorg. Chem., 1998, 37, 3071–3077 CrossRef CAS.
- W.-T. Jin, W.-Z. Weng and Z.-H. Zhou, Eur. J. Inorg. Chem., 2019, 2019, 1228–1235 CrossRef CAS.
- Richa, A. Kumar, I. Verma, M. Gautam, R. D. Erande, J. Kłak, D. Choquesillo-Lazarte, A. J. Mota, A. Rajput and H. Arora, J. Mol. Struct., 2022, 1269, 133805 CrossRef CAS.
- Q. Chen, D. P. Goshorn, C. P. Scholes, X. L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667–4681 CrossRef CAS.
- S. P. E. Smith and J. B. Christian, Electrochim. Acta, 2008, 53, 2994–3001 CrossRef CAS.
- C. F. Wise, R. G. Agarwal and J. M. Mayer, J. Am. Chem. Soc., 2020, 142, 10681–10691 CrossRef CAS PubMed.
- C. J. Fecenko, H. H. Thorp and T. J. Meyer, J. Am. Chem. Soc., 2007, 129, 15098–15099 CrossRef CAS PubMed.
- O. Snir, Y. Wang, M. E. Tuckerman, Y. V. Geletii and I. A. Weinstock, J. Am. Chem. Soc., 2010, 132, 11678–11691 CrossRef CAS PubMed.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarström, Nat. Chem., 2015, 7, 140–145 CrossRef CAS PubMed.
- W. D. Morris and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 10312–10319 CrossRef CAS PubMed.
- J. W. Darcy, S. S. Kolmar and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 10777–10787 CrossRef CAS PubMed.
- L. E. VanGelder, A. M. Kosswattaarachchi, P. L. Forrestel, T. R. Cook and E. M. Matson, Chem. Sci., 2018, 9, 1692–1699 RSC.
- J. Lee, K. Shizu, H. Tanaka, H. Nakanotani, T. Yasuda, H. Kaji and C. Adachi, J. Mater. Chem. C, 2015, 3, 2175–2181 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR of complexes and reactions, kinetic analyses, X-ray crystal structures and parameters, bond valence sum calculation results, CV, ESI-MS, and FTIR of complexes. CCDC 2215270 and 2215271. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05928b |
| This journal is © The Royal Society of Chemistry 2023 |