
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVacancy-cluster-mediated surface activation for boosting CO2 chemical fixation†
Wenxiu
Liu‡
a,
Lei
Li‡
a,
Wei
Shao‡
a,
Hui
Wang
*ab,
Yun
Dong
c,
Ming
Zuo
a,
Jiandang
Liu
c,
Hongjun
Zhang
 *c,
Bangjiao
Ye
c,
Xiaodong
Zhang
*ab and
Yi
Xie
ab
*c,
Bangjiao
Ye
c,
Xiaodong
Zhang
*ab and
Yi
Xie
ab
aHefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: wanghuig@ustc.edu.cn; zhxid@ustc.edu.cn
bInstitute of Energy, Hefei Comprehensive National Science Center, Hefei, Anhui 230031, P. R. China
cState Key Laboratory of Particle Detection and Electronics, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: hjzhang8@ustc.edu.cn
First published on 13th December 2022
Abstract
The cycloaddition of CO2 with epoxides towards cyclic carbonates provides a promising pathway for CO2 utilization. Given the crucial role of epoxide ring opening in determining the reaction rate, designing catalysts with rich active sites for boosting epoxide adsorption and C–O bond cleavage is necessary for gaining efficient cyclic carbonate generation. Herein, by taking two-dimensional FeOCl as a model, we propose the construction of electron-donor and -acceptor units within a confined region via vacancy-cluster engineering to boost epoxide ring opening. By combing theoretical simulations and in situ diffuse reflectance infrared Fourier-transform spectroscopy, we show that the introduction of Fe–Cl vacancy clusters can activate the inert halogen-terminated surface and provide reactive sites containing electron-donor and -acceptor units, leading to strengthened epoxide adsorption and promoted C–O bond cleavage. Benefiting from these, FeOCl nanosheets with Fe–Cl vacancy clusters exhibit enhanced cyclic carbonate generation from CO2 cycloaddition with epoxides.
Introduction
The conversion of carbon dioxide (CO2) into high-value-added chemicals has attracted worldwide attention, owing to its great potential in dealing with the challenge of global warming.1–3 Among various conversions, the cycloaddition of CO2 with epoxides exhibits 100% atomic utilization and offers a promising synthetic route for high-value-added multi-carbon (i.e., cyclic carbonates) products.4–7 As for the reaction, ring opening of epoxides is recognized to be the rate-determining step, and hence, exploring catalysts to boost such a step is crucial.8–10 Over the past few decades, heterogeneous catalysts have been drawing great attention due to their advantages in stability and reusability.11,12 However, compared with the homogeneous ones, heterogeneous catalysts generally suffer from low activity due to their limited number of active sites,13 and thus molecular co-catalysts (like tetra-alkylammonium halides) are usually needed.14 Besides, the complicated surface structures make it difficult to identify reactive sites in heterogeneous catalysts, thereby hindering the understanding of the involved structure–activity relationship.15 Therefore, designing heterogeneous catalysts with definite reactive sites to boost epoxide ring opening is of great significance in pursuing highly efficient cyclic carbonate generation.16Traditionally, two dominant processes are involved in ring opening of epoxides: the adsorption of epoxides and the cleavage of the C–O bond.17,18 As for ring opening of epoxides, a Lewis acid providing coordination sites toward an oxygen atom is typically needed. The coordination can effectively decrease the energy barrier for epoxide ring-opening, which provides a good leaving group and hence accelerates the subsequent C–O bond cleavage. Besides, given the electrophilic feature of a carbon atom in an epoxy ring, nucleophile species is required to attack the carbon atom for boosting epoxide ring-opening.19 In this case, constructing confined reactive sites with both electron-donor and -acceptor units on the surface of heterogeneous catalysts is expected to facilitate ring opening of epoxides. Meanwhile, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) can provide information on the dynamics of the reaction process and key intermediates, helping to enrich the understanding of the reaction pathway. This provides important support for understanding the catalytic reaction process and determining the active site of the reaction.20–22 Herein, we focus our attention on layered metal oxyhalide MOX (M = Fe, Bi; X = Cl, Br, I), whose heterogeneous structure with alternating [M2O2]2+ and halogen layers might enable the presence of electron-donor and -acceptor units in a confined region.23–26 Although halogens are considered to be robust nucleophilic species for attacking an electrophilic carbon atom of an epoxy ring, the halogen-terminated surface of MOX would inhibit the exposure of metal atoms serving as electron-acceptor units. Additionally, a halogen atom (Br, Cl, I) existing in the crystal lattice would be advantageous for catalyst recovery and separation.27–29 Note that the pair of electron-donor and -acceptor units proposed here is distinguished from a frustrated Lewis pair that underlies the steric hindrance between Lewis-acid and Lewis-base species. Therefore, structural regulation for realizing effective metal exposure should be implemented. Bearing this in mind, we anticipate the construction of reactive sites containing electron-donor and -acceptor units by introducing vacancies on a halogen-coated surface to facilitate the ring opening of epoxides (Scheme 1). We report on the synergistic construction of electron-donor and -acceptor units at one active site.
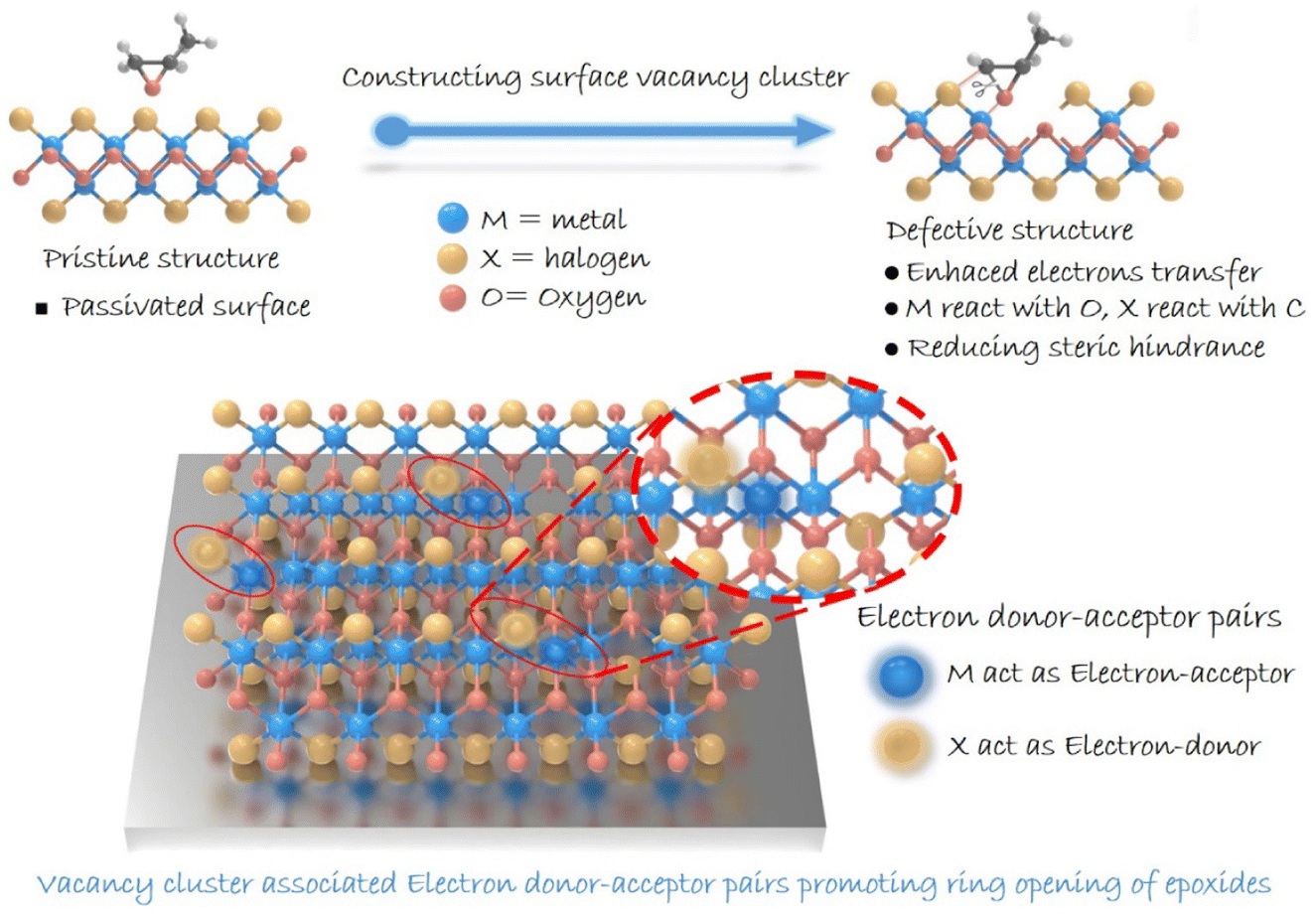 | ||
| Scheme 1 Illustration of the ring opening of epoxides in an MOX system. | ||
Results and discussion
Herein, by taking two-dimensional FeOCl as an example, we investigate the feasibility of vacancy engineering for constructing reactive sites with electron-donor and -acceptor units confined therein. Density functional theory (DFT) calculations were employed to investigate the impact of different vacancies on the electronic structure of an FeOCl system. Here, three types of models, i.e., pristine, Cl-vacancy-containing and Fe–Cl-vacancy-cluster-containing FeOCl single layers were built. As illustrated in Fig. 1a and S1,† there were notable defective states in FeOCl with Cl vacancy and Fe–Cl vacancy clusters, which would be beneficial for electron transfer during the catalytic processes. Furthermore, the adsorption behaviors of propylene oxide (PO) on FeOCl slabs were studied. As shown in Fig. 1b and S2b,† a PO molecule can be effectively adsorbed on the two defective FeOCl models. Besides, the steric hindrance involved in epoxide ring opening30 tends to be overcome or reduced at the Fe–Cl vacancy cluster owing to its larger size. Moreover, FeOCl with an Fe–Cl vacancy cluster exhibits promoted electron transfer between the slab and PO molecule, compared with FeOCl with Cl vacancies (Fig. S2b†).31,32 Such a feature suggests that an Fe–Cl vacancy cluster would strengthen the adsorption of a PO molecule, which would be favorable for the following ring opening of epoxides.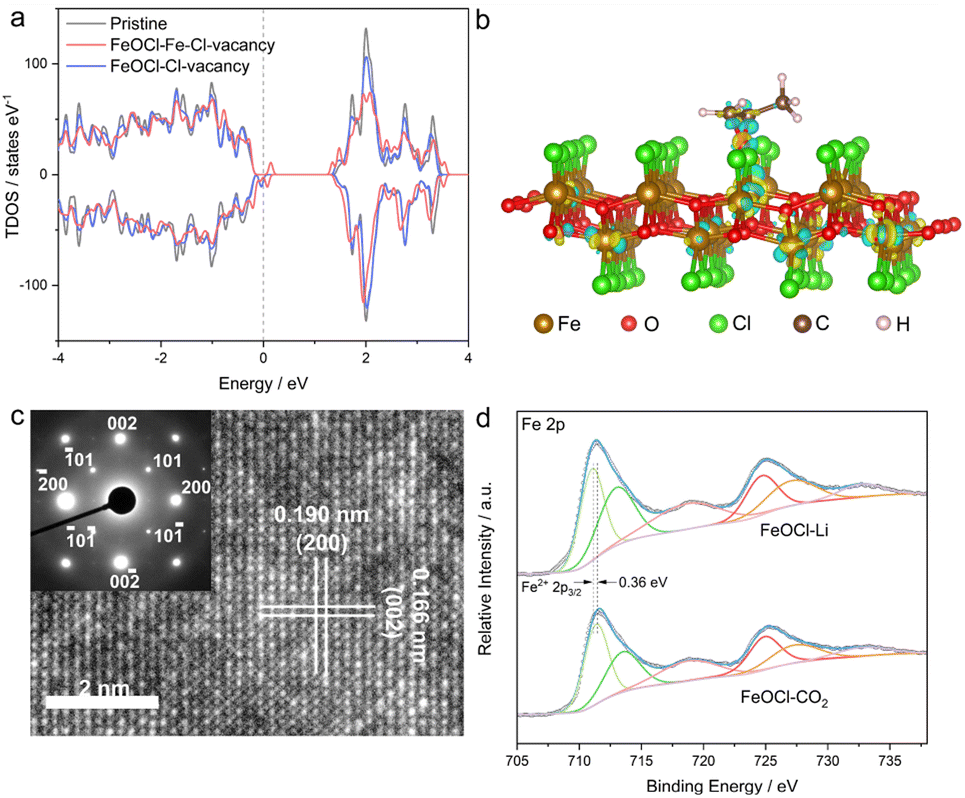 | ||
| Fig. 1 (a) Density of states of various FeOCl slabs. (b) The charge density of PO molecule absorption onto FeOCl with an Fe–Cl vacancy cluster. (c) HRTEM image and the corresponding SAED pattern of FeOCl–Li nanosheets. (d) Fe 2p XPS spectra of FeOCl–Li and FeOCl–CO2. | ||
In this study, defective FeOCl nanosheets were obtained by liquid exfoliation of N-butyllithium- and CO2-intercalated FeOCl bulk precursors (XRD pattern and SEM image are shown in Fig. S3†), and the obtained samples are denoted as FeOCl–Li and FeOCl–CO2, respectively (see details in the ESI†). The structures of the as-exfoliated FeOCl samples were investigated by X-ray diffraction (XRD) measurement. As shown in Fig. S4a and S5a,† XRD patterns of the samples match well with the typical FeOCl structure (JCPDS card no. 72-0619).33 After the liquid exfoliation process, the XRD pattern of FeOCl–Li exhibited strong {010} diffraction peaks ((010), (030), (040) and (050)) and faint diffraction peaks for other planes, suggesting high purity with predominant [010] orientation of the sample. Their similar structures are further confirmed by the Raman spectra (Fig. S6a†) that display the characteristic bands of an FeOCl system. Besides, Fourier transform infrared spectra (FTIR, Fig. S6b†) confirm the surface cleanliness of the samples, where a new peak located at 806 cm−1 in FeOCl–Li nanosheets would be assigned to the promoted stretching of the Fe–O bond.34
The morphologies of the as-exfoliated samples were characterized by transmission electron microscopy (TEM) and atomic force microscopy (AFM). The TEM images (Fig. S4b and S5b†) show that both samples exhibit free-standing sheet-like morphologies with micrometer-level lateral sizes. The AFM images (Fig. S7 and S8†) and the corresponding height profiles confirm the thicknesses of the as-prepared FeOCl nanosheets to be ∼0.9–2.7 nm, corresponding to one- to three-unit-cell thickness along the b axis. The high-resolution transmission electron microscope (HRTEM) images are displayed in Fig. 1c and S9,† where the lattice-fringe spacings of 1.90 Å and 1.66 Å correspond to the plane distances of (200) and (002), respectively. The HRTEM images as well as the corresponding selected-area electron diffraction (SAED; inset of Fig. 1c) patterns further confirm the [010] orientation of the sample. The scanning transmission electron microscopy (STEM) and the corresponding energy dispersive spectroscopy (EDS) mapping images (Fig. S10†) clearly reveal the homogeneous distributions of iron, oxygen, and chlorine in both samples. The two samples exhibit similar BET surface areas (Fig. S11†), which enables the parallel comparison of their catalytic performances. The chemical components and chemical states of the samples were investigated by X-ray photoelectron spectroscopy (XPS). Both samples exhibit the signals of Fe, O, and Cl (Fig. S12†). Compared with FeOCl–CO2 nanosheets, FeOCl–Li nanosheets exhibits a redshift of ∼0.36 eV in the Fe 2p XPS spectrum (Fig. 1d), suggesting the presence of low-valence Fe species that might be related to the reduction effect of N-butyllithium.35 As shown in Fig. S13,† Fe–O and Fe–Cl bonds are confirmed by the O 1s peak (∼530.5 eV) and Cl 2p peak (198.9 eV). The XPS spectra for all elements in FeOCl–Li nanosheets exhibit different degrees of redshift compared with those of the elements in FeOCl–CO2 nanosheets, which might have originated from their different defect types.
As an effective technique for identifying defective structures in solids, positron annihilation spectroscopy (PAS) was employed to determine the defect types in the two samples. As displayed in Fig. 2, the different positron annihilation lifetime spectra suggest the distinct defect types in the two samples. Here, a tri-exponential decay function was found to be more suitable for fitting the lifetime curves, and the corresponding fitting results are listed in Table S1.† For both samples, there is a nanosecond scale positron annihilation lifetime component (τ3), which would be related to the pick-off annihilation of ortho-positronium at interfaces.36,37 According to the calculated lifetimes (shown in Table S2†), the first two lifetime components could be assigned as follows: the shortest τ1 values (249.0 ps vs. 228.0 ps) would be related to free-state or trapping-state annihilation, while the different τ2 values (374.0 ps vs. 408.0 ps) suggest the dominant defect types to be a Cl vacancy (VCl) and Fe–Cl vacancy cluster (VFe–Cl) in FeOCl–CO2 nanosheets and FeOCl–Li nanosheets, respectively. For simplicity, we re-denote the samples containing a Cl vacancy and Fe–Cl vacancy cluster as FeOCl–VCl and FeOCl–VFe–Cl, respectively.
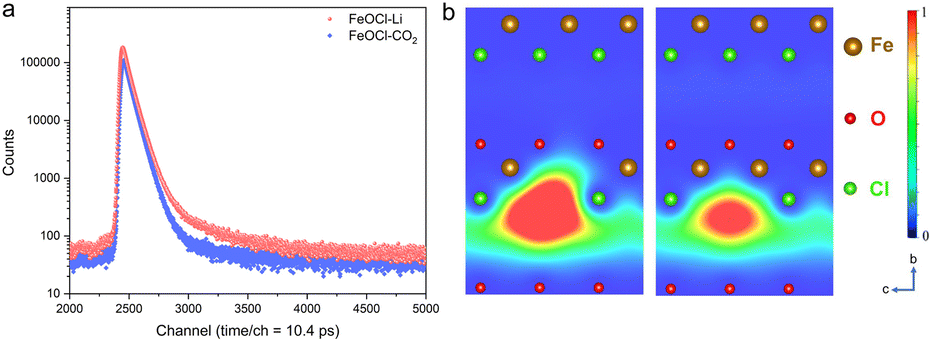 | ||
| Fig. 2 Positron annihilation spectroscopy (PAS) results of FeOCl–CO2 and FeOCl–Li. (a) Positron annihilation lifetime spectra of FeOCl–CO2 and FeOCl–Li nanosheets. (b) Schematic representations of trapped positrons at the Fe–Cl vacancy cluster (left) and Cl vacancy (right) in FeOCl. | ||
Considering that a vacancy cluster leads to promoted epoxide adsorption and electron transfer, we anticipate FeOCl–VFe–Cl to exhibit promoted activity for cycloaddition of CO2 with epoxides. We first synthesized FeOCl–VFe–Cl nanosheets by using different N-butyllithium-intercalation times to adjust the defect concentration (2 h, 4 h, 6 h and 8 h named FeOCl–VFe–Cl-2, FeOCl–VFe–Cl-4, FeOCl–VFe–Cl-6 and FeOCl–VFe–Cl-8). According to the results of ICP-AES and EPMA (Tables S3 and S4†), we found that the defect concentration of FeOCl–VFe–Cl gradually increased with the N-butyllithium-intercalation reaction time. Herein, the cycloaddition reaction of CO2 with PO into propylene carbonate (PC) was conducted to evaluate the catalytic performance of FeOCl systems. The FeOCl nanosheets with a butyllithium-intercalation reaction time of 8 h had the highest defect concentration and their catalytic conversion rate was also the highest (Fig. S14†). As shown in Fig. 3a, compared with FeOCl–VFe–Cl and FeOCl–VCl nanosheets, FeOCl-bulk exhibits extremely low catalytic activity in the high temperature region. It is surprising that both FeOCl–VFe–Cl and FeOCl–VCl nanosheets could catalyze the cycloaddition reaction, where FeOCl–VFe–Cl exhibits much higher catalytic activity than FeOCl–VCl throughout the temperature range (333–423 K; the products were detected by nuclear magnetic resonance spectroscopy, Fig. S15†). The reaction yield was calculated according to: yield = mol of propylene carbonate/mol of epoxide × 100%. Owing to the residues trapped in the slots of a stainless steel autoclave, the calculated results are typically lower than ∼90%. However, no obvious by-products could be detected. As shown in Fig. 3b, Arrhenius plots were obtained based on the linear fitting of ln TOF vs. 1000/T.38 Remarkably, the activation energy for FeOCl–VFe–Cl nanosheets is 55.5 ± 1.6 kJ mol−1, which is significantly less than that for FeOCl–VCl nanosheets (130.2 ± 7.8 kJ mol−1), further indicating the positive role of vacancy clusters in boosting the ring opening of epoxides. The recyclability and stability of FeOCl–VFe–Cl nanosheets was examined by 5 consecutive reaction cycles, in which the catalytic performance of FeOCl–VFe–Cl nanosheets was well maintained (Fig. 3c). Meanwhile, the corresponding Raman spectra, TEM image and HRTEM image verified the maintained phase and the morphology for the FeOCl–VFe–Cl nanosheets after a 5-cycle test, further indicating their long-term stability (Fig. S16–S18†). Besides, several other epoxides with different substituents (i.e., 1,2-epoxy-2-methylpropane, glycidol, epoxy bromopropane and 1,2-epoxybutane) were selected to investigate the scope of FeOCl–VFe–Cl-based catalytic CO2 cycloaddition reactions (Fig. 3d). For all the kinds of epoxides, FeOCl–VFe–Cl exhibited higher yields of target products than FeOCl–VCl. The relatively low conversion of 1,2-epoxybutane was attributed to the weak polarity of the carbon chain in the epoxide molecule.39 All the above results confirmed that FeOCl–VFe–Cl nanosheets can be an efficient catalyst for cycloaddition of CO2 with epoxides towards cyclic carbonates.
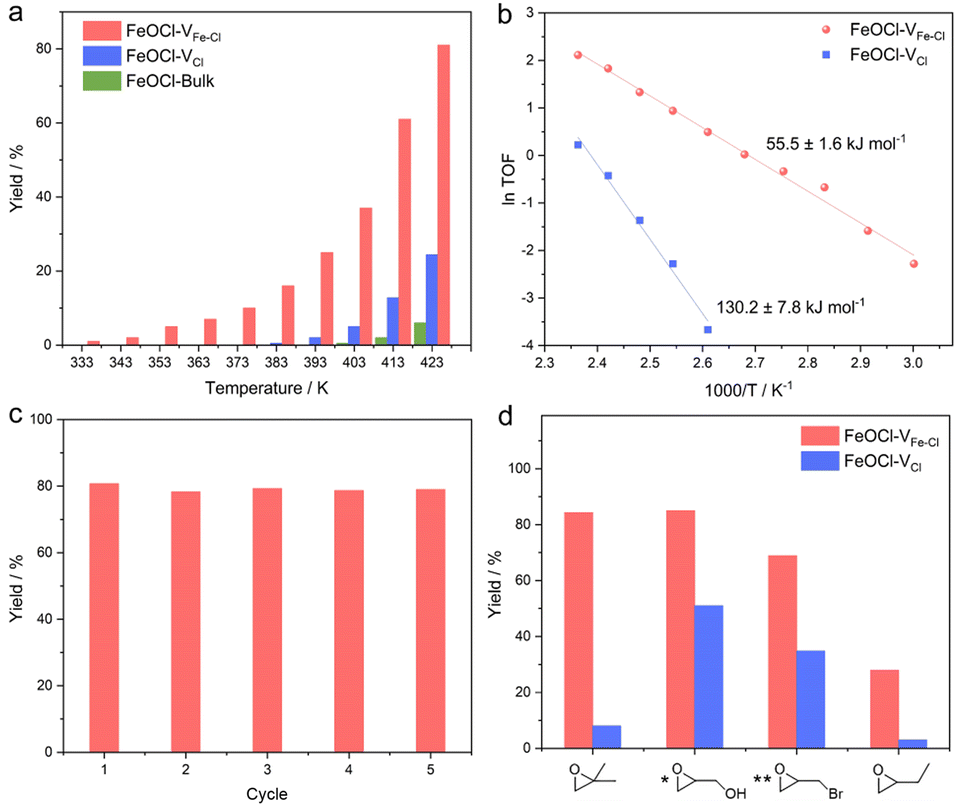 | ||
| Fig. 3 Evaluation of catalytic performance of FeOCl–VCl and FeOCl–VFe–Cl nanosheets. (a) The yields of propylene carbonate under 0.3 MPa of CO2 at difference temperatures. (b) The Arrhenius plots of different FeOCl nanosheets on the basis of figure (a). (c) Catalytic recyclability of FeOCl–VFe–Cl nanosheets. (d) Catalytic performance comparison of FeOCl nanosheets toward CO2 cycloaddition with different epoxides ([*] glycidol at 393 K; [**] epoxy bromopropane at 353 K). | ||
To gain in-depth understanding on the effects of a Fe–Cl vacancy cluster on cycloaddition of CO2 with epoxides, in situ diffuse reflectance infrared Fourier-transform spectroscopy (in situ DRIFTS) measurements were carried out,40 where the catalyst and PO were mixed in a reaction chamber under a 0.3 MPa CO2 atmosphere at 413 K. According to Fig. S19,† the ring-opening of PO is evidenced by a gradually increase in the peak at around ∼670 cm−1 that could be assigned to the formation of a C–Cl bond.41–43 Such a feature clearly confirms that a Cl atom at an Fe–Cl vacancy cluster could serve as an electron-donor unit for attacking the electrophilic carbon atom of PO. As shown in Fig. 4a and b, the peak at around 1047 cm−1, which could be assigned to symmetric stretching vibrations of C–O species,44 was improved with the increase in the reaction time, confirming the significant ring opening of PO by FeOCl–VFe–Cl. The peaks at around 1409 cm−1 and 1650 cm−1 correspond to symmetric stretching vibrations of the C–O bond and the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species of the COO group,45,46 whose increments confirm the formation of a carbonyl intermediate. The in situ DRIFTS results suggest that the cycloaddition of CO2 with PO involves the attack of a Cl site to the carbon atom of PO, resulting in the formation of a ring-opened intermediate; and CO2 insertion leads to the formation of a carbonate intermediate.
O species of the COO group,45,46 whose increments confirm the formation of a carbonyl intermediate. The in situ DRIFTS results suggest that the cycloaddition of CO2 with PO involves the attack of a Cl site to the carbon atom of PO, resulting in the formation of a ring-opened intermediate; and CO2 insertion leads to the formation of a carbonate intermediate.
 | ||
| Fig. 4 Mechanism exploration. (a) Time-dependent in situ DRIFTS spectra for the FeOCl–VFe–Cl nanosheets with CO2 and PO under an atmosphere of CO2 (0.3 MPa) at 413 K. (b) Amplifications of the selected areas of the full DRIFTS spectra. (c) The calculated energy profiles of the cycloaddition of CO2 with PO for different FeOCl nanosheets. | ||
To go further, DFT calculations of the reaction energy changes for each elementary reaction were performed to uncover the impacts of defect types on catalytic behaviors of the samples. Considering that the opening process of the C–O–C ring is the rate-determining step for CO2 cycloaddition with PO, the climbing-image nudged elastic band method was employed to explore the transition state (TS) reaction barrier for the ring-opening step of PO on FeOCl nanosheets.47 As demonstrated in Fig. 4c, the TS energy barriers are 106.6 and 66.7 kJ mol−1 for FeOCl slabs with a Cl vacancy and Fe–Cl vacancy cluster, respectively, manifesting the greater kinetics reaction activity of FeOCl–VFe–Cl nanosheets. The energy barriers in the addition of CO2 requires an uphill energy of 56.5 kJ mol−1 for FeOCl–VFe–Cl, which is less than that for FeOCl–VCl (84.9 kJ mol−1). This feature confirms that an Fe–Cl vacancy cluster could promote the insertion of CO2. From the perspective of DFT simulations, the enhanced activity of CO2 cycloaddition with PO on FeOCl–VFe–Cl nanosheets could be attributed to lower energy barriers arising from the optimized electron-donor and -acceptor units on the halogen-coated surface.
Conclusions
In summary, we have shown that constructing confined reactive sites with both electron-donor and -acceptor units on the surface could facilitate the ring opening of epoxides. By taking FeOCl nanosheets as an example, we demonstrated that Fe–Cl vacancy clusters, introduced by an N-butyllithium-assisted intercalation–exfoliation process, would significantly activate the halogen-terminated surface. Fe–Cl vacancy clusters could adjust a localized electron configuration, thus leading to the exposure of electron-donor (Cl sites) and -acceptor (Fe sites) units at one active site. By combining in situ DRIFTS and theoretical simulations, we highlight that the exposed Fe and Cl atoms at the defective sites serve as electron-acceptor and -donor units, respectively, which could facilitate the absorption of epoxides and the subsequent nucleophilic attack of a carbon atom, leading to promoted cleavage of a C–O bond. Benefiting from these, FeOCl–VFe–Cl nanosheets exhibit a reduced energy barrier of 55.5 ± 1.6 kJ mol−1 for the cycloaddition of CO2 with propylene oxide, which is less than half of that for the FeOCl–VCl case. This work not only provides a practical way for designing an efficient catalyst for CO2 cycloaddition, but also gives deep insights into the role of defective structures in the ring opening of epoxides.Data availability
Additional experimental details and data are provided in the ESI,† including XRD, optical images, Raman, FTIR, XPS, AFM, EDS, BET, NMR, EPMA and ICP-AES.Author contributions
W. L., L. L. and W. S. contributed equally to this work. W. L. conceived the idea, performed the experiments and collected the data; L. L. conducted and analyzed the DFT calculations; M. Z. conducted the HRTEM experiments; Y. D. worked on the PAS experiments; J. L., H. Z. and B. Y. supervised the PAS experiments; X. Z., H. W., W. S. and W. L. co-wrote the manuscript; X. Z., H. W. and Y. X. supervised the project. All authors contributed to the overall scientific interpretation and editing of the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1502903, 2019YFA0210004, and 2021YFA1501502), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36000000), the National Natural Science Foundation of China (21922509, 92163105, T2122004, 21905262, 21890754, U2032212, U2032160, and 22275179), the Anhui Provincial Key Research and Development Program (2022a05020054), the Anhui Provincial Natural Science Foundation (2108085J07 and 2008085QB88), the Youth Innovation Promotion Association of the CAS (Y2021123), the Fundamental Research Funds for the Central Universities (WK2060000039), and the University Synergy Innovation Program of Anhui Province (GXXT-2020-005 and GXXT-2021-020). The numerical calculations in this paper have been performed on the supercomputing system in the Supercomputing Center of the University of Science and Technology of China. We also thank the Infrared spectroscopy and microspectroscopy beamline (BL01B), Beamline BL12B MCD-A and MCD-B (Soochow Beamline for Energy Materials) and the Photoemission Endstation (BL10B) staff at the National Synchrotron Radiation Laboratory (NSRL) for characterization help.References
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS.
- Z. Fang, Z. Deng, X. Wan, Z. Li, X. Ma, S. Hussain, Z. Ye and X. Peng, Appl. Catal., B, 2021, 296, 120329 CrossRef CAS.
- G. Zhai, Y. Liu, Y. Mao, H. Zhang, L. Lin, Y. Li, Z. Wang, H. Cheng, P. Wang and Z. Zheng, Appl. Catal., B, 2022, 301, 120793 CrossRef CAS.
- L. Wang, W. Zhang, X. Zheng, Y. Chen, W. Wu, J. Qiu, X. Zhao, X. Zhao, Y. Dai and J. Zeng, Nat. Energy, 2017, 2, 869–876 CrossRef CAS.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin and Y. Luo, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS PubMed.
- Q. Yang, H. Peng, Q. Zhang, X. Qian, X. Chen, X. Tang, S. Dai, J. Zhao, K. Jiang and Q. Yang, Adv. Mater., 2021, 33, 2103186 CrossRef CAS PubMed.
- L. Gong, J. Sun, Y. Liu and G. Yang, J. Mater. Chem. A, 2021, 9, 21689–21694 RSC.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS.
- X. Zhang, H. Liu, P. An, Y. Shi, J. Han, Z. Yang, C. Long, J. Guo, S. Zhao and K. Zhao, Sci. Adv., 2020, 6, eaaz4824 CrossRef CAS.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS.
- S. Ghazali-Esfahani, H. Song, E. Păunescu, F. D. Bobbink, H. Liu, Z. Fei, G. Laurenczy, M. Bagherzadeh, N. Yan and P. J. Dyson, Green Chem., 2013, 15, 1584–1589 RSC.
- X. Tan, X. Sun and B. Han, Natl. Sci. Rev., 2021, 1–41 Search PubMed.
- M. Ding and H. Jiang, ACS Catal., 2018, 8, 3194–3201 CrossRef CAS.
- R. J. Wei, X. H. Zhang, B. Y. Du, Z. Q. Fan and G. R. Qi, RSC Adv., 2013, 3, 17307–17313 RSC.
- S. Zhao, Y. Wang, J. Dong, C. T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao and L. Zhang, Nat. Energy, 2016, 1, 1–10 Search PubMed.
- J. P. Cao, Y. S. Xue, N. F. Li, J. J. Gong, R. K. Kang and Y. Xu, J. Am. Chem. Soc., 2019, 141, 19487–19497 CrossRef CAS.
- Z. Z. Yang, Y. Zhao, G. Ji, H. Zhang, B. Yu, X. Gao and Z. Liu, Green Chem., 2014, 16, 3724–3728 RSC.
- F. D. Bobbink, D. Vasilyev, M. Hulla, S. Chamam, F. Menoud, G. Laurenczy, S. Katsyuba and P. J. Dyson, ACS Catal., 2018, 8, 2589–2594 CrossRef CAS.
- T. Marino, F. Ponte, G. Mazzone, E. Sicilia, M. Toscano and N. Russo, Dalton Trans., 2017, 46, 9030–9035 RSC.
- L. Li, W. Liu, R. Chen, S. Shang, X. Zhang, H. Wang, H. Zhang, B. Ye and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e2022144 Search PubMed.
- T. Yan, W. Bing, M. Xu, Y. Li, Y. Yang, G. Cui, L. Yang and M. Wei, RSC Adv., 2018, 8, 4695–4702 RSC.
- G. S. Foo, F. Polo-Garzon, V. Fung, D. e. Jiang, S. H. Overbury and Z. Wu, ACS Catal., 2017, 7, 4423–4434 CrossRef CAS.
- H. Wang, S. Chen, D. Yong, X. Zhang, S. Li, W. Shao, X. Sun, B. Pan and Y. Xie, J. Am. Chem. Soc., 2017, 139, 4737–4742 CrossRef CAS PubMed.
- T. Yu, X. Zhao, L. Ma and X. Shen, Mater. Res. Bull., 2017, 96, 485–490 CrossRef CAS.
- A. E. ElMetwally, G. Eshaq, F. Z. Yehia, A. M. Al-Sabagh and S. Kegnæs, ACS Catal., 2018, 8, 10668–10675 CrossRef CAS.
- J. Wang, K. P. Hou, Y. Wen, H. Liu, H. Wang, K. Chakarawet, M. Gong and X. Yang, J. Am. Chem. Soc., 2022, 144, 4294–4299 CrossRef CAS PubMed.
- D. Xing, B. Lu, H. Wang, J. Zhao and Q. Cai, New J. Chem., 2017, 41, 387–392 RSC.
- Y. Wang, S. Li, Y. Yang, X. Shen, H. Liu and B. Han, Chem. Commun., 2019, 55, 6942–6945 RSC.
- W. Gao, C. L. Wang, L. Chen, C. Y. Zhu, P. Li, J. Y. Li, J. P. Liu and X. M. Zhang, Appl. Organomet. Chem., 2022, 36 Search PubMed.
- L. Li, B. Li, H. Guo, Y. Li, C. Sun, Z. Tian and L. Chen, Nanoscale, 2020, 12, 15880–15887 RSC.
- X. Li, J. Lin, L. Li, Y. Huang, X. Pan, S. E. Collins, Y. Ren, Y. Su, L. Kang, X. Liu, Y. Zhou, H. Wang, A. Wang, B. Qiao, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19983–19989 CrossRef CAS.
- D. Chen, W. Yang, L. Jiao, L. Li, S. H. Yu and H. L. Jiang, Adv. Mater., 2020, 32, e2000041 CrossRef.
- M. Sun, C. Chu, F. Geng, X. Lu, J. Qu, J. Crittenden, M. Elimelech and J. H. Kim, Environ. Sci. Technol. Lett., 2018, 5, 186–191 CrossRef CAS.
- J. Wei, X. Feng, X. Hu, J. Yang, C. Yang and B. Liu, Colloids Surf., A, 2021, 631, 127754 CrossRef CAS.
- J. Wu, J. Peng, Y. Zhou, Y. Lin, X. Wen, J. Wu, Y. Zhao, Y. Guo, C. Wu and Y. Xie, J. Am. Chem. Soc., 2018, 141, 592–598 CrossRef PubMed.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao and S. Wei, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- L. Luo, M. Wang, Y. Cui, Z. Chen, J. Wu, Y. Cao, J. Luo, Y. Dai, W. X. Li and J. Bao, Angew. Chem., Int. Ed., 2020, 132, 14542–14550 CrossRef.
- B. H. Cheng, L. J. Deng, J. Jiang and H. Jiang, Chem. Eng. J., 2022, 442, 136265 CrossRef CAS.
- G. Li, X. Sui, X. Cai, W. Hu, X. Liu, M. Chen and Y. Zhu, Angew. Chem., Int. Ed., 2021, 133, 10667–10670 CrossRef.
- B. Cao, Y. Sun, J. Guo, S. Wang, J. Yuan, S. Esakkimuthu, B. B. Uzoejinwa, C. Yuan, A. E. F. Abomohra and L. Qian, Fuel, 2019, 246, 319–329 CrossRef CAS.
- Y. Sun, W. Zheng, X. Ding and R. P. Singh, Adsorpt. Sci. Technol., 2020, 38, 271–285 CrossRef CAS.
- H. Cicek, G. Kocak, Ö. Ceylan and V. Bütün, Macromol. Res., 2019, 27, 640–648 CrossRef CAS.
- Y. Ikezawa and T. Ariga, Electrochim. Acta, 2007, 52, 2710–2715 CrossRef CAS.
- W. P. Voo, B. B. Lee, A. Idris, A. Islam, B. T. Tey and E.-S. Chan, RSC Adv., 2015, 5, 36687–36695 RSC.
- H. Bekhti, Y. Boucheffa, A. A. Blal and A. Travert, Vib. Spectrosc., 2021, 117, 103313 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details (calculation method and experimental section), characterization studies, supplementary figures, and tables. See DOI: https://doi.org/10.1039/d2sc05596a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |