
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Volcanic ash ice nucleation activity is variably reduced by aging in water and sulfuric acid: the effects of leaching, dissolution, and precipitation†
William D.
Fahy‡
 a,
Elena C.
Maters‡
*bc,
Rona
Giese Miranda
d,
Michael P.
Adams
b,
Leif G.
Jahn§
a,
Ryan C.
Sullivan
a and
Benjamin J.
Murray
b
a,
Elena C.
Maters‡
*bc,
Rona
Giese Miranda
d,
Michael P.
Adams
b,
Leif G.
Jahn§
a,
Ryan C.
Sullivan
a and
Benjamin J.
Murray
b
aCenter for Atmospheric Particle Studies, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
bSchool of Earth and Environment, University of Leeds, Leeds LS2 9JT, UK
cDepartment of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: ecm63@cam.ac.uk
dFaculty of Geosciences, Geoengineering, and Mining, Technische Universität Bergakademie Freiberg, 09599 Freiberg, Germany
First published on 22nd December 2021
Abstract
Volcanic ash nucleates ice when immersed in supercooled water droplets, giving it the potential to influence weather and climate from local to global scales. This ice nucleation activity (INA) is likely derived from a subset of the crystalline mineral phases in the ash. The INA of other mineral-based dusts can change when exposed to various gaseous and aqueous chemical species, many of which also interact with volcanic ash in the eruption plume and atmosphere. However, the effects of aqueous chemical aging on the INA of volcanic ash have not been explored. We show that the INA of two mineralogically distinct ash samples from Fuego and Astroni volcanoes is variably reduced following immersion in water or aqueous sulfuric acid for minutes to days. Aging in water decreases the INA of both ash samples by up to two orders of magnitude, possibly due to a reduction in surface crystallinity and cation availability accompanying leaching. Aging in sulfuric acid leads to minimal loss of INA for Fuego ash, which is proposed to reflect a quasi-equilibrium between leaching that removes ice-active sites and dissolution that reveals or creates new sites on the pyroxene phases present. Conversely, exposure to sulfuric acid reduces the INA of Astroni ash by one to two orders of magnitude, potentially through selective dissolution of ice-active sites associated with surface microtextures on some K-feldspar phases. Analysis of dissolved element concentrations in the aged ash leachates shows supersaturation of certain mineral species which could have precipitated and altered the INA of the ash. These results highlight the key role that leaching, dissolution, and precipitation likely play in the aqueous aging of volcanic ash with respect to its INA. Finally, we discuss the implications for understanding the nature and reactivity of ice-active sites on volcanic ash and its role in influencing cloud properties in the atmosphere.
Environmental significanceVolcanic ash nucleates ice in cloud droplets, influencing cloud properties and their role in climate. However, its ice nucleation activity (INA) may change when exposed to atmospheric aging processes, limiting understanding of the potentially evolving impact of ash on clouds as it disperses away from the volcano. Here we exposed two volcanic ash samples to water or sulfuric acid on atmospherically relevant time scales and observed a variable reduction of INA over time, likely through different mechanisms of chemical alteration acting on ice-active pyroxene and feldspar minerals. These mechanisms could limit the formation of ice in volcanically influenced clouds and be applied to future work studying the alteration of other mineralogical ice-nucleating materials common in the atmosphere. |
1 Introduction
Volcanic ash from explosive eruptions can serve as ice-nucleating particles (INPs) in the atmosphere and may be especially important far from mineral dust sources of INPs such as arid- and semi-arid regions.1–4 While global annual mean emissions of volcanic ash (176–256 Tg a−1)5 are an order of magnitude lower than those of mineral dust (1000–2000 Tg a−1),6 ash loadings in the atmosphere regularly meet or exceed mineral dust loadings on regional scales, and can even surpass the global annual mean dust emissions during a single large event (e.g., >5000 Tg released during the 1991 Pinatubo eruption, Philippines).7 Eruptions of various magnitudes can inject volcanic ash into the upper troposphere and lower stratosphere, and while coarse grains (>63 μm) rapidly sediment to the ground, smaller super- and submicron particles have been reported to remain airborne for up to 6 months following an eruption and spread 1000 s of kilometers from the source volcano.8,9Volcanic ash is a silicate-based material consisting of glass and crystalline components, which can include feldspar, pyroxene, olivine, biotite, amphibole, mica, quartz and iron(–titanium) oxide minerals,10 depending on the source magma composition and storage and ascent conditions.11 All natural ash samples tested in the laboratory to date have acted as INPs when immersed in supercooled water.12–18 The presence and properties of crystalline components in the ash are thought to exert a strong influence on its ice nucleation activity (INA), while the glass component has been shown to nucleate ice relatively poorly.16 Several primary minerals have been implicated as the ice-active phases in volcanic ash in previous studies, including alkali (Na/K-rich) feldspar, plagioclase (Na/Ca-rich) feldspar, and pyroxene.14–17 Similarly, K-feldspar (hereafter used to refer to K-rich alkali feldspar), Na/Ca-feldspar, and quartz are recognized as ice-active phases in mineral dust,19–27 while secondary minerals in dust such as kaolinite, illite and montmorillonite may also nucleate ice.20,24,28–32 Ice nucleation on mineral surfaces is thought to occur at specific nanoscale sites33–36 due to some combination of chemical and physical features, for instance relating to crystal structure, microscale texture and resulting geometry, and the presence and orientation of chemical functionalities such as hydroxyl groups.37–42
In addition to the mineralogy of the ash bulk, the history of a given ash sample following eruption is likely to influence its INA, potentially by modifying the properties of the ash surface. For example, the INA of ash might be enhanced or depressed by interaction with volcanic gases (e.g., H2O, SO2, HCl) at high temperatures in the eruption plume, with the effect influenced by the susceptibility of individual minerals in the ash to thermochemical alteration.43 As airborne ash emerges from the hot eruption plume core and begins cooling and dispersing with the entrainment of air, ash particles are likely to develop a liquid film and/or become immersed in liquid droplets.44,45 The liquid phase can be acidified by uptake of acids (e.g., SO2/H2SO4, HCl, HF, HNO3) released during the eruption and/or encountered during atmospheric transport.46–49 Studies on mineral dusts and their crystalline components have shown that such ‘chemical aging’ by exposure to water,25,50 aqueous acids,50–52 or acid vapor53–55 can sometimes reduce the INA of these materials. A recent study found the INA of Fuego volcanic ash to be unaltered or reduced by aqueous chemical aging.56 The variable effects on INA observed are strongly INP type- and aging method-dependent, and their underlying mechanisms are poorly understood, in part due to limited knowledge of how ice-active surfaces are altered by exposure to different species. While mechanistic understanding of the chemical aging of volcanic ash at ambient temperature is lacking in the context of atmospheric ice nucleation, insights into the geochemical reaction mechanisms involved may be gained from existing literature on aqueous alteration of silicate minerals commonly found in ash.
For many multi-oxide silicate minerals (e.g., feldspar, pyroxene, amphibole, mica) immersed in near-neutral or low pH aqueous solution, alteration begins with rapid leaching of metal cations (e.g., Na+, K+, Ca2+, Mg2+, Fe2+) from interstices of the silicate network by exchange with protons (H+/H3O+) in solution.57–61 This can produce a surface ‘leached layer’ depleted in metal cations and enriched in H+/H3O+, silicon (Si), and aluminum (Al) that is in substitution of Si in the silicate network tetrahedra. Alternatively or additionally, an amorphous, hydrated, cation-depleted and Si(/Al)-enriched layer can form through a dissolution–reprecipitation process at the mineral–solution interface.62–66 The precise nature and formation mechanisms of leached and/or reprecipitated layers on silicate surfaces are still debated (e.g., see reviews by Chardon et al., Wilson, Yuan et al.)67–69 but this discussion is beyond the scope of our study.
Generally, the extent of surface alteration depends on various factors including the mineral composition, the pH of the aqueous solution, the cation and anion concentrations in the solution, and the mineral–solution contact duration.70 Further, the growth of any leached layer may become limited by the rate of cation/proton diffusion through it and by dissolution of the silicate network itself at the mineral surface.57 While the mechanisms involved vary across minerals in ways that are still not fully understood, Fig. 1a provides a simplified summary proposing key steps of leaching and dissolution of several silicates, where in most cases the final step is cleavage of Si–O bonds between network tetrahedra by H+/H3O+.61 All else being equal, the relative rates of alteration of silicate minerals can broadly be considered in relation to their (in)stability at Earth surface conditions, as outlined in the Goldich dissolution series, which follows Bowen's reaction series, in Fig. 1b.71–73 Accordingly, minerals such as olivine, pyroxene and Ca-rich feldspar, which crystallize from cooling magma at higher temperatures (earlier), are expected to alter more readily in aqueous solution at ambient temperature compared to minerals such as K-feldspar and quartz, which crystallize from cooling magma at lower temperatures (later).
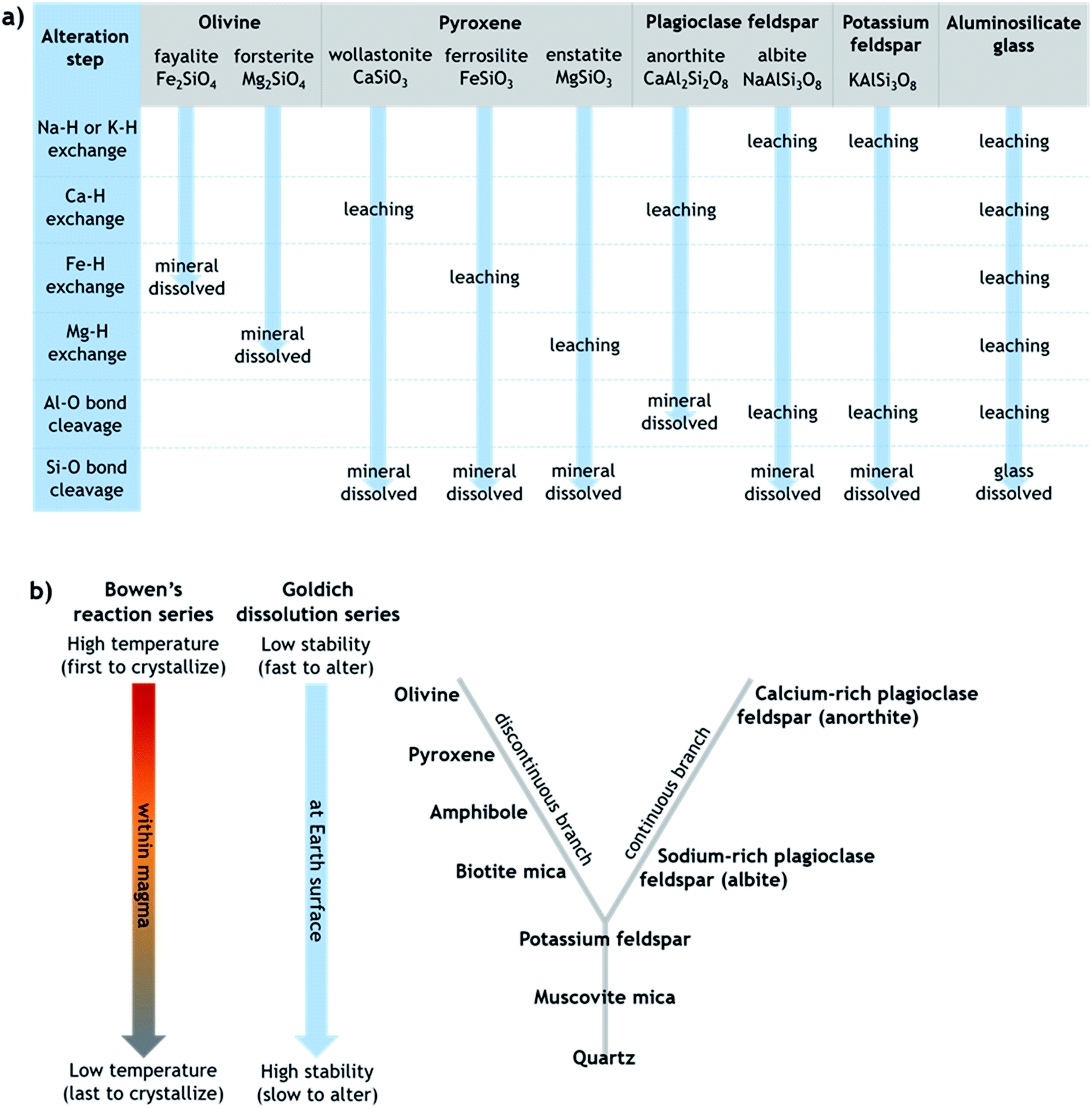 | ||
| Fig. 1 (a) Simplified schematic of the alteration steps of various silicates that can be present in volcanic ash, modified from Oelkers.61 Note that olivine dissolves directly by metal–proton exchange since silica tetrahedra occur as single units, whereas other silicates dissolve by Si–O (or Al–O in anorthite) bond cleavage between connected tetrahedra. (b) Schematic depicting the relative order of silicate minerals in terms of their crystallization from cooling magma (Bowen's reaction series) and their stability against alteration under Earth surface conditions (Goldich dissolution series), modified from Huddart and Stott.73 The unifying idea is that minerals that crystallized from magma at temperatures farthest from those at the Earth's surface are most susceptible to alteration when exposed at the Earth's surface. | ||
Considering this knowledge from the literature, depending on the mineralogical make-up of volcanic ash and the conditions of chemical aging encountered during atmospheric transport, leaching and dissolution will likely occur in parallel and at different rates on ash surfaces, and may influence the INA of the ash. To gain insight specifically into how volcanic ash interaction with an aqueous droplet at near-neutral or low pH affects its INA, here we conduct experiments suspending two mineralogically contrasting ash samples in water (H2O(l)) or sulfuric acid solution (H2SO4(aq)) over different atmospherically relevant time scales, prior to ice nucleation measurements. To explore how changes in INA between ‘non-aged’ and ‘aged’ samples might relate to alteration of specific mineral phases in the ash, dissolved element concentrations are analyzed in the aging solution from each experiment and compared with INA data using correlation coefficients and a learned predictive model between the two measurements. We then present a series of hypotheses on how combinations of geochemical alteration processes might explain complex trends in INA observed during aging of the ash in H2O or H2SO4, before proposing implications and further research to better constrain the potential impacts of volcanic ash on atmospheric ice nucleation.
2 Materials and methods
2.1 Volcanic ash samples
Ash samples from the Fuego (FUE) and Astroni (AST) volcanoes were chosen for study (Table 1) based on their contrasting composition and INA, as reported previously by Jahn et al. and Maters et al.14,16 The INA of the more crystalline basaltic–andesitic FUE ash is likely driven by pyroxene, while that of the more glassy trachyphonolitic AST ash is probably dominated by K-feldspar. Each specimen was mechanically milled prior to chemical aging experiments (see Section 2.2) using a zirconia ceramic mill ball and jar to produce atmospherically relevant particles sizes. Milling also exposed fresh surfaces for the purpose of testing the effect of chemical aging on INA under controlled conditions, independent of any influence on INA of prior alteration of ash surfaces while airborne, once deposited, or during storage. The Brunauer, Emmett, and Teller specific surface area (SSABET) of each milled sample (Table S1†), and of samples collected following chemical aging (Table S2†), was determined after overnight degassing from a ten-point N2 adsorption isotherm at −196 °C using a Micromeritics TriStar 3000 instrument.74| Ash sample (code) | Source eruption | Classificationc | Mineralogyd (wt%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Glass | Alk. Feld. | Plag. Feld. | Ortho pyrox. | Clino pyrox. | Oliv. | Crystal. SiO2 | Fe (–Ti) oxide | |||
| a Mineralogy reported in Jahn et al.14 b Mineralogy reported in Maters et al.16 c Based on bulk elemental composition (Table S1) according to the total alkali versus SiO2 classification scheme for igneous rocks.75 d Alk. feld. = Na/K-feldspar, Plag. feld. = Na/Ca-feldspar, orthopyrox. = orthopyroxene (compositions between Mg2Si2O6 and Fe2Si2O6 solid solution, with small amounts of Ca2+ substitution possible), clinopyrox. = clinopyroxene (other compositions of solid immiscibility, particularly with higher Ca2+ content), oliv. = olivine, crystal. SiO2 = crystalline SiO2, m. c. = minor component below ∼2 wt% quantification limit by X-ray diffraction. | ||||||||||
| Fuego ash (FUE)a | Volcán de Fuego, Guatemala, February 2015 | Basaltic–andesite | 20 | 3 | 68 | 3 | 4 | 2 | m. c | m. c. |
| Astroni ash (AST)b | Astroni volcano, Italy, 3.8–4.4 ka | Trachy-phonolite | 72 | 19 | 7 | — | 2 | — | — | m. c. |
2.2 Chemical aging experiments
Batch experiments of FUE or AST ash suspended in MilliQ H2O (18.2 MΩ cm) or pH 1.75 ± 0.5 (0.01 M) H2SO4 (93% VWR Normatom trace metal grade) at room temperature, at an ash-to-solution ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 by mass, and under constant gentle rotation at 1 rpm were carried out to explore the effects of ash–liquid interactions on ash INA (see Section 2.3). This ash-to-solution ratio falls within the wide range of estimated particle concentrations (10−4 to >103 g L−1) in liquid aerosols and cloud droplets.76 Aqueous aging was chosen to allow time-resolved chemical analysis of dissolved material, providing insight into the geochemistry occurring at the ash–liquid interface and in the aging solution. Moreover, this approach enables longer aging time scales to be studied compared to typical chamber or flow tube reactor experiments. As in previous atmospheric processing studies of ash,77,78 H2SO4 was selected as it is readily formed by oxidation of volcanic SO2 and is likely the dominant acid species to interact with ash in the atmosphere.79 While mixtures of species would more accurately mimic atmospheric conditions, these simpler experimental conditions facilitate isolation of key alteration mechanisms that may modify ash INA. Experiments were run in triplicate for different durations (minute to day scale) and then filtered by vacuum through a pre-rinsed 0.45 μm polyethersulfone membrane filter to achieve a desired ash–liquid contact time of 10 min, 1 h, 4 h, 24 h, or 120 h. The collected ash was dried in air for 24 h on a weighing boat beneath a plastic cover, and then stored at room temperature in a sealed plastic vial in a desiccator until ice nucleation measurements and subsequent SSABET analysis. The collected liquid was filtered further by syringe through a pre-rinsed 0.2 μm polyethersulfone membrane filter and acidified to ∼1.5 vol% HNO3 (67% Fisher Chemical trace metal grade) for storage at ∼4 °C prior to dissolved Si, Al, Fe, Mg, Ca, Na, K, Ti, and Mn analysis by inductively coupled plasma optical emission spectroscopy (ICP-OES; Thermo Fisher iCAP 7400 Radial). The corresponding limits of detection and quantification (LOD and LOQ) are given in Table S2.† Blank experiments with pure MilliQ H2O or pH 1.75 ± 0.5 H2SO4 (containing no added ash) were also run for 10 min, 1 h, 4 h, 24 h, or 120 h, and then filtered, acidified, and stored as above prior to analysis by ICP-OES.
:100 by mass, and under constant gentle rotation at 1 rpm were carried out to explore the effects of ash–liquid interactions on ash INA (see Section 2.3). This ash-to-solution ratio falls within the wide range of estimated particle concentrations (10−4 to >103 g L−1) in liquid aerosols and cloud droplets.76 Aqueous aging was chosen to allow time-resolved chemical analysis of dissolved material, providing insight into the geochemistry occurring at the ash–liquid interface and in the aging solution. Moreover, this approach enables longer aging time scales to be studied compared to typical chamber or flow tube reactor experiments. As in previous atmospheric processing studies of ash,77,78 H2SO4 was selected as it is readily formed by oxidation of volcanic SO2 and is likely the dominant acid species to interact with ash in the atmosphere.79 While mixtures of species would more accurately mimic atmospheric conditions, these simpler experimental conditions facilitate isolation of key alteration mechanisms that may modify ash INA. Experiments were run in triplicate for different durations (minute to day scale) and then filtered by vacuum through a pre-rinsed 0.45 μm polyethersulfone membrane filter to achieve a desired ash–liquid contact time of 10 min, 1 h, 4 h, 24 h, or 120 h. The collected ash was dried in air for 24 h on a weighing boat beneath a plastic cover, and then stored at room temperature in a sealed plastic vial in a desiccator until ice nucleation measurements and subsequent SSABET analysis. The collected liquid was filtered further by syringe through a pre-rinsed 0.2 μm polyethersulfone membrane filter and acidified to ∼1.5 vol% HNO3 (67% Fisher Chemical trace metal grade) for storage at ∼4 °C prior to dissolved Si, Al, Fe, Mg, Ca, Na, K, Ti, and Mn analysis by inductively coupled plasma optical emission spectroscopy (ICP-OES; Thermo Fisher iCAP 7400 Radial). The corresponding limits of detection and quantification (LOD and LOQ) are given in Table S2.† Blank experiments with pure MilliQ H2O or pH 1.75 ± 0.5 H2SO4 (containing no added ash) were also run for 10 min, 1 h, 4 h, 24 h, or 120 h, and then filtered, acidified, and stored as above prior to analysis by ICP-OES.
2.3 Ice nucleation measurements
The INA of the non-aged and aged ash samples was determined using the Microlitre Nucleation by Immersed Particles Instrument (μL-NIPI) described in detail by Whale et al.80 This instrument has previously been used to characterize a variety of ice-active materials relevant to this study including volcanic tephra and glass,15,16,43 K- and Na/Ca-feldspars,21,81 and other silicate minerals.25,82 For each non-aged and aged ash sample, a separate 1, 0.2, and 0.04 wt% suspension in MilliQ H2O was made gravimetrically and was vortexed to ensure an even distribution of particles. These three suspension concentrations were chosen to widen the temperature range over which to measure INA and observe potential aging effects. For each suspension, an array of 30 to 40 1 μL droplets was pipetted onto a hydrophobic silanized glass cover slip placed on a temperature-controlled stage (Grant-Asymptote EF600 Stirling Cryocooler) and cooled from room temperature to 0 °C at −5 °C min−1. The droplets were then exposed to a temperature ramp of −1 °C min−1 until all droplets were frozen, with freezing recorded by a digital camera for subsequent analysis. Uncertainty in temperature is estimated to be ±0.4 °C.80 A 0.2 L min−1 N2(g) flow over the droplets prevented condensation and frost formation. Background freezing spectra of pure MilliQ H2O (containing no added ash) were taken each day to characterize the INA of impurities in the H2O and interactions between droplets and the cover slip.83 Freezing spectra are reported as the ice nucleation active site density normalized to mass (nm) or surface area (ns) as a function of temperature (T) following eqn 1 to facilitate comparisons of INA within this study and with other experimental setups.84,85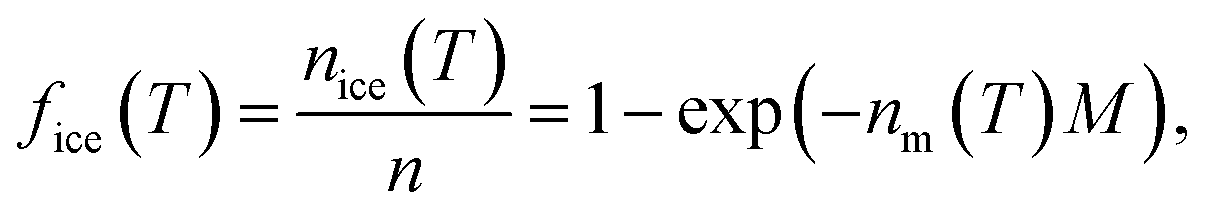 | (1) |
3 Results
3.1 Ice nucleation activities of non-aged and aged ash samples
The INA of the non-aged and aged FUE and AST ash samples is reported in Fig. 2. Aging in H2O or H2SO4 always reduced the INA of the ash, but the magnitude of this reduction appears to be sensitive to the specific ash, aging solution type, aging duration, and immersion freezing temperature being considered. The change in INA associated with each aging experiment is summarized in Tables S2 and S3.†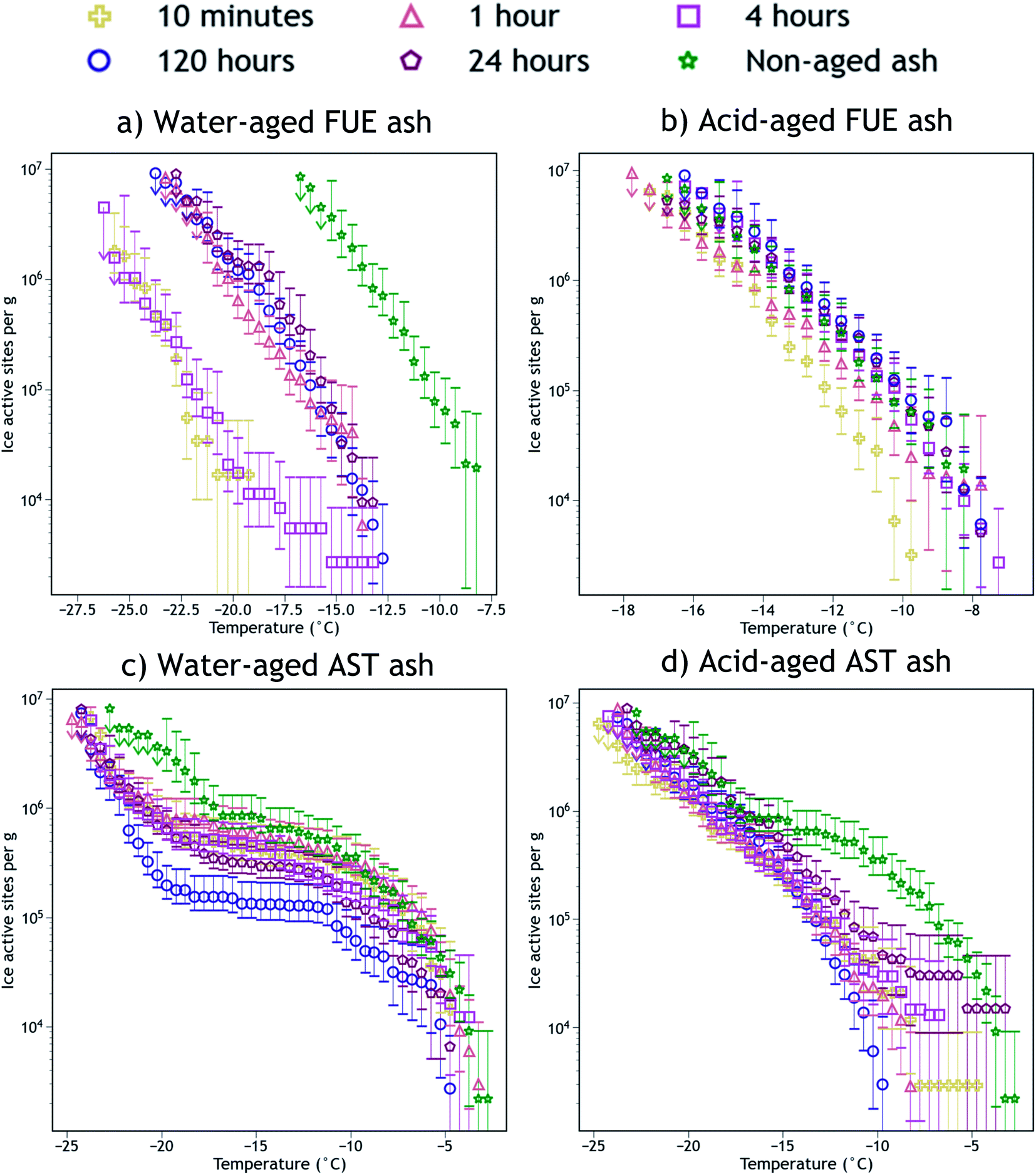 | ||
| Fig. 2 Ice nucleation active site density normalized to mass (nm) versus temperature spectra of FUE ash non-aged or aged for different durations in (a) H2O (water-aged) or (b) pH 1.75 H2SO4 (acid-aged), and AST ash non-aged or aged for different durations in (c) H2O (water-aged) or (d) pH 1.75 H2SO4 (acid-aged). The 95% confidence intervals were approximated as the 2.5th and 97.5th quantiles using Monte Carlo simulations based on a Poisson distribution of droplet freezing events. The background of each spectrum (a freezing experiment with pure water droplets measured the same day) has been subtracted and the data from the 1, 0.2, and 0.04 wt% suspension experiments have been averaged into a single spectrum. Further details of error analysis and background subtraction are presented in the ESI.† | ||
Exposure of FUE ash to H2O caused a reduction in INA of either 5 or 10 °C (a 10 or 1000× decrease in nm at any given temperature). The magnitude of this decrease is non-monotonic with respect to time, as after 10 min and 4 h in H2O the aging effect is markedly stronger (10 °C) than after 1, 24, and 120 h (5 °C). In contrast, exposure of FUE ash to H2SO4 caused only a slight reduction in INA of 1 to 2 °C (a ∼5× decrease in nm at any given temperature) after 10 min of aging, followed by a recovery of INA such that aging times of 1 to 120 h did not significantly reduce freezing temperatures relative to the non-aged sample.
Exposure of AST ash to H2O appeared to have a time-dependent effect on INA above −17 °C. After 10 min to 1 h of aging, little to no deactivation is observed, while after 4 h of aging, a borderline insignificant loss is observed. After 24 h, this reduction in INA becomes significant, and after 120 h a maximum reduction in nm by a factor of 5 to 10 occurs above −10 °C, persisting to −17 °C because relatively few sites become active between −10 °C and −17 °C. Below −17 °C, the early aging effects are different, as after 10 min in H2O, freezing temperatures are reduced by 3 to 4 °C (a ∼5× decrease in nm). This level of INA is not further altered until 120 h of aging in H2O, where the decrease in nm reaches a factor of 10 near −17 °C, although at the coldest temperatures (<−22 °C) this additional reduction in INA is not observed.
Again contrasting with the effects of H2O, exposure of AST ash to H2SO4 caused most of the ice nucleation sites active above −17 °C to be immediately and irreversibly lost from the freezing spectrum. While the freezing spectrum of the ash aged for 24 h does overlap with that of the non-aged ash at the warmest temperatures (>−8 °C), note the large error bars which indicate that there is not a statistically significant recovery of INA at these temperatures after aging in H2SO4. Only a few of these ice-active sites above −8 °C appear to remain from 10 min to 24 h of aging, represented by a switch from a negative concavity of the non-aged AST ash spectrum to a flat or slightly positive concavity of the aged ash spectrum. The portion of the spectrum below −17 °C remains essentially unaltered at all time points, similar to the observed stabilization of INA in FUE ash exposed to H2SO4.
The SSABET measurements of the ash samples (Tables S1 and S2†) enable detection of changes in their surface area due to chemical aging. Little change in SSABET is observed for FUE and AST ash samples exposed to H2O, except for a slight increase for AST ash aged for 120 h, whereas SSABET is raised by a factor of five for both ash samples exposed to H2SO4, meaning there is more ash surface area per unit mass post-aging. Since ice nucleation is localized to the ash surface, this change could impact the trends observed in INA in Fig. 2. To deconvolute surface area changes from other aging effects on INA, we also present surface area normalized ice nucleation active site density (ns) freezing spectra as a function of temperature in Fig. S1.† In the ns spectra of FUE ash, the slightly lower INA observed upon exposure to H2SO4 for 10 minutes relative to all later time points becomes less significant, while the overall loss of INA from non-aged to acid-aged FUE and AST (below −17 °C) becomes statistically significant. The other changes in INA presented in Fig. 2 are not altered significantly depending on the normalization scheme. Since nm reflects the aggregate of all aging effects that could influence volcanic ash while ns removes the effects of changing surface area, nm will be used for the remainder of this study, however, changes to SSABET as a result of the aging process will be discussed where appropriate.
3.2 Dissolved element concentrations in chemical aging solutions
Mean dissolved element signatures from the aging experiments are reported in Table S2† as the ratio of their molar abundance relative to Si in the aging solution (as measured by ICP-OES) over their molar abundance relative to Si in the non-aged bulk ash sample (as measured by XRF). A value for this metric above unity (i.e., X/Si s/b > 1, where X is a metal element, s refers to the aging solution, and b refers to the bulk ash) is presumed to reflect preferential release of X from the ash surface (e.g., by leaching/dissolution) and/or preferential removal of Si from the aging solution (e.g., by precipitation). The Si concentration in the aging solution normalized to the measured SSABET is also reported as an indicator of overall dissolution of the ash. Silicon was chosen as a reference element as it is typically the last to be released from the silicate network and is thus often used as a proxy for silicate dissolution.61The major trends in dissolved element signatures are generally consistent with the reaction steps shown in Fig. 1. That is, we infer fast leaching of elements that occur in interstices of the silicate network such as alkali and alkaline earth metals (Na, K, Ca, and Mg), followed by slower dissolution of elements such as Al and Si that exist in tetrahedra of the silicate network. However, without knowledge of the chemical composition and spatial distribution of glass and crystalline components at the ash surface as well as their individual rates of leaching and dissolution, the contributions of different phases in the ash to the dissolved element signatures observed are difficult to disentangle.
4 Discussion
The mechanisms by which chemical aging in aqueous solution impairs or reduces the INA of volcanic ash have not been previously investigated or reported in the literature. However, based on the available geochemical data (e.g., ash mineralogy, leachate chemistry), we present hypotheses to explain the observed ice nucleation phenomena. Variations in the effect depending on the specific ash sample, aging solution type, and aging duration are inferred to reflect parallel competing changes in INA of the ash during leaching and dissolution of its component phases and precipitation of secondary phases. In agreement with conclusions from previous studies,14,16 the INA of FUE ash is likely driven by pyroxene while that of AST ash is probably dominated by K-feldspar above −17 °C and pyroxene or Na/Ca-feldspar below −17 °C.4.1 Analysis of correlations between ice nucleation spectra and dissolved element signatures
To quantify the complex correlations between changes in INA (see Section 3.1, Tables S3 and S4†) and mean dissolved element signatures (see Section 3.2, Table S2†) of the aged ash samples, a vector representing a numerical approximation of the difference in INA of ash at each aging duration compared to the INA of the non-aged ash was calculated by projecting each cumulative freezing spectrum onto a 10th-order Chebyshev polynomial basis set.87 The INA difference vector was then determined by calculating the quotient of the polynomial coefficients of the aged ash and non-aged ash freezing spectra. Element-wise Pearson correlation matrices of all dissolved element signatures and vector coefficients for all experiments divided by ash/aging solution pair are reproduced in Fig. S2–S5† in the ESI.†88 Correlations and anticorrelations will be referred to as ‘strong’ (>0.67), ‘weak’ (between 0.67 and 0.33), or ‘no correlation’ (<0.33).For FUE ash aged in H2O, dissolved Ca, Na, and Mg concentrations are variably but positively correlated with one another (Fig. S2†), consistent with their release in particular by leaching of Na/Ca-feldspar, Mg-rich pyroxene, and/or olivine (as in Fig. 1; see Section 4.2). However, for FUE ash exposed to H2SO4, dissolved Mg is not correlated with other alkali and alkaline earth metals, instead having a weak relationship with Si (Fig. S3†), potentially reflecting leaching and dissolution of Mg-rich pyroxene and/or olivine (see Section 4.4). For AST ash aged in H2O or H2SO4, dissolved Ca, Na, and K are always strongly correlated (Fig. S4 and S5†), and are probably released readily by leaching of glass, feldspar, and pyroxene components of the ash surface. Note that correlations between ice nucleation difference descriptors (the coefficients of the divided Chebyshev polynomials) and dissolved element signatures indicate that the release of elements from the ash correlates positively with changes in INA, while the reverse is true for anticorrelations. Specific examples will be highlighted where relevant in the following sections.
Next, using the Python machine learning library scikit-learn89 a nonlinear Support Vector Machine regressor model90,91 was built and tested between the mean dissolved element signatures and the INA difference vectors corresponding to each aging experiment of a given duration. Permutation importances92 were calculated to quantify how important each element signature is for predicting the changes in INA observed for each ash/aging solution pair, shown in Fig. 3. A positive permutation importance means that the model uses some aspect of that element signature to predict changes in INA. Negative values indicate the model is improved by randomization, which could mean that the model incorrectly expects them to be important based on other data. A permutation importance of zero does not necessarily imply that a given element signature is uncorrelated with ice nucleation descriptors – it simply means the model does not use that signature to predict INA. Note that permutation importances are more sensitive than correlation coefficients, such that a single mineral phase contributing to an overall element signature could be the cause of a positive permutation importance in that element. Further discussion, testing, and validation of this method can be found in the ESI.† The correlation coefficients and permutation importances are quantitative metrics relating dissolved element signatures to changes in INA, and will be used to inform the following discussion of physical and chemical mechanisms by which the INA of FUE and AST ash might be altered in aqueous environments.
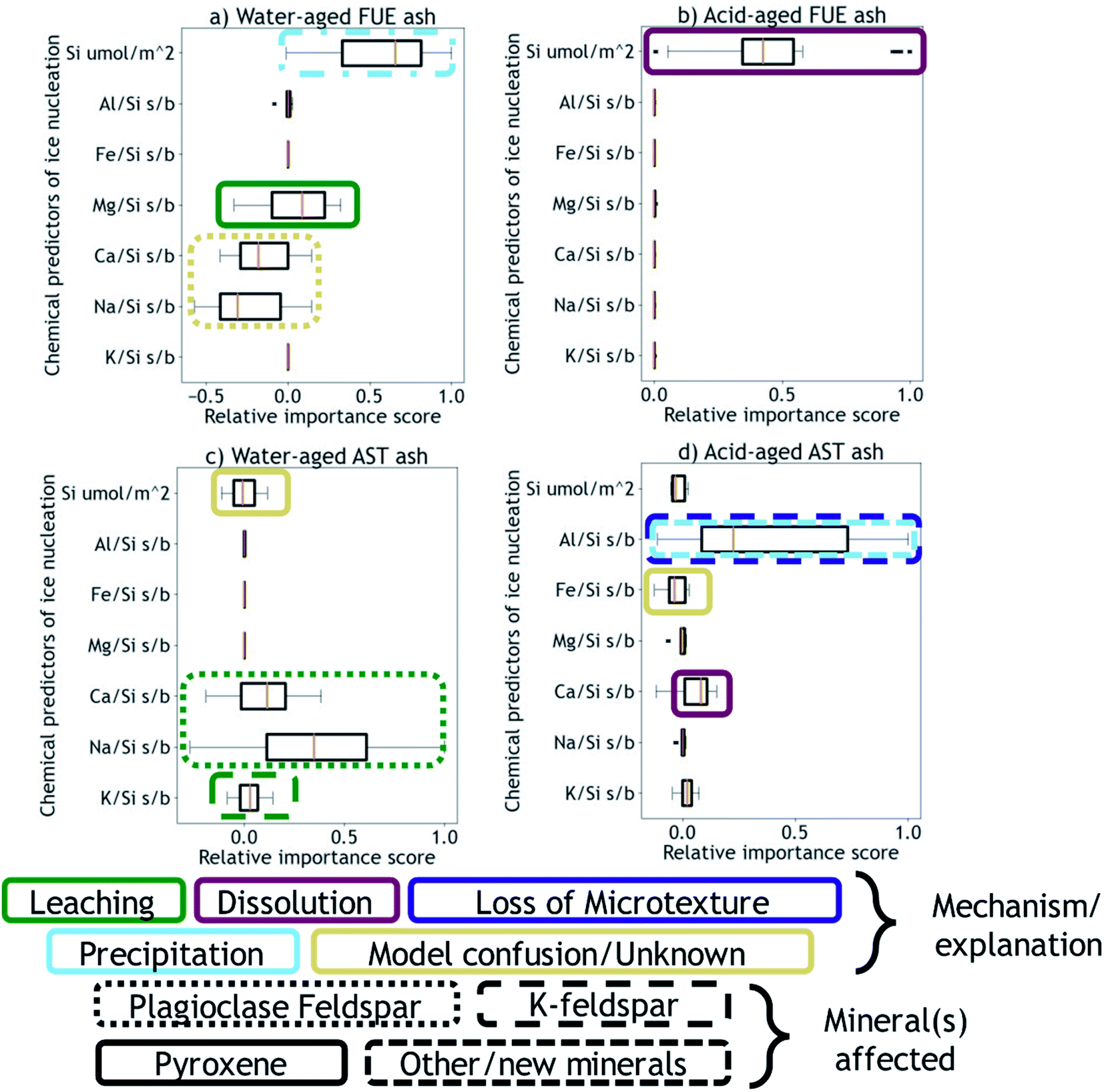 | ||
| Fig. 3 Permutation importances of mean element signatures to the predictive model of INA for FUE ash aged in (a) H2O (water-aged) or (b) pH 1.75 H2SO4 (acid-aged), and AST ash aged in (c) H2O (water-aged) or (d) pH 1.75 H2SO4 (acid-aged). All values are scaled such that 1.0 represents the maximum positive importance in each dataset. The proposed reaction mechanisms and the mineral(s) affected are indicated by the annotations using the box color and box line style, respectively. | ||
4.2 Leaching of mineral surfaces
We propose that formation of silica-like surface layers during aqueous leaching62,65,67 of volcanic ash is one of the main drivers of chemical aging deactivating INA in these ash samples. At 10 min of aging in H2O, the immediate drop in nm of FUE ash across the full range of temperatures and at colder temperatures in the freezing spectrum of AST ash (<−17 °C; Fig. 2a and c) is inferred to result from the development of a cation-depleted and Si(/Al)-enriched surface on pyroxene and feldspar minerals that would otherwise be more ice-active. This hypothesis is supported by high relative concentrations of alkali and alkaline earth metals in solution at early H2O aging time points (Table S2†) and by the positive correlation coefficients between these elements and ice nucleation descriptors for both ash samples (Fig. S2 and S4†). In addition, the positive permutation importance of Mg for FUE ash (Fig. 3a) may reflect the importance of leaching of Mg-rich pyroxene93,94 to changes in INA, while the negative permutation importances of Ca and Na may indicate that leaching of Na/Ca-feldspar (the dominant phase in FUE ash) is not a significant contributor to the aged ash's ice nucleation behavior. Cations are also likely released by leaching of glass surfaces on FUE and AST ash, but glass is poorly ice-active so its alteration is not expected to drive the changes in INA observed here.16,43 The freezing spectra of both FUE and AST ash show reduced activity after aging 10 min of aging in H2SO4, but little change after longer aging durations (Fig. 2b and d), so leaching of pyroxene and/or feldspar surfaces under acidic conditions may similarly impair ice nucleation on short time scales in those experiments.Formation of a cation-depleted and Si(/Al)-enriched surface layer is hypothesized to lower INA by reducing its capacity for further uptake of H+/H3O+ in H2O, since surface protonation has been proposed to be a key step in ice nucleation by silicate materials containing exchangeable cations.24 Moreover, if aging entails dissolution–reprecipitation alongside or instead of leaching at the mineral–solution interface (Yuan et al. and references therein69), the resulting Si(/Al)-enriched layer is likely to be more amorphous than the original mineral surface,67,69 which could also contribute to lowering INA by reducing the lattice match of the surface sites with ice polymorphs.16,24,37,41,43,95
At longer time scales in H2O, leaching is expected to slow as cations diffuse from progressively greater depths within the solid materials to exchange with H+/H3O+ in solution (Oelkers and references therein61), although the rate of leaching of different glass and mineral surfaces on FUE and AST ash will vary. For example, cations are likely released more readily from pyroxene and Na/Ca-feldspar than from K-feldspar, based on the Goldich dissolution series (Fig. 1). This disparity in leaching rates of different minerals may explain the INA trends of AST ash aged in H2O, where activity consistent with pyroxene and/or Na/Ca-feldspar at colder temperatures (<−17 °C) is lost after 10 min, while activity consistent with K-feldspar at warmer temperatures (>−17 °C) persists for longer, until it too is steadily suppressed. The positive permutation importances of Na, Ca, and K for AST ash aged in H2O (Fig. 3c) conform to this hypothesis. Eventually, the INA of the ash may be impaired by leaching of all mineral surfaces in H2O, although this effect may be convoluted by precipitation of secondary phases of variable INA (see Section 4.3). Others have found the INA of microcline (a K-feldspar) to be unaffected or only slightly reduced (freezing temperature lowered by up to 2 °C) following suspension in H2O from one week to several months.19,21,96 The samples tested in those studies might be more stable than the K-feldspar in AST ash, and/or less ice-active secondary phases might contribute to lowering the INA of AST ash (see Section 4.3).
4.3 Precipitation of secondary phases from solution
As dissolved element concentrations increase in the aging solutions, precipitation of secondary phases becomes possible in the free solution and/or on nucleation sites on ash surfaces, especially in the experiments in H2O where pH is expected to be near-neutral. This phenomenon has been reported previously to influence the INA of quartz and simple silicate materials,24,25 but has not previously been reported in the context of volcanic ash. Table 2 lists saturation indices associated with the aging of FUE and AST ash in H2O and H2SO4, calculated from measured dissolved element concentrations using the geochemical modeling software PHREEQC.97| Ash sample | Aging solution/duration | Saturation index of selected minerals | |||||
|---|---|---|---|---|---|---|---|
| Montmorillonite | Kaolinite | Gibbsite | K-mica | Crystalline SiO2a | Al-oxyhydroxideb | ||
| a Quartz/cristobalite/chalcedony. b Boehmite/diaspore. | |||||||
| FUE | H2O/10 min | −0.1 | 3.8 | 2.6 | — | −1.1/−1.5/−1.6 | 2.1/3.8 |
| H2O/1 h | 0.4 | 3.9 | 2.4 | — | −0.9/−1.3/−1.3 | 2.0/3.7 | |
| H2O/4 h | 1.7 | 4.8 | 2.7 | — | −0.7/−1.1/−1.1 | 2.2/3.9 | |
| H2O/24 h | 2.9 | 5.6 | 2.9 | — | −0.5/−0.9/−0.9 | 2.4/4.1 | |
| H2O/120 h | 4.2 | 6.5 | 3.1 | — | −0.3/−0.7/−0.7 | 2.6/4.4 | |
| AST | H2O/10 min | 1.6 | 4.7 | 2.6 | 7.9 | −0.6/−1.0/−1.1 | 2.1/3.8 |
| H2O/1 h | 2.7 | 5.4 | 2.7 | 9.0 | −0.4/−0.8/−0.9 | 2.2/4.0 | |
| H2O/4 h | 3.7 | 6.0 | 2.8 | 10.0 | −0.3/−0.6/−0.7 | 2.4/4.1 | |
| H2O/24 h | 5.4 | 7.2 | 3.2 | 11.9 | 0.0/−0.4/−0.4 | 2.7/4.4 | |
| H2O/120 h | 5.6 | 7.3 | 3.1 | 12.0 | 0.1/−0.3/−0.4 | 2.7/4.4 | |
| FUE | H2SO4/10 min | −16.4 | −11.1 | −6.7 | −21.1 | 0.7/0.3/0.3 | −7.1/−5.4 |
| H2SO4/1 h | −14.6 | −9.9 | −6.4 | −19.4 | 1.1/0.7/0.6 | −6.9/−5.2 | |
| H2SO4/4 h | −13.1 | −8.9 | −6.2 | −17.9 | 1.3/0.9/0.9 | −6.7/−5.0 | |
| H2SO4/24 h | −12.0 | −8.3 | −6.1 | −16.9 | 1.6/1.2/1.1 | −6.6/−4.9 | |
| H2SO4/120 h | −11.7 | −8.1 | −6.1 | −16.6 | 1.6/1.2/1.2 | −6.5/−4.8 | |
| AST | H2SO4/10 min | −18.3 | −12.1 | −6.6 | −21.2 | 0.2/−0.2/−0.3 | −7.1/−5.4 |
| H2SO4/1 h | −17.4 | −11.5 | −6.4 | −20.2 | 0.3/−0.1/−0.1 | −6.9/−5.2 | |
| H2SO4/4 h | −16.8 | −11.1 | −6.3 | −19.6 | 0.4/0.0/0.0 | −6.8/−5.1 | |
| H2SO4/24 h | −15.8 | −10.4 | −6.2 | −18.5 | 0.6/0.2/0.2 | −6.7/−5.0 | |
| H2SO4/120 h | −14.6 | −9.6 | −6.1 | −17.4 | 0.8/0.4/0.4 | −6.5/−4.8 | |
In aging experiments of both FUE and AST ash in H2O, secondary mineral phases such as montmorillonite, kaolinite, and gibbsite are found to be supersaturated in solution, indicating that these phases could have precipitated and been retained with the filtered ash particles after aging. All three of these minerals have previously been observed to nucleate ice,26,28,41 such that precipitation of ice-active phases may contribute to the observed recovery of INA of FUE ash from the leached state at aging durations longer than 10 min in H2O. The strongly positive permutation importance of Si for FUE ash aged in H2O (Fig. 3a) could reflect Si concentration representing the total quantity of elements released from the ash and contributing to precipitation of secondary phases, consistent with the steady increase in saturation indices over time. The non-monotonic change in INA of FUE ash aged in H2O overall might thus reflect competing influences of leaching and precipitation, but additional experiments and analyses would be needed to unravel their individual contributions. No recovery in INA is observed for AST ash aged in H2O over time, even though montmorillonite, gibbsite, and kaolinite are also supersaturated in the aging solutions; this conforms with these minerals being much less ice-active than the K-feldspar largely driving the INA of AST ash.
In addition to potentially introducing new ice-active sites, secondary mineral phases are often more disordered than the primary mineral phases from which they originate or are naturally weakly ice-active, such as K-mica which could precipitate onto AST ash aged in H2O. If such poorly crystalline or weakly ice-active secondary phases nucleate on or adsorb onto the aged ash surface,98 they could impair INA by covering more active ice nucleation sites. Relatedly, a decrease in INA of quartz observed over five days in H2O has been attributed to the adsorption of supersaturated silicic acid onto the quartz, covering a fraction of the ice-active sites on the quartz surface.27 This effect is expected to be time-dependent, with longer leaching durations leading to greater concentrations of secondary phases and hence more masking of original ice-active surfaces. As in FUE ash aged in H2O, a combination of leaching and precipitation might contribute to the overall changes in INA observed for AST ash aged in H2O. For example, the drop in nm at temperatures <−17 °C after 10 min of aging may be due to fast leaching of pyroxene and/or Na/Ca-feldspar, with a time-dependent decrease in nm thereafter (particularly at warmer temperatures) reflecting further leaching and/or masking of mineral surfaces by poorly ice-active aluminosilicate or Al-oxyhydroxide precipitates (Fig. 2c).
In aging experiments of both FUE and AST ash in H2SO4, most minerals are undersaturated except for some crystalline SiO2 phases, which are predicted to be slightly supersaturated. These phases could have precipitated and contributed to the loss of ice-active sites by nucleation or adsorption on ash surfaces aged in H2SO4, particularly in acid-aged FUE ash, where the positive permutation importance of Si (Fig. 3b) and positive correlation between Si and ice nucleation descriptors (Fig. S3†) indicate that a process affecting dissolved Si concentrations is the main driver of changes in INA. In acid-aged AST ash, the influence of precipitation of SiO2 phases on INA is expected to be minor compared to that of dissolution of strongly ice-active components (see Section 4.4), as these phases are barely saturated on the time scales of this experiment. For both ash samples, precipitation of Al-bearing secondary minerals is likely inhibited by low pH and by complexation between aqueous SO42− and Al in the experiments in H2SO4(aq).99
4.4 Dissolution of mineral surfaces
We hypothesize that variable dissolution of silicate surfaces also underlies the effects of chemical aging on INA, especially in acidic solution. Increasing dissolved Si concentrations in leachates of FUE and AST ash with increasing aging duration support dissolution of the ash over time, and higher dissolved Si concentrations from ash samples aged in H2SO4 compared to those aged in H2O are consistent with greater dissolution at lower pH.68,100 Acid-mediated breakdown of the silicate network is also facilitated by aqueous complexation of SO42− and Al.99 The increase in SSABET over time of FUE and AST ash aged in H2SO4 also supports extensive dissolution of these silicate materials, as a reduction in overall particle size and selective dissolution at etch pits and other surface textures69,101–103 could both contribute to increasing the SSABET of the ash samples.After 10 min of aging in H2SO4, the freezing spectra of FUE ash shows a slight decrease in INA relative to the non-aged ash (Fig. 2b), potentially due to accelerated leaching of pyroxene surfaces at low pH. This deactivation is followed by a partial recovery of nm after 1 h and then full recovery to the active site densities observed in the non-aged ash from 4 to 24 h, in parallel with the increase in SSABET. Similarly, the INA at <−17 °C of AST ash exposed to H2SO4 is largely preserved from 10 min to 120 h of aging (Fig. 2d), as SSABET increases.
As noted previously, pyroxene is inferred to drive ice nucleation by FUE ash at all temperatures and in part by AST ash at temperatures <−17 °C, so the observed preservation of INA in most acid-aged ash samples presumably relates to alteration of pyroxene in H2SO4. Zakaznova-Herzog et al. found that pyroxene alteration at low pH involves formation of a leached monolayer which is rapidly broken down, liberating Si to solution.104 Other studies have also observed very thin cation-depleted leached layers under acidic conditions which may not change crystal structure when cations are leached prior to Si release.61,103 Similarly, thin leached layers have been reported for some feldspars, for example on the Ca-rich endmember of Na/Ca-feldspar (anorthite), where an Al:Si ratio of one results in near-congruent dissolution.59,105–108 We hypothesize that the relatively stable INA maintained by the acid-aged FUE ash and AST ash (<−17 °C) reflects a quasi-steady-state density of ice-active sites across the ash surfaces, with some sites lost and others created during simultaneous leaching and dissolution in H2SO4. Dissolution of thin leached layers on pyroxene and potentially Na/Ca-feldspar might expose underlying surfaces characterized by a higher density of ice-active sites. Alternatively or additionally, surface –OH or –OH2+ groups that can enhance INA29,42,109 might be generated as Al–O and Si–O bonds are cleaved by H+ at the mineral–solution interface.24,67,110
The positive permutation importance of Si for acid-aged FUE ash (Fig. 3b) could indicate that Si release, particularly from the dissolution of pyroxene in H2SO4, is key for predicting the stability of this samples' INA. Additionally, Si and Mg are correlated with ice nucleation descriptors in acid-aged FUE ash (Fig. S3†) and Si is correlated with ice nucleation descriptors in acid-aged AST ash (Fig. S5†), which may correspond to small changes in the position of the quasi-equilibrium state of ice-active sites as the aging solution becomes more saturated. The small or zero permutation importances of Na and K for both ash samples aged in H2SO4 and the anticorrelations of these metals with ice nucleation descriptors may indicate that leaching of feldspars and other Na- and K-containing components is unimportant for predicting changes in INA for these ash samples, highlighting the role of other transformations in altering the INA of AST ash. The slightly positive permutation importance of Ca for acid-aged AST ash may reflect the contribution of more Ca-rich pyroxene dissolution in H2SO4 to INA trends (<−17 °C) for this sample, where Ca serves as the element descriptor of the leaching/dissolution quasi-equilibrium of pyroxene, instead of Si as in acid-aged FUE ash.
In contrast to the INA associated with pyroxene, the INA at >−17 °C of AST ash exposed to H2SO4 is strongly deactivated at 10 min and continues to decrease up to 120 h of aging, presumably reflecting a loss of ice nucleation active sites on K-feldspar at the ash surface. Like many aluminosilicate minerals, leaching and dissolution of K-feldspar is accelerated at low pH relative to neutral conditions.68,100 While K-feldspar is one of the most stable minerals in volcanic ash (Fig. 1), H2SO4 may selectively destroy high energy patches and/or perthitic microtextures thought to give rise to this mineral's exceptional INA.22,33,34,42 The high INA of AST ash has been attributed to similar features in its K-feldspar phases,16,111 so removal of these features could explain the drop in INA at >−17 °C of AST ash exposed to H2SO4 here. Numerous studies show that microtextures and dislocations dissolve preferentially or even control the dissolution rate of K-feldspar (Wilson and references therein68). Lastly, complexation of SO42− and Al on K-feldspar surfaces might also impair INA by blocking ice-active sites and/or removing –OH groups, as has been suggested for microcline immersed in various SO42−-containing solutions, with no such impairment observed for Na/Ca-feldspars.19,24,99 Both mechanisms of INA impairment of K-feldspar in H2SO4 may be reflected in the high permutation importance of Al for the acid-aged AST ash (Fig. 3d), where the distinct importance of Al rather than Si might point to the dominance of K-feldspar dissolution over pyroxene dissolution in predicting the overall changes in INA. There is also a direct correlation between Al and ice nucleation descriptors (Fig. S5†), as would be expected if dissolution of Al-containing phases such as feldspar was driving changes in INA and/or if Al/SO42− surface complexes are impairing the INA of the acid-aged AST ash. These correlations vary by ice nucleation descriptor, and since different descriptors represent different aspects of the curvature of the freezing spectrum, this is consistent with the change in shape of the spectrum observed in the K-feldspar regime of AST ash aged in H2SO4, further supporting the importance of Al.
The aging effects observed here contrast with those of previous aging experiments on volcanic ash, but the disparities are likely caused by differences in processing conditions. Maters et al.43 showed that exposure to H2O(g) on its own or mixed with HCl(g) reduced the INA of Astroni ash, while exposure to H2O(g) mixed with SO2(g) could enhance INA, but these high temperature (up to 800 °C) gas–ash interaction experiments are not comparable to aqueous aging at ambient temperature. Experiments performed by Jahn56 showed a reduction in Fuego ash INA after five days immersed in 0.01 M H2SO4(aq) and no change in INA upon immersion in H2O(l), but the slow drying procedure used may have altered the pH and saturation conditions within the aging solution, thereby influencing the relevant mechanisms of chemical processing.
Notably, previous experiments on mineral dust and/or individual mineral phases present in the ash samples studied here are broadly consistent with our findings. Arizona Test Dust (ATD, which contains feldspars) aged in H2O(l) and H2SO4(aq) by Perkins et al. shows similar sensitivities in its freezing spectrum depending on aging solution: ice nucleation sites active at colder temperatures (<−12 °C) are degraded in H2O(l) but largely preserved in H2SO4(aq) (as for FUE ash and partly for AST ash) and the most active ice nucleation sites associated with K-feldspar (>−12 °C) are largely preserved in H2O(l) but degraded in H2SO4(aq) (as for AST ash).50 Sullivan et al. also reported preservation of immersion-mode INA in aerosolized ATD exposed to HNO3 vapor,54 and Kulkarni et al. reported a similar preservation in immersion-mode INA in ATD, illite, montmorillonite, K-feldspar, and quartz exposed to H2SO4 vapor.112 In other cases, irreversible losses of immersion-mode INA in ATD,53,113 kaolinite,114 and feldspars115 were observed when exposed to H2SO4 vapor, although the presence of H2O(g) and use of heat to produce the H2SO4 vapor may play an important role in this process. Similarly, Yun et al. recently showed a pH-sensitive deactivation of K-feldspar INA when exposed to H2SO4(aq), HNO3(aq), HCl(aq), and aqueous organic acids.116 Finally, Losey et al. showed that formation and precipitation of gypsum (CaSO4·2H2O) on fly ash exposed to H2SO4(aq) can reduce the INA of the original minerals, while aging without the possibility of gypsum precipitation slightly enhances the INA of the fly ash samples.51 While these observations corroborate the explanations proposed for the changes in the INA of volcanic ash here, interplay between leaching, precipitation, dissolution, and possibly other mechanisms affecting the INA of the various materials hinders direct comparison to the observations for FUE and AST ash.
5 Conclusions and implications
We have shown that the effects of aqueous chemical aging in H2O or H2SO4 on the INA of two volcanic ash samples depend on the aging duration, ash mineralogy, and ice nucleation temperature regime. We infer that the INA of FUE and AST ash exposed to H2O was reduced on minute to hour time scales in the temperature regimes dominated by pyroxene and perhaps Na/Ca-feldspar (at all temperatures for FUE ash and <−17 °C for AST ash) due to formation of a Si(/Al)-rich layer on these mineral surfaces, while the temperature regime dominated by the more chemically stable K-feldspar (>−17 °C in AST ash) was less affected. Further enhancements and reductions with aging duration in the INA of FUE and AST ash, respectively, may be the result of precipitation of secondary phases of variable INA from the H2O aging solution. When exposed to H2SO4, in the temperature regime where ice nucleation may be controlled by pyroxene or Na/Ca-feldspar, a quasi-steady state INA is observed. This may reflect parallel leaching and dissolution of ash resulting in a competition between loss and exposure of new ice-active sites on mineral surfaces. Finally, the fast and dramatic loss of INA of AST ash in the temperature regime attributed to K-feldspar is hypothesized to result from fast dissolution of high-energy surface microtextures that have been shown to account for most of the INA of some types of K-feldspars. Fig. 4 provides a summary of the proposed aging effects reflected in the INA trends of FUE and AST ash aged in H2O or H2SO4.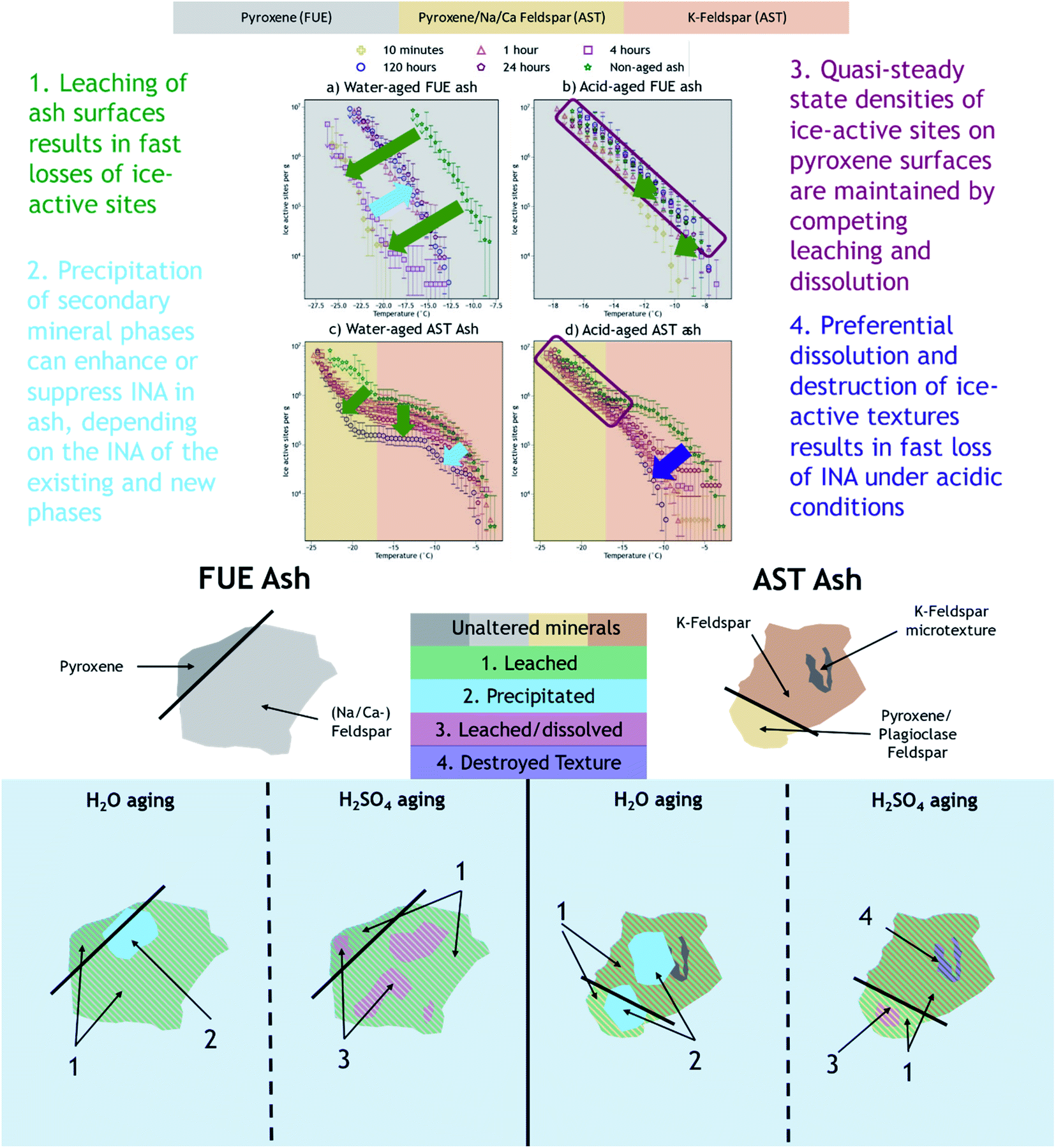 | ||
| Fig. 4 Attribution of ice nucleation spectral features to specific mineral phases and summary of proposed mechanisms by which chemical aging impacts the INA of FUE and AST ash. Note the full composition of each ash is not represented because not all minerals are expected to contribute to the observed ash INA. | ||
While previous studies on individual silicate minerals or mineral dust proxies often show small losses of INA in acid and sometimes report changes in INA from suspension in H2O,24,25,51,53–55,96,112,115 the 10 °C difference between the freezing spectra of the non-aged and water-aged FUE ash is striking and the non-monotonicity of its INA with aging duration in H2O is unprecedented. Clearly, more research is required to understand the complexities of the effects of chemical alteration on INA, especially for less-studied minerals such as pyroxenes and for multicomponent materials like volcanic ash, natural mineral dust, and biomass-burning aerosol117,118 that are present in the atmosphere.
In addition to aqueous chemical aging, exposure to gaseous acids, oxidants, and other pollutants can alter the INA of silicate materials,31,43,119 both in the wider atmosphere and – in the case of volcanic ash – at high temperatures in the eruption plume. When these processes occur in parallel or in series, they may change the INA of the airborne particles in unexpected ways, leading to additional uncertainty about their capacity for ice nucleation during atmospheric transport. For example, a potential enhancement of INA due to formation of ice-active anhydrite (CaSO4) on ash surfaces by reaction with gaseous SO2 in the hot eruption plume43 may be offset by a loss of INA during subsequent ash leaching and dissolution in aqueous H2SO4 and H2O in the cooling plume and wider atmosphere. Laboratory studies will play a key role in unravelling the complexity of such competing effects. Further investigations are needed on the influence of various factors (e.g., other mineralogies, other aging procedures, particle size, solution temperature and pH, presence of other solutes) that could affect the impact of aqueous aging on the INA of silicate materials.81,116,120,121 Also, the influence of gas-phase chemical processing on the INA of silicate aerosol via flow tube or chamber experiments is worthy of future study. Findings from such studies will have implications for understanding the INA of both primary volcanic ash emissions and resuspended ash material which has undergone further physicochemical alteration following deposition, including by leaching, dissolution and precipitation upon contact with aqueous solution (e.g., rainwater) in the environment.
Elucidating the influence on INA of chemical and physical variables individually and deconvoluting the molecular-scale mechanisms of aging and atmospheric pathways of solid–gas/liquid exposure will be important next steps towards a deeper understanding of ice nucleation by airborne particles. Many of these variables should be studied under laboratory conditions to control potential convoluting factors, and applying surface sensitive physicochemical analyses (e.g., Yun et al.121) to aged materials will be particularly helpful for studying the mechanisms of alteration of INA. Fieldwork and modelling studies will also be necessary to constrain the real-world impacts of aging via different processes on atmospheric INP concentrations and activity. Without further research, it is difficult to predict on the basis of our findings how the INA of volcanic ash will change as it ages in the environment, but understanding the geochemical mechanisms underlying aging is necessary for a more complete picture of heterogeneous ice nucleation and its evolution in the atmosphere.
Data availability
The dataset associated with this article is available in the Research Data Leeds Repository at https://doi.org/10.5518/1079.Author contributions
Conceptualization: all co authors; investigation & methodology: WDF, ECM, RGM and MPA; funding acquisition, resources & supervision: ECM, RCS and BJM; formal analysis: WDF; writing – original draft & visualization: WDF and ECM; writing – review & editing: WDF, ECM, MPA, LGJ, RCS and BJM.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
ECM was funded by the European Union's Horizon 2020 Research and Innovation Programme under Marie Skłodowska-Curie Actions grant agreement no. 746695, and currently holds an Early Career Fellowship funded jointly by the Leverhulme Trust and Isaac Newton Trust. WDF was funded by the Undergraduate Research Office at Carnegie Mellon University (ISURF) to conduct research at the University of Leeds in summer 2019. RGM benefitted from a Deutscher Akademischer Austauschdienst RISE Worldwide internship at the University of Leeds in summer 2019. RCS acknowledges the National Science Foundation of the United States of America (CAREER; CHM-1554941) for funding, and a supplement to that grant further supported WDF. BJM acknowledges the European Research Council (MarineIce; 648661) and the Natural Environment Research Council (NE/T00648X/1) for funding. ECM thanks Corrado Cimarelli, Donald Dingwell and Ulrich Küppers for providing the Astroni ash. WDF thanks Daniel B. Williams for providing the Volcán de Fuego ash. We are grateful to Andy Hobson, Stephen Reid, Lesley Neave and Fiona Keay for assistance with laboratory experiments and analyses.References
- B. J. Murray, D. O'Sullivan, J. D. Atkinson and M. E. Webb, Chem. Soc. Rev., 2012, 41, 6519–6554 RSC.
- Z. A. Kanji, L. A. Ladino, H. Wex, Y. Boose, M. Burkert-Kohn, D. J. Cziczo and M. Krämer, Meteorol. Monogr., 2017, 58, 1.1–1.33 CrossRef.
- P. V. Hobbs, C. M. Fullerton and G. C. Bluhm, Nat. Phys. Sci., 1971, 230, 90–91 CrossRef.
- A. Sanchez-Marroquin, O. Arnalds, K. J. Baustian-Dorsi, J. Browse, P. Dagsson-Waldhauserova, A. D. Harrison, E. C. Maters, K. J. Pringle, J. Vergara-Temprado, I. T. Burke, J. B. McQuaid, K. S. Carslaw and B. J. Murray, Sci. Adv., 2020, 6, 8137–8161 CrossRef PubMed.
- A. J. Durant, C. Bonadonna and C. J. Horwell, Elements, 2010, 6, 235–240 CrossRef CAS.
- N. M. Mahowald, A. R. Baker, G. Bergametti, N. Brooks, R. A. Duce, T. D. Jickells, N. Kubilay, J. M. Prospero and I. Tegen, Global Biogeochem. Cycles, 2005, 19, GB4025 Search PubMed.
- M. G. Wiesner, A. Wetzel, S. G. Catane, E. L. Listanco and H. T. Mirabueno, Bull. Volcanol., 2004, 66, 226–242 CrossRef.
- J. P. Vernier, T. D. Fairlie, T. Deshler, M. Natarajan, T. Knepp, K. Foster, F. G. Wienhold, K. M. Bedka, L. Thomason and C. Trepte, J. Geophys. Res., 2016, 121, 11104–11118 Search PubMed.
- E. J. Jensen, S. Woods, R. P. Lawson, T. P. Bui, L. Pfister, T. D. Thornberry, A. W. Rollins, J. P. Vernier, L. L. Pan, S. Honomichl and O. B. Toon, Geophys. Res. Lett., 2018, 45, 11483–11489 CrossRef.
- M. Nakagawa and T. Ohba, Global Environ. Res., 2003, 6, 41–51 Search PubMed.
- N. Rogers, in The Encyclopedia of Volcanoes, ed. H. Sigurdsson, B. Houghton, S. R. McNutt, H. Rymer and J. Stix, Academic Press, London, 2nd edn, 2015, ch. 4, pp. 93–112 Search PubMed.
- A. Gibbs, M. Charman, W. Schwarzacher and A. C. Rust, GeoResJ, 2015, 7, 66–69 CrossRef.
- C. R. Hoyle, V. Pinti, A. Welti, B. Zobrist, C. Marcolli, B. Luo, A. Höskuldsson, H. B. Mattsson, O. Stetzer, T. Thorsteinsson, G. Larsen and T. Peter, Atmos. Chem. Phys., 2011, 11, 9911–9926 CrossRef CAS.
- L. G. Jahn, W. D. Fahy, D. B. Williams and R. C. Sullivan, ACS Earth Space Chem., 2019, 3, 626–636 CrossRef CAS.
- T. P. Mangan, J. D. Atkinson, J. W. Neuberg, D. O’Sullivan, T. W. Wilson, T. F. Whale, L. Neve, N. S. Umo, T. L. Malkin and B. J. Murray, PLoS One, 2017, 12, e0169720 CrossRef CAS PubMed.
- E. C. Maters, D. B. Dingwell, C. Cimarelli, D. Müller, T. F. Whale and B. J. Murray, Atmos. Chem. Phys., 2019, 19, 5451–5465 CrossRef CAS.
- G. P. Schill, K. Genareau and M. A. Tolbert, Atmos. Chem. Phys., 2015, 15, 7523–7536 CrossRef CAS.
- I. Steinke, O. Möhler, A. Kiselev, M. Niemand, H. Saathoff, M. Schnaiter, J. Skrotzki, C. Hoose and T. Leisner, Atmos. Chem. Phys., 2011, 11, 12945–12958 CrossRef CAS.
- A. Kumar, C. Marcolli, B. Luo and T. Peter, Atmos. Chem. Phys., 2018, 18, 7057–7079 CrossRef CAS.
- J. D. Yakobi-Hancock, L. A. Ladino and J. P. D. Abbatt, Atmos. Chem. Phys., 2013, 13, 11175–11185 CrossRef CAS.
- A. D. Harrison, T. F. Whale, M. A. Carpenter, M. A. Holden, L. Neve, D. Osullivan, J. Vergara Temprado and B. J. Murray, Atmos. Chem. Phys., 2016, 16, 10927–10940 CrossRef CAS.
- T. F. Whale, M. A. Holden, A. N. Kulak, Y.-Y. Kim, F. C. Meldrum, H. K. Christenson and B. J. Murray, Phys. Chem. Chem. Phys., 2017, 19, 31186–31193 RSC.
- Y. Boose, A. Welti, J. Atkinson, F. Ramelli, A. Danielczok, H. G. Bingemer, M. Plötze, B. Sierau, Z. A. Kanji and U. Lohmann, Atmos. Chem. Phys., 2016, 16, 15075–15095 CrossRef CAS.
- A. Kumar, C. Marcolli and T. Peter, Atmos. Chem. Phys., 2019, 19, 6059–6084 CrossRef CAS.
- A. D. Harrison, K. Lever, A. Sanchez-Marroquin, M. A. Holden, T. F. Whale, M. D. Tarn, J. B. McQuaid and B. J. Murray, Atmos. Chem. Phys., 2019, 19, 11343–11361 CrossRef CAS.
- L. Kaufmann, C. Marcolli, J. Hofer, V. Pinti, C. R. Hoyle and T. Peter, Atmos. Chem. Phys., 2016, 16, 11177–11206 CrossRef CAS.
- A. Kumar, C. Marcolli and T. Peter, Atmos. Chem. Phys., 2019, 19, 6035–6058 CrossRef CAS.
- V. Pinti, C. Marcolli, B. Zobrist, C. R. Hoyle and T. Peter, Atmos. Chem. Phys., 2012, 12, 5859–5878 CrossRef CAS.
- G. C. Sosso, G. A. Tribello, A. Zen, P. Pedevilla and A. Michaelides, J. Chem. Phys., 2016, 145, 211927 CrossRef PubMed.
- S. L. Broadley, B. J. Murray, R. J. Herbert, J. D. Atkinson, S. Dobbie, T. L. Malkin, E. Condliffe and L. Neve, Atmos. Chem. Phys., 2012, 12, 287–307 CrossRef CAS.
- A. Salam, G. Lesins and U. Lohmann, Air Qual., Atmos. Health, 2008, 1, 135–142 CrossRef CAS.
- B. J. Murray, S. L. Broadley, T. W. Wilson, J. D. Atkinson and R. H. Wills, Atmos. Chem. Phys., 2011, 11, 4191–4207 CrossRef CAS.
- M. A. Holden, T. F. Whale, M. D. Tarn, D. Osullivan, R. D. Walshaw, B. J. Murray, F. C. Meldrum and H. K. Christenson, Sci. Adv., 2019, 5(2), eaav4316 CrossRef PubMed.
- A. Kiselev, F. Bachmann, P. Pedevilla, S. J. Cox, A. Michaelides, D. Gerthsen and T. Leisner, Science, 2017, 355, 367–371 CrossRef CAS PubMed.
- M. A. Holden, J. M. Campbell, F. C. Meldrum, B. J. Murray and H. K. Christenson, Proc. Natl. Acad. Sci. U.S.A., 2021, 118(18), e2022859118 CrossRef CAS PubMed.
- E. Pach and A. Verdaguer, J. Phys. Chem. C, 2019, 123(34), 20998–21004 CrossRef CAS.
- B. Glatz and S. Sarupria, Langmuir, 2018, 34(3), 1190–1198 CrossRef CAS PubMed.
- G. C. Sosso, T. Li, D. Donadio, G. A. Tribello and A. Michaelides, J. Phys. Chem. Lett., 2016, 7, 16 Search PubMed.
- Y. Bi, B. Cao and T. Li, Nat. Commun., 2017, 8, 15372 CrossRef CAS PubMed.
- M. A. Freedman, J. Phys. Chem. Lett., 2015, 6, 3850–3858 CrossRef CAS PubMed.
- E. Chong, M. King, K. E. Marak and M. A. Freedman, J. Phys. Chem. A, 2019, 123, 2447–2456 CrossRef CAS PubMed.
- P. Pedevilla, M. Fitzner and A. Michaelides, Phys. Rev. B, 2017, 96, 115441 CrossRef.
- E. C. Maters, C. Cimarelli, A. S. Casas, D. B. Dingwell and B. J. Murray, Earth Planet. Sci. Lett., 2020, 551, 116587 CrossRef CAS.
- P. Delmelle, F. Villiéras and M. Pelletier, Bull. Volcanol., 2005, 67, 160–169 CrossRef.
- T. L. Lathem, P. Kumar, A. Nenes, J. Dufek, I. N. Sokolik, M. Trail and A. Russell, Geophys. Res. Lett., 2011, 38, L11802 CrossRef.
- R. C. Sullivan, S. A. Guazzotti, D. A. Sodeman and K. A. Prather, Atmos. Chem. Phys., 2007, 7, 1213–1236 CrossRef CAS.
- P. Ayris and P. Delmelle, Phys. Chem. Earth, 2012, 45–46, 103–112 CrossRef.
- P. Delmelle, F. B. Wadsworth, E. C. Maters and P. M. Ayris, Rev. Mineral. Geochem., 2018, 84, 285–308 CrossRef CAS.
- T. Jurkat, C. Voigt, F. Arnold, H. Schlager, H. Aufmhoff, J. Schmale, J. Schneider, M. Lichtenstern and A. Dörnbrack, J. Geophys. Res., 2010, 115, D00L17 CrossRef.
- R. J. Perkins, S. M. Gillette, T. C. J. Hill and P. J. DeMott, ACS Earth Space Chem., 2020, 4, 133–141 CrossRef CAS.
- D. J. Losey, S. K. Sihvonen, D. P. Veghte, E. Chong and M. A. Freedman, Environ. Sci.: Processes Impacts, 2018, 20, 1581–1592 RSC.
- S. K. Sihvonen, G. P. Schill, N. A. Lyktey, D. P. Veghte, M. A. Tolbert and M. A. Freedman, J. Phys. Chem. A, 2014, 118, 8787–8796 CrossRef CAS PubMed.
- R. C. Sullivan, M. D. Petters, P. J. Demott, S. M. Kreidenweis, H. Wex, D. Niedermeier, S. Hartmann, T. Clauss, F. Stratmann, P. Reitz, J. Schneider and B. Sierau, Atmos. Chem. Phys., 2010, 10, 11471–11487 CrossRef CAS.
- R. C. Sullivan, L. Miñambres, P. J. Demott, A. J. Prenni, C. M. Carrico, E. J. T. Levin and S. M. Kreidenweis, Geophys. Res. Lett., 2010, 37, L24805 CrossRef.
- D. J. Cziczo, K. D. Froyd, S. J. Gallavardin, O. Moehler, S. Benz, H. Saathoff and D. M. Murphy, Environ. Res. Lett., 2009, 4, 044013 CrossRef.
- L. G. I. Jahn, Biomass-Burning and Volcanic Ash Aerosols: Atmospheric Aging, Chemical Reactivity, and Ice Nucleation Ability, PhD thesis, Carnegie Mellon University, 2020.
- J. Schott, R. A. Berner and E. L. Sjöberg, Geochim. Cosmochim. Acta, 1981, 45, 2123–2135 CrossRef CAS.
- W. H. Casey, H. R. Westrich, G. W. Arnold and J. F. Banfield, Geochim. Cosmochim. Acta, 1989, 53, 821–832 CrossRef CAS.
- E. H. Oelkers and J. Schott, Geochim. Cosmochim. Acta, 1995, 59, 5039–5053 CrossRef CAS.
- E. H. Oelkers, S. V. Golubev, C. Chairat, O. S. Pokrovsky and J. Schott, Geochim. Cosmochim. Acta, 2009, 73, 4617–4634 CrossRef CAS.
- E. H. Oelkers, Geochim. Cosmochim. Acta, 2001, 65, 3703–3719 CrossRef CAS.
- W. H. Casey, H. R. Westrich, J. F. Banfield, G. G. Ferruzzi and G. W. G. W. Arnold, Nature, 1993, 366, 253–256 CrossRef CAS.
- R. Hellmann, J. M. Penisson, R. L. Hervig, J. H. Thomassin and M. F. Abrioux, Phys. Chem. Miner., 2003, 30, 192–197 CrossRef CAS.
- R. Hellmann, R. Wirth, D. Daval, J. P. Barnes, J. M. Penisson, D. Tisserand, T. Epicier, B. Florin and R. L. Hervig, Chem. Geol., 2012, 294–295, 203–216 CrossRef CAS.
- E. Ruiz-Agudo, C. V. Putnis, C. Rodriguez-Navarro and A. Putnis, Geology, 2012, 40, 947–950 CrossRef CAS.
- E. Ruiz-Agudo, H. E. King, L. D. Patiño-Ĺpez, C. V. Putnis, T. Geisler, C. Rodriguez-Navarro and A. Putnis, Geology, 2016, 44, 567–570 CrossRef CAS.
- E. S. Chardon, F. R. Livens and D. J. Vaughan, Earth-Sci. Rev., 2006, 78, 1–26 CrossRef CAS.
- M. J. Wilson, Clay Miner., 2004, 39, 233–266 CrossRef CAS.
- G. Yuan, Y. Cao, H. M. Schulz, F. Hao, J. Gluyas, K. Liu, T. Yang, Y. Wang, K. Xi and F. Li, Earth-Sci. Rev., 2019, 191, 114–140 CrossRef CAS.
- I. J. Muir and H. W. Nesbitt, Geochim. Cosmochim. Acta, 1997, 61, 265–274 CrossRef CAS.
- S. S. Goldich, J. Geol., 1938, 46, 17–58 CrossRef CAS.
- A. F. White and S. L. Brantley, Rev. Mineral. Geochem., 1995, 31, 1–22 CAS.
- D. Huddart and T. A. Stott, Earth Environments: Past, Present, and Future, Wiley, Hoboken, 2010 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- Igneous Rocks: a Classification and Glossary of Terms: Recommendations of the International Union of Geological Sciences, Subcommission on the Systematics of Igneous Rocks, ed. R. Le Maitre, A. Streckeisen, B. Zanettin, M.B. Le BasBonin and P. Bateman, Cambridge University Press, Cambridge, 2002 Search PubMed.
- Z. Shi, M. D. Krom, T. D. Jickells, S. Bonneville, K. S. Carslaw, N. Mihalopoulos, A. R. Baker and L. G. Benning, Aeolian Res., 2012, 5, 21–42 CrossRef.
- E. C. Maters, P. Delmelle and S. Bonneville, Environ. Sci. Technol., 2016, 50, 5033–5040 CrossRef CAS PubMed.
- E. C. Maters, P. Delmelle and H. P. Gunnlaugsson, Chem. Geol., 2017, 449, 73–81 CrossRef CAS.
- A. Robock, Rev. Geophys., 2000, 38, 191–219 CrossRef CAS.
- T. F. Whale, B. J. Murray, T. W. Wilson, N. S. Umo, K. J. Baustian, J. D. Atkinson, D. A. Workneh and G. J. Morris, Atmos. Meas. Tech., 2015, 8, 2437–2447 CrossRef CAS.
- T. F. Whale, M. A. Holden, T. W. Wilson, D. O'Sullivan and B. J. Murray, Chem. Sci., 2018, 9, 4142–4151 RSC.
- J. D. Atkinson, B. J. Murray, M. T. Woodhouse, T. F. Whale, K. J. Baustian, K. S. Carslaw, S. Dobbie, D. O'Sullivan and T. L. Malkin, Nature, 2013, 498, 355–358 CrossRef CAS PubMed.
- M. Polen, T. Brubaker, J. Somers and R. C. Sullivan, Atmos. Meas. Tech., 2018, 11, 5315–5334 CrossRef CAS.
- G. Vali, Atmos. Chem. Phys., 2014, 14, 5271–5294 CrossRef.
- G. Vali, J. Atmos. Sci., 1971, 28, 402–409 CrossRef.
- G. Vali, Atmos. Meas. Tech., 2019, 12, 1219–1231 CrossRef CAS.
- J. P. Boyd, R. Petschek, J. P. Boyd and R. Petschek, J. Sci. Comput., 2014, 59, 1–27 CrossRef.
- P. Schober and L. A. Schwarte, Anesth. Analg., 2018, 126, 1763–1768 CrossRef PubMed.
- P. Fabian, M. Vincent, G. Olivier, B. Mathieu, P. Peter, W. Ron, V. Jake and D. Cournapeau, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- N. Abramson, D. Braverman and G. Sebestyen, IEEE Trans. Inf. Theor., 1963, 9, 257–261 CrossRef.
- A. J. Smola and B. Schölkopf, Stat. Comput., 2004, 14, 199–222 CrossRef.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- R. W. Luce, R. W. Bartlett and G. A. Parks, Geochim. Cosmochim. Acta, 1972, 36, 35–50 CrossRef CAS.
- R. A. Eggleton and J. N. Boland, Clays Clay Miner., 1982, 30, 11–20 CrossRef CAS.
- Y. Qiu, N. Odendahl, A. Hudait, R. Mason, A. K. Bertram, F. Paesani, P. J. DeMott and V. Molinero, J. Am. Chem. Soc., 2017, 139, 3052–3064 CrossRef CAS PubMed.
- A. Peckhaus, A. Kiselev, T. Hiron, M. Ebert and T. Leisner, Atmos. Chem. Phys., 2016, 16, 11477–11496 CrossRef CAS.
- D. L. Parkhurst and C. A. J. Appelo, Description of input and examples for PHREEQC version 3: a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, in Modeling Techniques, U.S. Geological Survey, Denver, 2013, pp. 1-497 Search PubMed.
- C. B. Richardson and T. D. Snyder, Langmuir, 1994, 10, 2462–2465 CrossRef CAS.
- Y. Min, J. D. Kubicki and Y.-S. Jun, Environ. Sci. Technol., 2015, 49, 1946–1954 CrossRef CAS PubMed.
- S. L. Brantley, in Kinetics of Water-Rock Interaction, ed. J. Kubicki and A. F. White, Springer, New York, 2008, Ch. 5, pp. 151–210 Search PubMed.
- L. L. Stillings and S. L. Brantley, Geochim. Cosmochim. Acta, 1995, 59, 1483–1496 CrossRef CAS.
- R. J. Smith, B. H. N. N. Horgan, P. Mann, E. A. Cloutis and P. R. Christensen, J. Geophys. Res.: Planets, 2017, 122, 203–227 CAS.
- J. Schott and R. A. Berner, in The Chemistry of Weathering, ed. J. I. Drever, Springer, Dordrecht, 1985, Ch. 3, pp. 35–53 Search PubMed.
- V. P. Zakaznova-Herzog, H. W. Nesbitt, G. M. Bancroft and J. S. Tse, Geochim. Cosmochim. Acta, 2008, 72, 69–86 CrossRef CAS.
- G. Jordan, S. R. Higgins, C. M. Eggleston, S. M. Swapp, D. E. Janney and K. G. Knauss, Geochim. Cosmochim. Acta, 1999, 63, 3183–3191 CrossRef CAS.
- R. A. Nixon, Geology, 1979, 7, 221–224 CrossRef CAS.
- R. A. Berner and G. R. Holdren, Geology, 1977, 5, 369–372 CrossRef CAS.
- A. E. Blum, in Feldspars and their Reactions, ed. I. Parsons, Springer, Dordrecht, 1994, Ch. 15, pp. 595–630 Search PubMed.
- H. Xue, Y. Lu, H. Geng, B. Dong, S. Wu, Q. Fan, Z. Zhang, X. Li, X. Zhou and J. Wang, J. Phys. Chem. Lett., 2019, 2458–2462 CrossRef CAS PubMed.
- R. Hellmann, C. M. Eggleston, M. F. Hochella and D. A. Crerar, Geochim. Cosmochim. Acta, 1990, 54, 1267–1281 CrossRef CAS.
- R. L. Astbury, M. Petrelli, T. Ubide, M. J. Stock, I. Arienzo, M. Dantonio and D. Perugini, Lithos, 2018, 318–319, 464–477 CrossRef CAS.
- G. Kulkarni, K. Zhang, C. Zhao, M. Nandasiri, V. Shutthanandan, X. Liu, J. Fast and L. Berg, J. Geophys. Res. Atmos., 2015, 120, 7682–7698 CAS.
- D. Niedermeier, S. Hartmann, T. Clauss, H. Wex, A. Kiselev, R. C. Sullivan, P. J. Demott, M. D. Petters, P. Reitz, J. Schneider, E. Mikhailov, B. Sierau, O. Stetzer, B. Reimann, U. Bundke, R. A. Shaw, A. Buchholz, T. F. Mentel and F. Stratmann, Atmos. Chem. Phys., 2011, 11, 11131–11144 CrossRef CAS.
- Y. Tobo, P. J. DeMott, M. Raddatz, D. Niedermeier, S. Hartmann, S. M. Kreidenweis, F. Stratmann and H. Wex, Geophys. Res. Lett., 2012, 39, L19803 CrossRef.
- S. Augustin-Bauditz, H. Wex, S. Kanter, M. Ebert, D. Niedermeier, F. Stolz, A. Prager and F. Stratmann, Geophys. Res. Lett., 2014, 41, 7375–7382 CrossRef.
- J. Yun, A. Kumar, N. Removski, A. Shchukarev, N. Link, J.-F. Boily and A. K. Bertram, ACS Earth Space Chem., 2021, 5, 1212–1222 CrossRef CAS.
- L. G. Jahn, M. J. Polen, L. G. Jahl, T. A. Brubaker, J. Somers and R. C. Sullivan, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 201922128 CrossRef PubMed.
- L. G. Jahl, T. A. Brubaker, M. J. Polen, L. G. Jahn, K. P. Cain, B. B. Bowers, W. D. Fahy, S. Graves and R. C. Sullivan, Sci. Adv., 2021, 7, eabd3440 CrossRef CAS PubMed.
- Z. A. Kanji, A. Welti, C. Chou, O. Stetzer and U. Lohmann, Atmos. Chem. Phys., 2013, 13, 9097–9118 CrossRef.
- B. Zobrist, C. Marcolli, T. Peter and T. Koop, J. Phys. Chem. A, 2008, 112, 3965–3975 CrossRef CAS PubMed.
- J. Yun, N. Link, A. Kumar, A. Shchukarev, J. Davidson, A. Lam, C. Walters, Y. Xi, J. F. Boily and A. K. Bertram, ACS Earth Space Chem., 2020, 4, 873–881 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00071c |
| ‡ Authors contributed equally to this work. |
| § Now at Department of Chemical Engineering, University of Texas at Austin, Austin, Texas 78705, USA |
| This journal is © The Royal Society of Chemistry 2022 |