
Diastereoselective synthesis of cyclopentanone-fused spirooxindoles by N-heterocyclic carbene-catalyzed homoenolate annulation with isatilidenes†
Atanu
Patra
a,
Anup
Bhunia
a,
Santhivardhana Reddy
Yetra
a,
Rajesh G.
Gonnade
b and
Akkattu T.
Biju
*a
aOrganic Chemistry Division, CSIR-National Chemical Laboratory (CSIR-NCL), Dr. Homi Bhabha Road, Pune-411008, India. E-mail: at.biju@ncl.res.in; Fax: +91-20-25902629; Tel: +91-20-25902441
bCentre for Materials Characterization, CSIR-National Chemical Laboratory (CSIR-NCL), Dr. Homi Bhabha Road, Pune-411008, India
First published on 29th September 2015
Abstract
N-Heterocyclic carbene (NHC)-catalyzed formal [3 + 2] annulation of α,β-unsaturated aldehydes with N-substituted isatilidenes resulting in the diastereoselective synthesis of cyclopentanone-fused spirooxindoles is demonstrated. Mechanistically, the reaction proceeds via the generation of homoenolate equivalent intermediates from NHC and enals, which on interception with isatilidenes afford spiro-heterocyclic compounds bearing an all-carbon quaternary spiro-center in moderate to good yields and generally with high diastereoselectivity. Moreover, the functionalization of the spirooxindoles as well as the initial studies on the enantioselective version of this reaction are presented.
Introduction
N-Heterocyclic carbene (NHC)-based organocatalysis has been widely explored for the umpolung of aldehydes resulting in a variety of unique organic transformations.1 Ever since the seminal discovery independently by Glorius2 and Bode3 in 2004 on NHC-induced generation of homoenolate equivalents from α,β-unsaturated aldehydes followed by the formal [3 + 2] annulation with aldehydes leading to γ-butyrolactones, the NHC-homoenolate concept has been utilized for the construction of several acyclic compounds, carbocycles, heterocycles, and even spiroheterocycles.4 In 2006, Nair and co-workers demonstrated the NHC-catalyzed homoenolate formal [3 + 2] annulation with 1,2-dicarbonyl compounds for the synthesis of spiro γ-butyrolactones (Scheme 1).5 Interestingly, the enantioselective version of the spirooxindole γ-butyrolactone synthesis was disclosed by Ye and co-workers6 using chiral NHCs and later by Scheidt and co-workers using a cooperative NHC/Lewis acid strategy.7 Moreover, the enantioselective synthesis of spirooxindole γ-butyrolactams by NHC-catalyzed homoenolate annulation with isatin-derived ketimines was disclosed by Chi and co-workers.8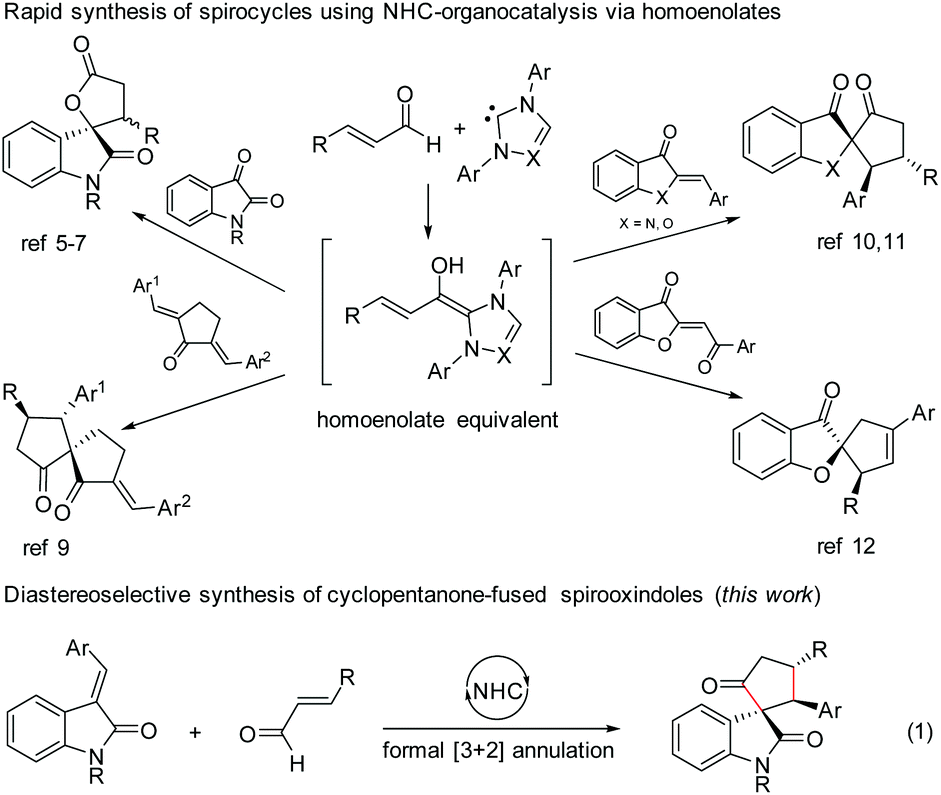 | ||
| Scheme 1 NHC-catalyzed routes to spirocyclic systems. | ||
The stereoselective and formal [3 + 2] annulation route to spirocyclopentanones by the NHC-catalyzed reaction of enals with cyclic dienones was demonstrated by the Nair group in 2008.9 Moreover, highly enantioselective formal [3 + 2] annulation reaction of enals with azaaurones/aurones leading to the synthesis of spiroheterocycles was recently disclosed by the Glorius group10 and Zhao group.11 Very recently, NHC-catalyzed reaction of enals with benzoylidene benzofuran 3-ones (aurone analogs) resulting in the synthesis of cyclopentene-fused spirobenzofuran 3-ones was uncovered by the Nair group.12 Furthermore, the enantioselective [4 + 3] annulation reaction of NHC-bound homoenolate equivalents with o-quinone methides to access 2-benzoxopinones was developed independently by the Ye group13 and the Scheidt group.14
In the context of our interest in the reaction of NHC-bound homoenolate equivalents with electrophilic systems,15 we have recently reported the NHC-homoenolate annulation with 2′-hydroxy chalcones15b and 2-enoylpyridines/2-enoyl pyridine N-oxides15a resulting in the diastereoselective synthesis of cyclopentane-fused coumarins and β-lactone-fused cyclopentanes respectively. Inspired by these results, we envisioned that homoenolate annulation with isatilidenes could result in a straightforward synthesis of spirooxindoles. Herein, we report the highly diastereoselective formal [3 + 2] annulation reaction of enals with isatilidenes resulting in the formation of cyclopentanone-fused spirooxindole derivatives possessing an all-carbon quaternary spiro-stereogenic center (Scheme 1, eqn (1)).16,17 It is noteworthy that spirooxindoles are associated with interesting biological properties and this core structure can be found in various natural products and medicinally relevant molecules.18
Results and discussion
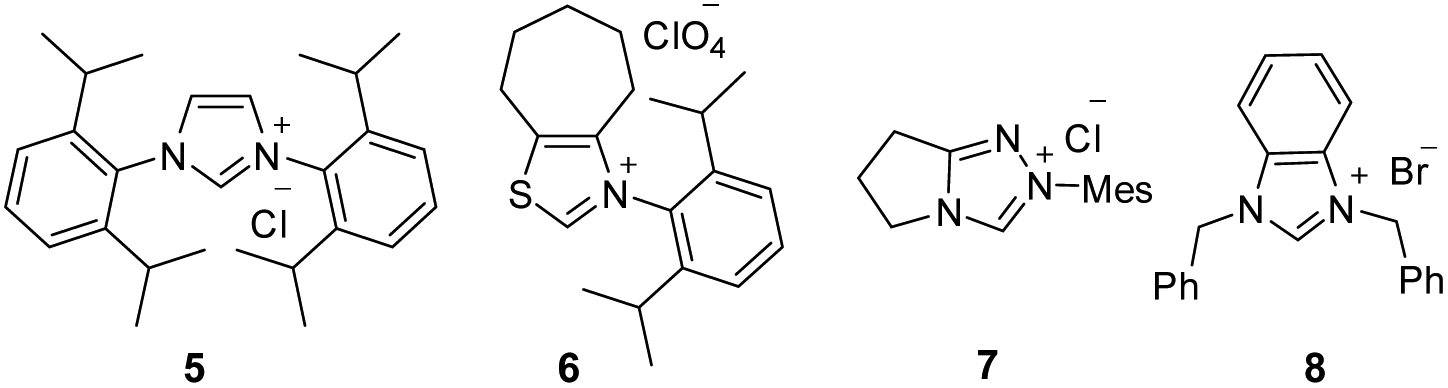
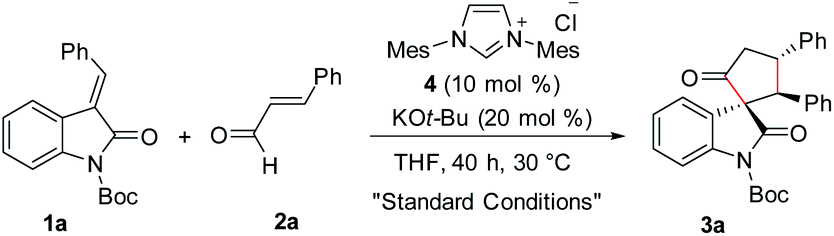
Given the importance of spirooxindole-based heterocycles and in view of our interest in NHC-organocatalysis, the present study was initiated by treating the N-Boc isatilidene 1a with cinnamaldehyde 2a in the presence of NHC generated from the imidazolium salt 4 using KO-tBu as the base. To our delight, under these conditions, the cyclopentanone-fused spirooxindole 3a was formed in 51% yield and an excellent diastereoselectivity of >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 1). The N-protection of isatilidene 1a was mandatory for the reaction and the attempted experiments with N-unprotected 1a were not successful. The reactions performed using sterically demanding NHCs derived from the precursors 5–7 furnished inferior results (entries 2–4). However, the reaction attempted using the imidazolium salt 8 afforded 3a in 59% yield, but with reduced diastereoselectivity of 3:1 (entry 5). Hence, further studies were carried out using NHC generated from 4. A rapid screening of bases revealed that other organic (including DBU and Et3N) and inorganic (including K2CO3 and Cs2CO3) bases are not beneficial for this spiroannulation reaction (entries 7–10). Variation of solvents indicated that except THF, other solvents furnished very low yield of the desired product 3a (entries 11–14). Interestingly, when the reaction was performed using 15 mol% of 4 and 30 mol% of KOt-Bu, the yield of 3a was improved to 59% maintaining excellent diastereoselectivity (entry 15). Under this condition, the use of 1.5 equiv. of enal 2a afforded 3a in 71% yield and >20:1 diastereoselectivity (entry 16).19 Further attempts to improve the yield of 3a by the use of Lewis acids and Brønsted acids as additives were unsuccessful (not shown in Table 1). It may be mentioned that under the present reaction conditions, the spirocyclopentene12 as well as the [4 + 3] annulation products were not observed.13,14
:1 (Table 1, entry 1). The N-protection of isatilidene 1a was mandatory for the reaction and the attempted experiments with N-unprotected 1a were not successful. The reactions performed using sterically demanding NHCs derived from the precursors 5–7 furnished inferior results (entries 2–4). However, the reaction attempted using the imidazolium salt 8 afforded 3a in 59% yield, but with reduced diastereoselectivity of 3:1 (entry 5). Hence, further studies were carried out using NHC generated from 4. A rapid screening of bases revealed that other organic (including DBU and Et3N) and inorganic (including K2CO3 and Cs2CO3) bases are not beneficial for this spiroannulation reaction (entries 7–10). Variation of solvents indicated that except THF, other solvents furnished very low yield of the desired product 3a (entries 11–14). Interestingly, when the reaction was performed using 15 mol% of 4 and 30 mol% of KOt-Bu, the yield of 3a was improved to 59% maintaining excellent diastereoselectivity (entry 15). Under this condition, the use of 1.5 equiv. of enal 2a afforded 3a in 71% yield and >20:1 diastereoselectivity (entry 16).19 Further attempts to improve the yield of 3a by the use of Lewis acids and Brønsted acids as additives were unsuccessful (not shown in Table 1). It may be mentioned that under the present reaction conditions, the spirocyclopentene12 as well as the [4 + 3] annulation products were not observed.13,14
|
|
||
|---|---|---|
| Entry | Variation of the standard conditionsa | Yield of 3ab,c (%) |
|
a Standard conditions: 1a (0.25 mmol), 2a (0.25 mmol), 4 (10 mol%), KOt-Bu (20 mol%), THF (1.0 mL), 30 °C and 40 h.
b Isolated yield of the product.
c The diastereoselectivity observed by 1H NMR of crude products was >20:1 unless indicated.
d The dr of 3:1 was observed.
|
||
| 1 | None | 51 |
| 2 | 5 instead of 4 | <5 |
| 3 | 6 instead of 4 | 21 |
| 4 | 7 instead of 4 | <5 |
| 5 | 8 instead of 4 | 59d |
| 6 | 50 °C instead of 30 °C | 41 |
| 7 | DBU instead of KOt-Bu | 29 |
| 8 | Et3N instead of KOt-Bu | <5 |
| 9 | K2CO3 instead of KOt-Bu | 23 |
| 10 | Cs2CO3 instead of KOt-Bu | <5 |
| 11 | DME instead of THF | <5 |
| 12 | 1,4-Dioxane instead of THF | 11 |
| 13 | CH2Cl2 instead of THF | <5 |
| 14 | Toluene instead of THF | 26 |
| 15 | 15 mol% of 4 and 30 mol% of KOt-Bu | 59 |
| 16 | 15 mol% of 4 , 30 mol% of KOt-Bu and 1.5 equiv. of 2a | 71 |
|
||
The spirooxindole derivative 3a was characterized using routine spectroscopic techniques. Finally, the structure and the relative stereochemistry of the three chiral centers in 3a was confirmed by using single-crystal X-ray analysis (Fig. 1).20
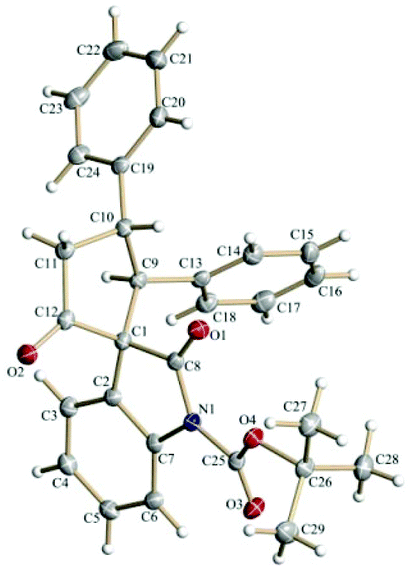 | ||
| Fig. 1 ORTEP diagram of 3a (50% probability factor for thermal ellipsoids). | ||
With the reaction conditions for the diastereoselective synthesis of cyclopentanone-fused spirooxindoles, we then examined the scope and limitations of this annulation reaction. First, we studied the variation of the isatilidene moiety (Scheme 2). When acetyl protection on nitrogen was used, the product 3b was formed in 58% yield, but with reduced diastereoselectivity of 3:1. In addition, the N-benzyl protection afforded the desired product 3c in 60% yield in 3:1 dr. A series of isatilidenes with different substitutions on the β-aryl ring underwent smooth annulation reaction resulting in the formation of the spirooxindoles in moderate to good yields (3d–3h). The substitution at the β-aryl ring of 1 did not affect the diastereoselectivity of the reaction and in all cases the spirocompound was isolated in >20:1 dr. It is noteworthy that the pentafluoroaryl substitution on the β-aryl ring furnished the expected product 3h in 55% yield. Moreover, electron-releasing and -withdrawing substituents are tolerated at the indolin-2-one moiety, and the desired products are formed in moderate to good yields (3i, 3j). In addition, β-heteroaryl substituted isatilidenes also afforded the spiroheterocycles in moderate to good yields (3k, 3l). Notably, in the case of β-furyl substrate, the product 3l was formed in 56% yield and 1:1 dr. Disappointingly, β-alkyl substituted isatilidenes did not undergo the present homoenolate annulation reaction under the optimized reaction conditions.
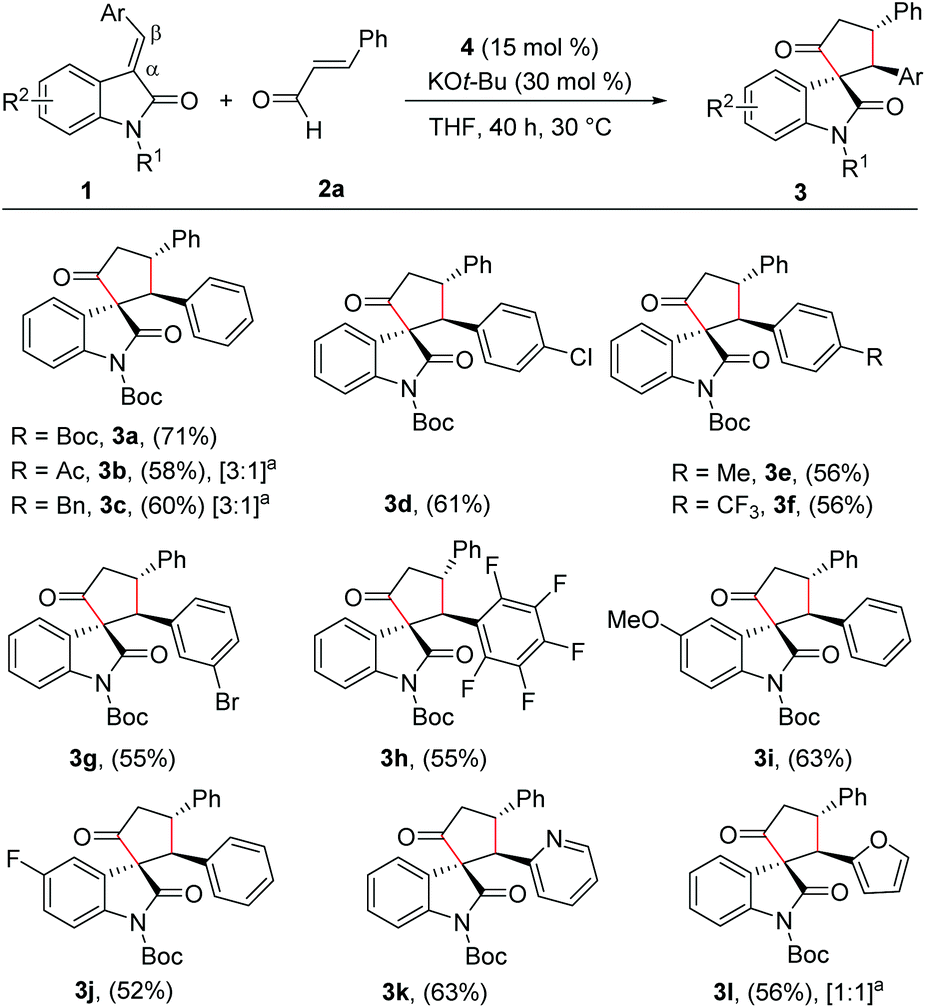 | ||
| Scheme 2 Substrate scope for the synthesis of cyclopentanone-fused spirooxindoles. Variation of isatilidenes. General reaction conditions: 1 (0.50 mmol), 2a (0.75 mmol), 4 (15 mol%), KOt-Bu (30 mol%), THF (2.0 mL) at 30 °C for 40 h. Isolated yields of products after flash column chromatography are provided and dr is >20:1 unless indicated. aThe diastereomeric ratio determined by 1H NMR of the crude reaction mixture. | ||
Next, we evaluated the scope of the reaction with various α,β-unsaturated aldehyde derivatives. Interestingly, electron-releasing and -withdrawing groups at the 4-position of the β-aryl ring are well-tolerated and the corresponding cyclopentanone-fused spiroheterocycles are formed in moderate yields (3m–3o). Moreover, 2-methoxy cinnamaldehyde afforded the desired product 3p in 61% yield. Additionally, β-furyl enal afforded the desired spiroheterocycle 3q in 51% yield. Gratifyingly, an alkyl substituent at the β-position of the enal was also tolerated and the target product 3r was isolated in 52% yield. In all cases, the cyclopentanone-fused spiroheterocycles were formed in high diastereoselectivity of >20:1 (Scheme 3).
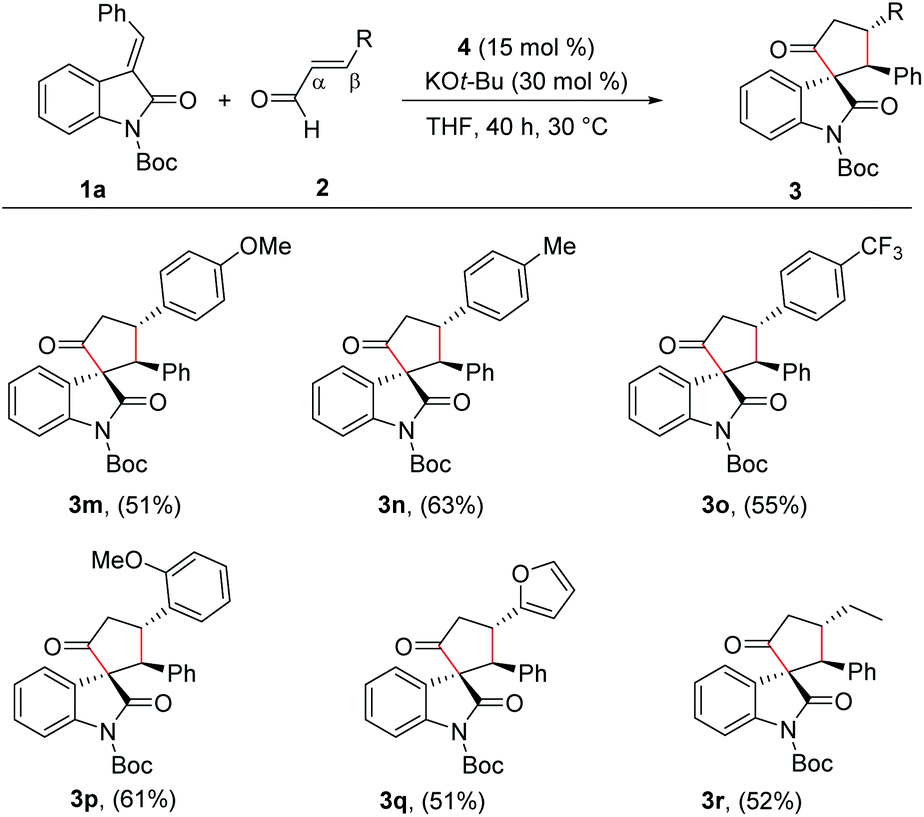 | ||
| Scheme 3 Variation of enals. General reaction conditions: 1a (0.50 mmol), 2 (0.75 mmol), 4 (15 mol%), KOt-Bu (30 mol%), THF (2.0 mL) at 30 °C for 40 h. Isolated yields of products are provided and dr is >20:1 in all cases. | ||
The tentative mechanism of this transformation is shown in Scheme 4. The free carbene generated from the imidazolium salt 4 undergoes nucleophilic addition to the enal followed by a proton transfer which allows the formation of the nucleophilic Breslow intermediate (A).21 This is in resonance with the homoenolate equivalent B. The selective conjugate addition of homoenolate equivalent to isatilidene 1 generates the enol intermediate C, which on tautomerization forms the acyl azolium intermediate D. An intramolecular C-acylation can result in the formation of the spirocyclic compound 3 regenerating the free carbene.
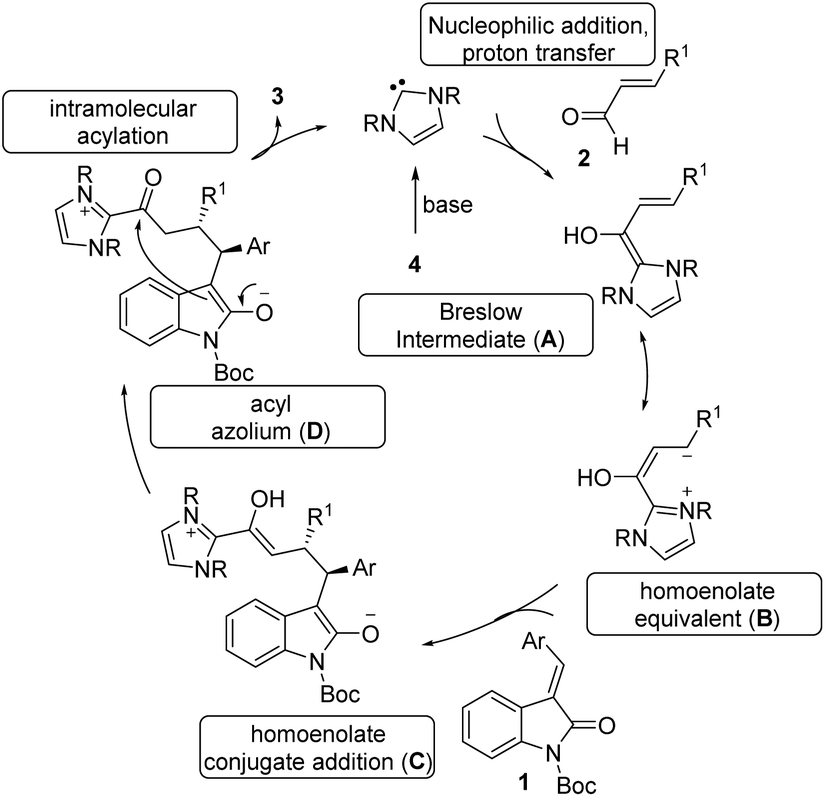 | ||
| Scheme 4 Proposed mechanism of the reaction. | ||
We also carried out functionalization of the cyclopentanone-fused spirooxindole 3a. Treatment of 3a with hydroxylamine hydrochloride under basic conditions afforded the corresponding spirooxindole oxime 9a in 64% yield and in an E:Z ratio of 3:2 (Scheme 5). The product 9a could be a substrate for the Beckmann rearrangement leading to the spiro δ-lactams. Moreover, N-Boc deprotection under trifluoroacetic acid (TFA) conditions furnished N-unprotected spirooxindole derivative 10a in 81% yield. Selective reduction of the keto group in 10a using NaBH4 resulted in the formation of the cyclopentanol-fused spirooxindole 11a in 77% yield and a moderate dr of 6:1.
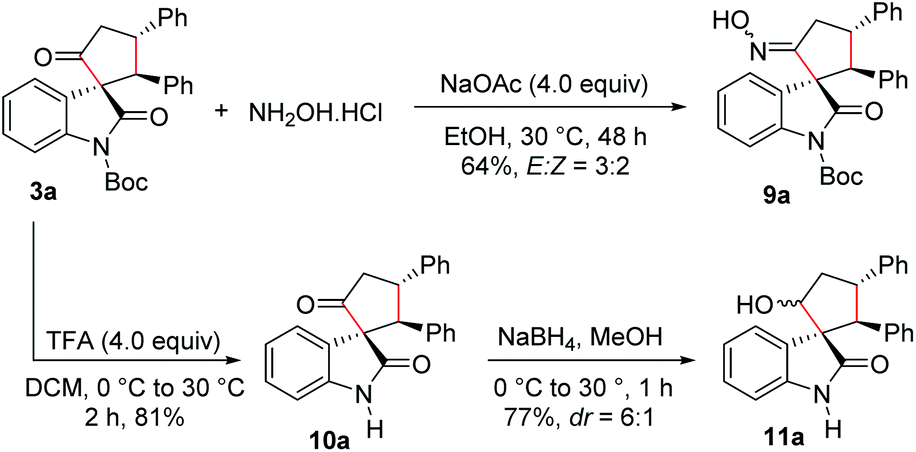 | ||
| Scheme 5 Functionalization of cyclopentanone-fused spirooxindoles. | ||
Furthermore, we performed experiments on the enantioselective version of this reaction.22 Reaction of N-Boc isatilidene 1a with enal 2a in the presence NHC generated from the chiral amino indanol-derived triazolium salt 8 using KO-tBu as the base resulted in the enantioselective synthesis of the cyclopentanone-fused spirooxindole chiral-3a in 22% yield, and excellent diastereoselectivity of >20:1 and in 97% ee (Scheme 6). Although the yield of chiral-3a is low, the high diastereoselectivity and enantioselectivity observed in this reaction is noteworthy. Notably, when the reaction of 1a was performed with 2-methoxy cinnamaldehyde 2p under the present reaction conditions, the desired product chiral-3p was isolated in 24% yield and in high diastereoselectivity of >20:1, but the ee value dropped to 54%.
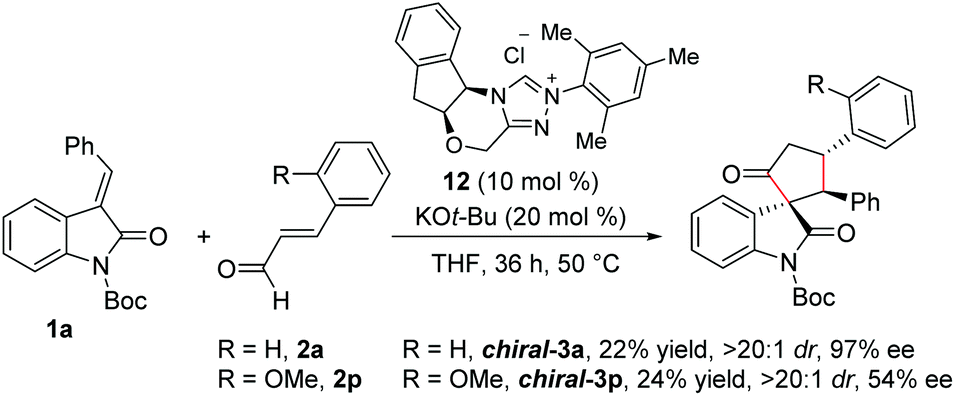 | ||
| Scheme 6 Enantioselective synthesis of cyclopentanone-fused spirooxindoles. | ||
Conclusion
In conclusion, we have developed the NHC-catalyzed reaction of α,β-unsaturated aldehydes with isatilidene derivatives resulting in the diastereoselective synthesis of cyclopentanone-fused spirooxindoles bearing an all-carbon quaternary spiro-stereogenic center. The reaction proceeds via the generation of homoenolate intermediates, which underwent a formal [3 + 2] annulation reaction to afford the desired products. In view of the interesting biological properties of spirooxindoles, and their ubiquity in various natural products and medicinally important molecules, it is anticipated that the cyclopentanone-fused spirooxindoles synthesized herein may have potential biological properties.Experimental section
Procedure for the synthesis of 3a
To a flame-dried screw-capped test tube equipped with a magnetic stir bar were added the imidazolium salt 4 (0.027 g, 0.075 mmol) and the (E)-3-benzylidene-2-oxoindoline-1-carboxylate 1a (0.160 g, 0.5 mmol). Then the screw-capped tube was evacuated and backfilled with argon. To this mixture was added THF (2.0 mL) under an argon atmosphere. The resultant reaction mixture was kept stirring at 30 °C. To this mixture was added trans cinnamaldehyde 2a (0.099 g, 94 μL, 0.75 mmol) followed by KOt-Bu (0.017 gm, 0.15 mmol). Then the reaction mixture was stirred at 30 °C for 40 h. After 40 h, the solvent was evaporated and the crude residue was purified by flash column chromatography to afford tert-butyl-2′,5-dioxo-2,3-diphenyl spiro[cyclopentane-1,3′-indoline]-1′-carboxylate 3a as a white solid (0.161 g, 71% yield).R f (Pet. ether/EtOAc = 80/20): 0.61; 1H NMR (400 MHz, CDCl3)δ 7.66 (d, J = 8.1 Hz, 1H), 7.35–7.25 (m, 7H), 7.20–7.16 (m, 1H), 7.09–7.02 (m, 5H), 4.89–4.81 (m, 1H), 4.01 (d, J = 12.3 Hz, 1H), 3.45–3.38 (m, 1H), 2.84–2.77 (m, 1H), 1.52 (s, 9H). 13C NMR (100 MHz, CDCl3)δ 209.27, 170.84, 148.42, 141.00, 140.58, 134.27, 129.42, 128.93, 128.37, 127.81, 127.54, 127.18, 126.76, 125.06, 123.21, 115.30, 84.50, 70.97, 60.67, 47.66, 41.06, 28.09. HRMS calculated [M + Na]+ for C29H27O4NNa: 476.1832, found: 476.1833. FTIR (cm−1) 3023, 2403, 1742, 1661, 1607, 1482, 1354, 1258, 1216, 1150, 1092, 1026, 928, 842, 767, 670.
Acknowledgements
Generous financial support by CSIR-New Delhi (as part of 12th Five-Year plan program under ORIGIN-CSC0108) is greatly acknowledged. A. P and A. B. thank CSIR-New Delhi for the senior research fellowship. We thank Dr P. R. Rajamohanan for the excellent NMR support and Mrs B. Santhakumari for the HRMS data.Notes and references
- For recent reviews on NHCs in organocatalysis, see: (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (c) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696 CrossRef CAS PubMed; (d) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem. – Eur. J., 2013, 19, 4664 CrossRef CAS PubMed; (e) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC; (f) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS PubMed; (g) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 351 RSC; (h) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686 CrossRef CAS PubMed; (i) D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53 RSC; (j) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617 CrossRef CAS PubMed; (k) C. E. I. Knappke, A. Imami and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 937 CrossRef CAS PubMed; (l) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed.
- C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS PubMed.
- S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS PubMed.
- For reviews on NHC-homoenolate chemistry, see: (a) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040 RSC; (b) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336 RSC; (c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC.
- (a) V. Nair, S. Vellalath, M. Poonoth, R. Mohan and E. Suresh, Org. Lett., 2006, 8, 507 CrossRef CAS PubMed. For a related annulation of homoenolates with benzofuran 2,3-diones, see: (b) K. C. Seetha Lakshmi, R. R. Paul, E. Suresh and V. Nair, Synlett, 2014, 25, 853 CrossRef CAS. For a report on homoenolate annulation with chalcones, see: (c) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, J. Am. Chem. Soc., 2006, 128, 8736 CrossRef CAS PubMed.
- L. H. Sun, L. T. Shen and S. Ye, Chem. Commun., 2011, 47, 10136 RSC.
- J. Dugal-Tessier, E. A. O'Bryan, T. B. H. Schroeder, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 4963 CrossRef CAS PubMed.
- H. Lv, B. Tiwari, J. Mo, C. Xing and Y. R. Chi, Org. Lett., 2012, 14, 5412 CrossRef CAS PubMed.
- (a) V. Nair, B. P. Babu, S. Vellalath and E. Suresh, Chem. Commun., 2008, 747 RSC. For the enantioselective version of this reaction using chiral NHCs, see: (b) J. R. Struble, J. Kaeobamrung and J. W. Bode, Org. Lett., 2008, 10, 957 CrossRef CAS PubMed.
- C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232 CrossRef CAS PubMed.
- M. Wang, Z.-Q. Rong and Y. Zhao, Chem. Commun., 2014, 50, 15309 RSC.
- K. C. S. Lakshmi, J. Krishnan, C. R. Sinu, S. Varughese and V. Nair, Org. Lett., 2014, 16, 6374 CrossRef PubMed.
- H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS PubMed.
- (a) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS PubMed. For a related NHC-catalyzed formal [4 + 3] annulation of enals with in situ generated azoalkenes, see: (b) C. Guo, B. Sahoo, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17402 CrossRef CAS PubMed.
- (a) S. Mukherjee, S. Mondal, A. Patra, R. G. Gonnade and A. T. Biju, Chem. Commun., 2015, 51, 9559 RSC; (b) A. Bhunia, A. Patra, V. G. Puranik and A. T. Biju, Org. Lett., 2013, 15, 1756 CrossRef CAS PubMed. For related reports on NHC-catalysis from our group, see: (c) S. R. Yetra, S. Mondal, E. Suresh and A. T. Biju, Org. Lett., 2015, 17, 1417 CrossRef CAS PubMed; (d) S. Mondal, S. R. Yetra, A. Patra, S. S. Kunte, R. G. Gonnade and A. T. Biju, Chem. Commun., 2014, 50, 14539 RSC; (e) S. R. Yetra, T. Roy, A. Bhunia, D. Porwal and A. T. Biju, J. Org. Chem., 2014, 79, 4245 CrossRef CAS PubMed; (f) S. R. Yetra, T. Kaicharla, S. S. Kunte, R. G. Gonnade and A. T. Biju, Org. Lett., 2013, 15, 5202 CrossRef CAS PubMed; (g) S. R. Yetra, A. Bhunia, A. Patra, M. V. Mane, K. Vanka and A. T. Biju, Adv. Synth. Catal., 2013, 355, 1089 CrossRef CAS PubMed.
- For reviews on synthesis of quaternary carbon stereocentres, see: (a) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed; (b) J. Christoffers and A. Mann, Angwe. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS; (c) K. Fuji, Chem. Rev., 1993, 93, 2037 CrossRef CAS; (d) B. M. Trost and C. Jiang, Synthesis, 2006, 369 CrossRef CAS.
- For the synthesis of cyclopentanone-fused spirooxindoles using phosphine catalysis, see: D. B. Ramachary, C. Venkaiah and P. M. Krishna, Org. Lett., 2013, 15, 4714 CrossRef CAS PubMed.
- For reviews on spirooxindoles, see: (a) B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673 CrossRef CAS PubMed; (b) N. R. Ball Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC; (c) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed; (d) R. Dalpozzo, G. Bartolib and G. Bencivennib, Chem. Soc. Rev., 2012, 41, 7247 RSC.
- For details, see the ESI†.
- CCDC 1413690 (3a) contain the supplementary crystallographic data for this paper.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- For details on optimization studies towards the enantioselective version, see the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental procedure, characterization data of all compounds, and single crystal X-ray data of 3a. CCDC 1413690. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00242g |
| This journal is © the Partner Organisations 2015 |