
Donor–acceptor interaction-driven folding of linear naphthalene–glycol oligomers templated by a rigid bipyridinium rod†
Tian-Guang
Zhan‡
a,
Ben-Ye
Lu‡
a,
Feng
Lin
a,
Tian-You
Zhou
a,
Xin
Zhao
*a and
Zhan-Ting
Li
*ab
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xzhao@sioc.ac.cn
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: ztli@sioc.ac.cn
First published on 18th September 2015
Abstract
This paper reports the construction of folded and helical supramolecular structures through the self-assembly of a series of flexible linear donor oligomers induced by a rigid rod-like acceptor template. The donor oligomers were constructed by connecting two, four, six, and eight 1,5-dioxynaphthalene (DAN) units with polyether chains, respectively, and the acceptor template was generated by incorporating 4,4′-bipyridine moieties at the 3,6-positions of a pyridazine skeleton. 1H NMR, UV-vis, and fluorescence spectroscopy studies indicated that the formation of the folded and helical structures was mainly driven by the intermolecular donor–acceptor interactions between the electron-rich DAN units and the electron-deficient template, which was further supported by DFT calculations. It was also found that the strength of the interactions between the donor oligomers and the acceptor template remarkably increased with the elongation of the oligomers.
Introduction
Artificial molecules that adopt folded secondary structures, widely known as foldamers,1 represent a very rich and active field of research.2 Since the twists and folds of biomolecules explicitly underpin their functions, the task of imparting synthetic molecules with folded secondary structures should have enormous potential to fabricate functional materials with novel and emergent properties. Currently the most general strategy to construct a foldamer is to incorporate all bonding sites into a linear structure in a way that these bonding sites intramolecularly bind together to induce a folded or helical structure, which can be defined as internal induction. On the basis of this principle, a variety of helical or folded architectures have been established through the use of different non-covalent interactions such as hydrogen-bonding,3 coordination interaction,4 donor–acceptor interaction,5 and radical dimerization.6 On the other hand, a flexible linear structure could also evolve into a well-defined secondary structure through an external induction approach if a suitable template is provided. Such externally induced formation of secondary structures has already been well demonstrated by nature. A common example is the twisted growth of some lianas around tree trunks. However, artificial folded and helical architectures constructed through this approach are quite limited.7 It could be due to the challenge of designing templates which can efficiently induce the formation of well-defined secondary structures. For the foldamer−rod host–guest structures, only a few examples have been reported by Jiang and Huc et al. and Moore and co-workers, in which the foldamers were constructed by hydrogen-bonding and the solvophobic effect, respectively.8As one of the most important non-covalent interactions in the field of supramolecular chemistry, the donor–acceptor interaction, which occurs between π-electron-rich and π-electron-deficient aromatic systems, has been widely used in the design of various supramolecular architectures. Stoddart et al. have utilized the donor–acceptor interaction between DAN and viologen derivatives to induce the formation of interlocked supermolecules,9 oligomeric pseudorotaxanes,10 and three-dimensional frameworks.11 By using aromatic stacking between the π-electron-deficient diimide segments and the π-electron-rich pyrenyl units, Colquhoun and co-workers have established the motif of molecular tweezers12 and further used them for the recognition of sequence information13 and healable supramolecular polymers.14 In spite of these progress, using the donor–acceptor interaction as the main driven force to construct folded/helical structures is still a great challenge. Compared to hydrogen-bonding which has been widely used to construct foldamers, the donor–acceptor interaction exhibits weak bonding strength and less directivity. These weak points dramatically limit its application in the construction of well-defined secondary structures.
In this work, we demonstrate the induced formation of folded and helical structures from a series of flexible linear oligomers driven by the donor–acceptor interaction. The linear oligomers were constructed through connecting different numbers of electron-rich 5-dialkoxynaphthalene (DAN) units by oligoethyleneglycol (D1–D4, Scheme 1). In order to enhance inducing efficiency, a rigid rod-like tetracationic pyridazine-bridged diviologen (PBDV) was designed as a template, in which the two viologen units make PBDV a highly electron-deficient molecule. The complexes formed between PBDV and the donor oligomers D1–D4 have been studied by (1D and 2D) 1H NMR, UV-vis and fluorescence spectroscopy, as well as DFT calculations, which confirmed the formation of supramolecular donor–acceptor complexes in which the flexible linear donor oligomers twined around the rigid rod-like acceptor. Furthermore, an increased stability of the as-formed complexes with the increasing number of DAN units in the oligomers was also observed.
 | ||
| Scheme 1 Structures of PBDV and oligomers D1–D4. | ||
Results and discussion
The synthetic route to PBDV is illustrated in Scheme 2. Commercially available reactants 3,6-dichloropyridazine and 4,4′-bipyridine were sealed in a Shrek tube without a solvent and then heated to 120 °C for 2 days under an argon atmosphere. The resulting dicationic intermediate 1 was obtained as a chloride salt, which was further treated with iodomethane in anhydrous CH3CN to yield the target compound PBDV in 80% yield after anion exchange with ammonium hexafluorophosphate.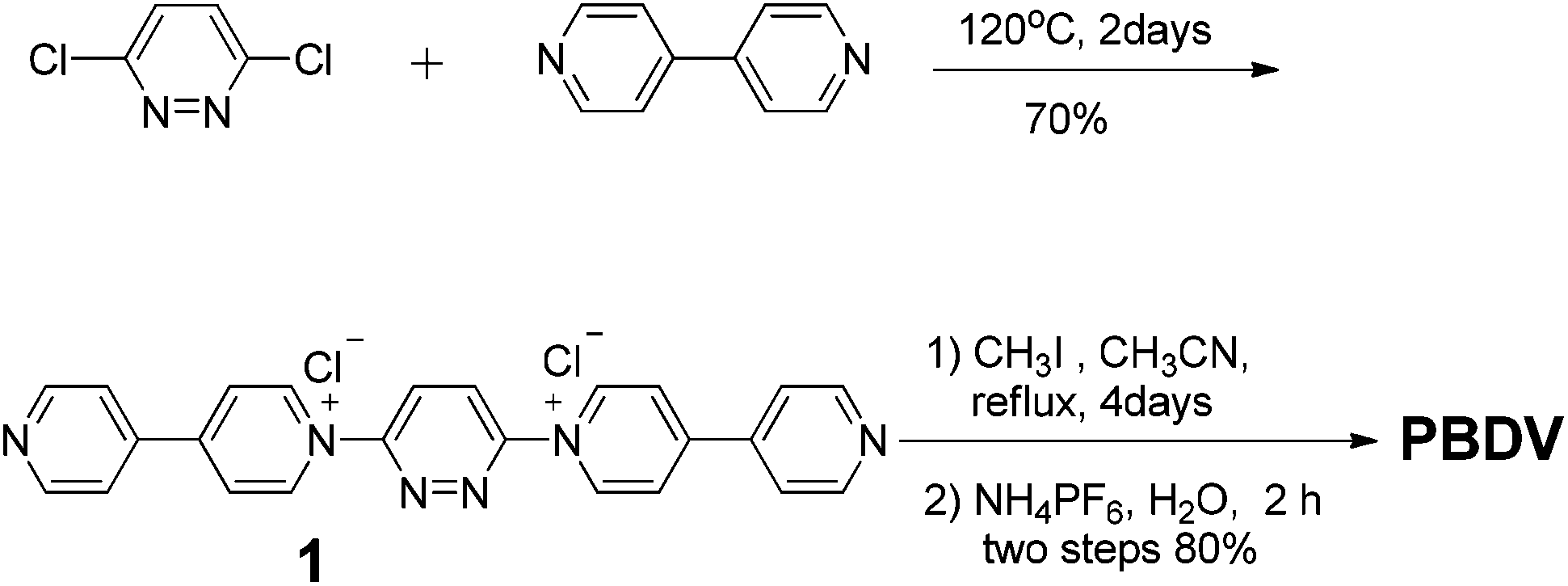 | ||
| Scheme 2 The synthetic route to PBDV. | ||
For the synthesis of donor oligomers D1–D4 (Scheme 3), their corresponding intermediates 3, 5 and 6 were firstly obtained in the yields of 37%, 40% and 65%, respectively, starting from 1,5-dioxynaphthalene and compound 4.
 | ||
| Scheme 3 The synthetic routes to donor oligomers D1–D4. | ||
Refluxing a mixture of 3 and 4 in the presence of K2CO3 afforded D1 in 10% yield while compound 7 was obtained as the major product. Compound 7 was further converted to 8 upon the treatment of 2. Similarly, intermediates 9 and 10 were obtained by the reactions of 6 with 3 and 8, respectively. With these intermediates in hand, oligomers D2–D4 were prepared through refluxing compound 5 with 7, 9, and 10, respectively. All the compounds have been fully characterized by 1H and 13C NMR, and (HR) mass spectroscopy, on the basis of which their chemical structures were unambiguously assigned (see the ESI† for the data).
The interactions between PBDV and D1–D4 were firstly investigated by 1H NMR spectroscopy. Since D4 displayed a poor solubility in CD3CN, a mixture of CD3CN and CDCl3 (v/v, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used as the solvent. Upon mixing PBDV and different DAN oligomers, the signals of the aromatic protons (except Hc) of the viologen unit and the methyl protons (Hf) in PBDV were shifted upfield (Fig. 1a and Fig. S1–S4, ESI†). The changes of the chemical shifts of the protons are summarized in Fig. 1b. It revealed that the chemical shift changes become significant when the number of DAN units increased in the oligomers, indicating an enhancement of donor–acceptor interactions between the PBDV and DAN upon the elongation of the donor molecules. This result indicated the formation of a complex driven by the multiple donor–acceptor interactions between PBDV and DAN. Similar trends for the changes of chemical shifts of the protons were also observed when the DAN unit of the oligomers was kept at the same concentration (Fig. S5, ESI†), indicating again that the more DAN units were involved, the stronger the donor–acceptor interaction generated.
:1) was used as the solvent. Upon mixing PBDV and different DAN oligomers, the signals of the aromatic protons (except Hc) of the viologen unit and the methyl protons (Hf) in PBDV were shifted upfield (Fig. 1a and Fig. S1–S4, ESI†). The changes of the chemical shifts of the protons are summarized in Fig. 1b. It revealed that the chemical shift changes become significant when the number of DAN units increased in the oligomers, indicating an enhancement of donor–acceptor interactions between the PBDV and DAN upon the elongation of the donor molecules. This result indicated the formation of a complex driven by the multiple donor–acceptor interactions between PBDV and DAN. Similar trends for the changes of chemical shifts of the protons were also observed when the DAN unit of the oligomers was kept at the same concentration (Fig. S5, ESI†), indicating again that the more DAN units were involved, the stronger the donor–acceptor interaction generated.
 | ||
| Fig. 1 (a) Partial 1H NMR spectra of PBDV and a 1:1 mixture of PBDV and D4 in CD3CN/CDCl3 (4:1), and (b) chemical shift changes of the protons in PBDV after it being mixed with equivalent D1–D4 in a binary solvent of CD3CN–CDCl3 (v/v, 4:1) at 20 °C. The concentration of PBDV was 3.0 mM. | ||
It is well known that a charge–transfer (CT) complex could be formed between the electro-deficient viologen unit and electro-rich species such as DAN, and it could be identified by the characteristic CT absorption band usually appearing in the visible range of a UV-vis spectrum.15 UV-vis spectroscopy was thus used to confirm the formation of donor–acceptor complexes. As can be seen in Fig. 2, while PBDV and D1–D4 only exhibit absorptions below 450 nm, new absorption peaks centred at 570 nm could be observed for their mixtures. These broad peaks could be rationally assigned to the CT absorptions originating from the charge transfer between PBDV and DAN. Their absorption intensity increased as the number of DAN units in the oligomers increased, indicating that the donor–acceptor interaction between them was enhanced by the multiple donor–acceptor interaction sites as well as synergistic effect. Such a trend could also be deduced by the observation of the color change upon mixing PBDV and donor oligomers, which varied from light yellow (with D1) to deep purple (with D4) with the increase in the number of DAN units in the oligomers (Fig. S6, ESI†).
 | ||
| Fig. 2 UV-vis spectra of solutions of PBDV with D1–D4 in CH3CN/CHCl3 (4:1) at 20 °C. The concentration of PBDV was 3.0 mM and the molar ratio for PBDV and the oligomers was 1:1. | ||
In order to get more insight into the supramolecular donor–acceptor complexes formed in solution, fluorescence experiments were further performed. First of all, the fluorescence emission intensities of oligomers D1–D4 were recorded at different concentrations, respectively (Fig. S7, ESI†). Their plots of intensity–concentration displayed a linear relationship, suggesting that the intermolecular interactions between the DAN units in the oligomers were negligible. In the next step fluorescence titration experiments of D1–D4 with PBDV were carried out. Upon incremental introduction of PBDV into the solutions of the oligomers, the fluorescence emission of DAN was gradually quenched (Fig. 3a and S8 in ESI†). Job's plots were generated by using fluorescence spectroscopy, which indicated a stoichiometry of 1:1 for PBDV and the oligomers (Fig. 3b and S9†). The binding constants (Ka) between PBDV and the DAN oligomers were estimated from Benesi–Hildebrand (B–H) plots17 by fitting their UV-vis spectroscopy titration data (Fig. S10†). The results are provided in Table 1, which revealed that the Ka value of the complex formed between PBDV and D4 was about 54 times larger than that of the complex formed between PBDV and D1, indicating again that the stability of complexes increased with the increasing number of DAN units, as a result of the multivalent donor–acceptor interactions and the synergistic effect.
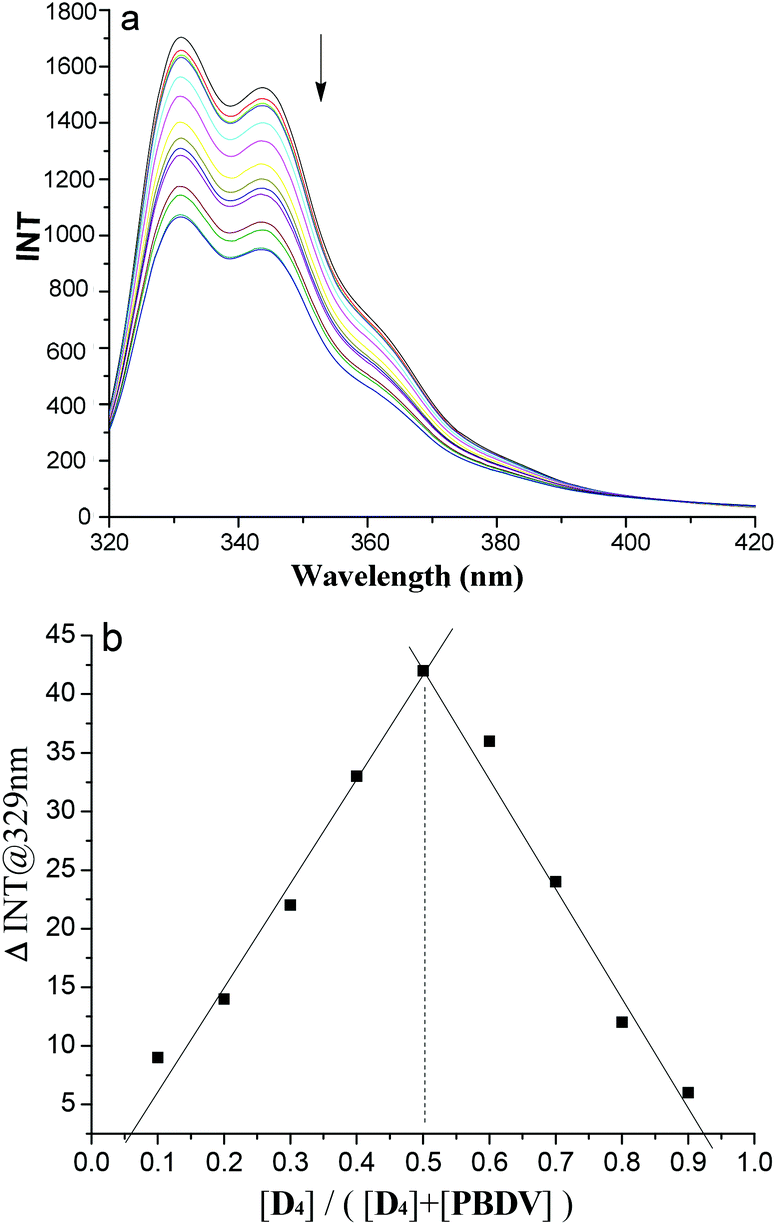 | ||
| Fig. 3 (a) Fluorescence titration spectra of D4 (1.0 × 10−8 M) with PBDV (0 to 5.0 × 10−8 M) in CH3CN at 25 °C. Excited at 290 nm. (b) The Job's plot of PBDV with D4 generated by using fluorescence emission. | ||
| PBDV + D1 | PBDV + D2 | PBDV + D3 | PBDV + D4 | |
|---|---|---|---|---|
| Calculated from B–H plots by fitting the UV-vis titration data of DAN oligomers with PBDV in CH3CN/CHCl3 (4:1) at 25 °C. |
||||
| K a/M−1 | (7.13 ± 0.06) × 102 | (5.16 ± 0.07) × 103 | (1.55 ± 0.03) × 104 | (3.48 ± 0.08) × 104 |
On the basis of the above results, the formation of stable complexes from PBDV and DAN oligomers D1–D4 in 1:1 stoichiometry driven by donor–acceptor interactions between PBDV and DAN, could be confirmed. We anticipated that the flexible DAN oligomers adopted folded or helical conformations by wrapping on the rigid rod-like PBDV. In order to get the exact structures of the supramolecular complexes, we tried to grow their single crystals suitable for X-ray crystallographic analysis. However, all the attempts to grow single crystals were unsuccessful. As an alternative, DFT calculations at the B3LYP/6-31G level for optimized structures were conducted to gain insight into their structural features. To obtain the energy minimum folded/helical conformations, Monte Carlo conformational searches were firstly carried out to explore possible folded/helical structures which could be generated by PBDV and each DAN oligomers. For each case, a total of 20000 structures were accumulated and the ten lowest-energy conformers were further optimized by DFT calculations with the Gaussian 09 package at the B3LYP/6-31G level without consideration of solvation. In this way, several energetically minimized representative structures were obtained for each complex (Fig. S10–13, ESI†). Among these structures, the energetically lowest ones are illustrated in Fig. 4 from different views. For the complex formed from PBDV and D1, the energetically lowest optimized structure was the one in which two DAN units acted as a molecular tweezer to clip the pyridazine ring of PDBV (Fig. 4a). In the case of PBDV and D2, the whole donor oligomer twined around the backbone of PBDV to adopt a helical conformation, in which all the four DAN units stacked on the aromatic ring of PBDV in a face-to-face manner to form a helical structure.
 | ||
| Fig. 4 The optimized energetically lowest structures of the complexes formed between PBDV and (a) D1, (b) D2, (c) D3, and (d) D4 which were generated by DFT calculations at the B3LYP/6-31G level. | ||
In the case of PBDV and D3, as the donor oligomer extended to include six DAN units, its flexible chain was induced by the rigid linear backbone of PBDV to wrap around PBDV, driven by the strong stacking between all the six DAN units and the aromatic ring of PBDV (Fig. 4c). However, the complex formed from PBDV and D4 displayed some differences from the above ones. As revealed by the simulations (Fig. 4d), although a helical structure of the donor oligomer still generated under the induction of PBDV, the arrangement between PBDV and DAN units is different from the above complexes because the space on the both sides of the aromatic ring of PBDV was too congested to accommodate the eight DAN units of D4 for the stacking between them. Alternatively, some of the DAN units were sticking to the side of PBDV to form C–H⋯π interactions. In all the cases, it should be noted that the C–H⋯O hydrogen bonding between the hydrogen atoms of the pyridinium moieties of PBDV and the oxygen atoms of oligo-oxyethylene linkers in D1–D4 may act as a second force to stabilize the helical folding supramolecular structures.18
The formation of folded and helical structures of D1–D4 induced by PBDV was further supported by 2D 1H NMR NOESY experiments. The NOESY spectrum of the 1:1 mixture of PBDV and D4 is provided as a representative (Fig. 5). As revealed in Fig. 5, significant intermolecular NOE connections (in the dashed box region) were observed between all the aromatic protons of PBDV and protons of DAN units of D4, clearly indicating that they approached each other at a very close distance, which is consistent with the helical structure as illustrated in Fig. 4d. The NOESY spectra of the other complexes (Fig. S15–S17, ESI†) displayed similar through-space correlations, also suggesting the formation of the wrapped structures between PBDV and D1–D3.
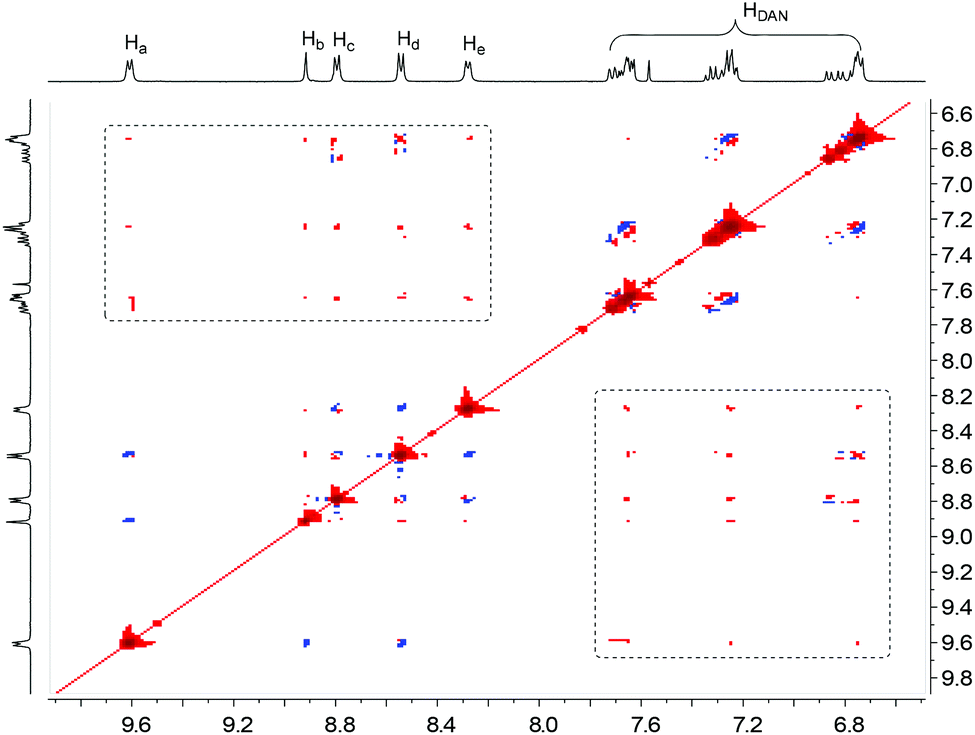 | ||
| Fig. 5 Partial 2D NOESY spectrum of PBDV + D4 in CD3CN/CDCl3 (4:1, 3.0 mM). | ||
Conclusions
In summary, a rigid rod-like electron-deficient acceptor molecule and a series of electro-rich flexible linear donor oligomers have been designed and synthesized. Upon mixing the acceptor and each of the donors, the flexible linear oligomers could be induced by the acceptor to adopt folded or helical conformations through twining round the skeleton of the rod-like acceptor in 1:1 stoichiometry, driven by the donor–acceptor interaction. While currently most foldamers are constructed by internal induction of the folded structure through intramolecular interactions, this work demonstrates that the construction of folded/helical architectures by means of the induction of external templates is also an efficient approach. Although in the present work the folded/helical structures of the donor oligomers will collapse once the template is removed, this issue could be addressed by designing linear oligomers that can be post-cross-linked within the as-formed secondary structures. In such a way, the hierarchy of the structures can be retained even the template is removed. In this context, the externally inducing approach should also be a very promising approach to fabricate complicated artificial secondary structures.
Acknowledgements
We thank the National Natural Science Foundation of China (No. 91127007 and 21172249) for financial support.Notes and references
- (a) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS; (b) W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399 CrossRef CAS PubMed; (c) Z.-T. Li, J.-L. Hou and C. Li, Acc. Chem. Res., 2008, 41, 1343 CrossRef CAS PubMed; (d) B. Gong, Acc. Chem. Res., 2008, 41, 1376 CrossRef CAS PubMed; (e) X. Zhao and Z.-T. Li, Chem. Commun., 2010, 46, 1601 RSC; (f) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS PubMed; (g) D.-W. Zhang, X. Zhao and Z.-T. Li, Acc. Chem. Res., 2014, 47, 1961 CrossRef CAS PubMed.
- (a) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS PubMed; (b) X. Li, Y.-D. Wu and D. Yang, Acc. Chem. Res., 2008, 41, 1428 CrossRef CAS PubMed; (c) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC; (d) K.-D. Zhang, X. Zhao, G.-T. Wang, Y. Liu, Y. Zhang, H.-J. Lu, X.-K. Jiang and Z.-T. Li, Angew. Chem., Int. Ed., 2011, 50, 9866 CrossRef CAS PubMed; (e) L.-Y. You, S.-G. Chen, X. Zhao, Y. Liu, W.-X. Lan, Y. Zhang, H.-J. Lu, C.-Y. Cao and Z.-T. Li, Angew. Chem., Int. Ed., 2012, 51, 1657 CrossRef CAS PubMed; (f) Z. Yu and S. Hecht, Angew. Chem., Int. Ed., 2013, 52, 13740 CrossRef CAS PubMed; (g) Z. Yu and S. Hecht, Angew. Chem., Int. Ed., 2011, 50, 1614 Search PubMed; (h) R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926 CrossRef CAS PubMed; (i) A. Khan, C. Kaiser and S. Hecht, Angew. Chem., Int. Ed., 2006, 45, 1878 CrossRef CAS PubMed.
- (a) J.-L. Hou, X.-B. Shao, G.-J. Cheng, Y.-X. Zhou, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2004, 126, 12386 CrossRef CAS PubMed; (b) A. Violette, M. C. Averlant-Petit, V. Semetey, C. Hemmerlin, R. Casimir, R. Graff, M. Marraud, J.-P. Briand, D. Rognan and G. Guichard, J. Am. Chem. Soc., 2005, 127, 2156 CrossRef CAS PubMed; (c) B. Gong, H. Zeng, J. Zhu, L. Yuan, Y. Han, S. Cheng, M. Furukawa, R. D. Parra, A. Y. Kovalevsky, J. L. Mills, E. Skrzypczak-Jankun, S. Martinovic, R. D. Smith, C. Zheng, T. Szyperski and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11583 CrossRef CAS PubMed; (d) H. Zhao, W. Q. Ong, F. Zhou, X. Fang, X. Chen, S. F. Y. Li, H. Su, N.-J. Cho and H. Zeng, Chem. Sci., 2012, 3, 2042 RSC.
- (a) A.-M. Stadler, N. Kyritsakas and J.-M. Lehn, Chem. Commun., 2004, 2024 RSC; (b) N. Delsuc, M. Hutin, V. E. Campbell, B. Kauffmann, J. R. Nitschke and I. Huc, Chem. – Eur. J., 2008, 14, 7140 CrossRef CAS PubMed.
- (a) A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038 CrossRef CAS PubMed; (b) R. S. Lokey and B. L. Iverson, Nature, 1995, 375, 303 CrossRef CAS PubMed; (c) S. Ghosh and S. Ramakrishnan, Angew. Chem., Int. Ed., 2004, 43, 3264 CrossRef CAS PubMed; (d) X. Zhao, M.-X. Jia, X.-K. Jiang, Z.-T. Li and G.-J. Cheng, J. Org. Chem., 2004, 69, 270 CrossRef CAS PubMed; (e) S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2009, 48, 7635 CrossRef CAS PubMed.
- (a) D.-W. Zhang, J. Tian, L. Chen, L. Zhang and Z.-T. Li, Chem. – Asian J., 2015, 10, 56 CrossRef CAS PubMed; (b) L. Chen, H. Wang, D.-W. Zhang, Y. Zhou and Z.-T. Li, Angew. Chem., Int. Ed., 2015, 54, 4028 CrossRef CAS PubMed; (c) Y. Wang, M. Frasconi, W.-G. Liu, Z. Liu, A. A. Sarjeant, M. S. Nassar, Y. Y. Botros, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 876 CrossRef CAS PubMed.
- (a) Y.-X. Xu, G.-T. Wang, X. Zhao, X.-K. Jiang and Z.-T. Li, J. Org. Chem., 2009, 74, 7267 CrossRef CAS PubMed; (b) Z.-M. Shi, S.-G. Chen, X. Zhao, X.-K. Jiang and Z.-T. Li, Org. Biomol. Chem., 2011, 9, 8122 RSC; (c) K.-J. Chang, B.-N. Kang, M.-H. Lee and K.-S. Jeong, J. Am. Chem. Soc., 2005, 127, 12214 CrossRef CAS PubMed; (d) J.-m. Suk and K.-S. Jeong, J. Am. Chem. Soc., 2008, 130, 11868 CrossRef CAS PubMed; (e) H. Juwarker and K.-S. Jeong, Chem. Soc. Rev., 2010, 39, 3664 RSC.
- (a) Q. Gang, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172 CrossRef PubMed; (b) T. Nishinaga, A. Tanatani, K. Oh and J. S. Moore, J. Am. Chem. Soc., 2002, 124, 5934 CrossRef CAS PubMed; (c) A. Tanatani, T. S. Hughes and J. S. Moore, Angew. Chem., Int. Ed., 2002, 41, 325 CrossRef CAS.
- C. J. Bruns and J. F. Stoddart, Adv. Polym. Sci., 2013, 261, 271 CrossRef CAS.
- (a) W. Zhang, W. R. Dichtel, A. Z. Stieg, D. Benítez, J. K. Gimzewski, J. R. Heath and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6514 CrossRef CAS PubMed; (b) S. Basu, A. Coskun, D. C. Friedman, M. A. Olson, D. Benítez, E. Tkatchouk, G. Barin, J. Yang, A. C. Fahrenbach, W. A. Goddard III and J. F. Stoddart, Chem. – Eur. J., 2011, 17, 2107 CrossRef CAS PubMed; (c) Z. Zhu, H. Li, Z. Liu, J. Lei, H. Zhang, Y. Y. Botros, C. L. Stern, A. A. Sarjeant, J. F. Stoddart and H. M. Colquhoun, Angew. Chem., Int. Ed., 2012, 51, 7231 CrossRef CAS PubMed; (d) C. M. Gothard, C. J. Bruns, N. A. Gothard, B. A. Grzybowski and J. F. Stoddart, Org. Lett., 2012, 14, 5066 CrossRef CAS PubMed; (e) Z. Zhu, C. J. Bruns, H. Li, J. Lei, C. Ke, Z. Liu, S. Shafaie, H. M. Colquhoun and J. F. Stoddart, Chem. Sci., 2013, 4, 1470 RSC.
- D. Cao, M. Juríček, Z. J. Brown, A. C.-H. Sue, Z. Liu, J. Lei, A. K. Blackburn, S. Grunder, A. A. Sarjeant, A. Coskun, C. Wang, O. K. Farha, J. T. Hupp and J. F. Stoddart, Chem. – Eur. J., 2013, 19, 8457 CrossRef CAS PubMed.
- H. M. Colquhoun, Z. Zhu and D. J. Williams, Org. Lett., 2003, 5, 4353 CrossRef CAS PubMed.
- (a) H. M. Colquhoun and Z. Zhu, Angew. Chem., Int. Ed., 2004, 43, 5040 CrossRef CAS PubMed; (b) H. M. Colquhoun, Z. Zhu, C. J. Cardin, Y. Gan and M. G. B. Drew, J. Am. Chem. Soc., 2007, 129, 16163 CrossRef CAS PubMed; (c) Z. Zhu, C. J. Cardin, Y. Gan and H. M. Colquhoun, Nat. Chem., 2010, 2, 653 CrossRef PubMed.
- (a) S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051 CrossRef CAS PubMed; (b) J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362 CrossRef CAS PubMed; (c) L. R. Hart, J. H. Hunter, N. A. Nguyen, J. L. Harries, B. W. Greenland, M. E. Mackay, H. M. Colquhoun and W. Hayes, Polym. Chem., 2014, 5, 3680 RSC.
- N. C. Yang and Y. Gaoni, J. Am. Chem. Soc., 1964, 86, 5022 CrossRef CAS.
- The binding constants were also estimated by fluorescence titration data. However, it was found that the values obtained from fluorescence titration experiments were 2–3 orders of magnitude larger than those obtained from UV-vis titration data. This discrepancy pushed us to verify the values by another approach. Since the CT-band represents only the complexes, concentration dependent variation of the CT-band was further used to estimate the binding constants. Although the changes of absorbance of the CT band of the complexes formed from PBDV and D1 and D2 were too small to generate accurate binding constants, in the cases of the complexes formed from PBDV and D3 and D4, the binding constants derived from CT absorption were quite close to those generated from UV absorption. It indicated that UV-vis titration was a more reliable technique than fluorescence titration for these complexes. This result suggests that fluorescence titration, especially that based on fluorescence quenching, should be used with caution to estimate binding constant, as fluorescence intensity may be sensitive to many other unavoidable factors.
- A. H. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef.
- F. M. Raymo, M. D. Bartberger, K. N. Houk and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 9264 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, additional 1H NMR, fluorescence spectra, Job's plots, and energy diagrams of the optimized structures. See DOI: 10.1039/c5qo00244c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2015 |