
Modulation of the assembly fashion among metal–organic frameworks for enantioretentive epoxide activation†
Jun
Guo
 *a,
Xiaomin
Xue
a,
Fangfang
Li
a,
Meiting
Zhao
b,
Youcong
Xing
a,
Yanmin
Song
c,
Chang
Long
d,
Tingting
Zhao
a,
Yi
Liu
*ae and
Zhiyong
Tang
*d
*a,
Xiaomin
Xue
a,
Fangfang
Li
a,
Meiting
Zhao
b,
Youcong
Xing
a,
Yanmin
Song
c,
Chang
Long
d,
Tingting
Zhao
a,
Yi
Liu
*ae and
Zhiyong
Tang
*d
aState Key Laboratory of Separation Membranes and Membrane Processes, School of Chemistry, Tiangong University, Tianjin 300387, China. E-mail: junguo@tiangong.edu.cn; yiliuchem@whu.edu.cn
bTianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, Institute of Molecular Aggregation Science, Tianjin University, Tianjin 300072, China
cCosychem Technology (Tianjin) Co., Ltd, Tianjin 300457, China
dCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: zytang@nanoctr.cn
eSchool of Chemical and Environmental Engineering, Wuhan Polytechnic University, Wuhan 430023, China
First published on 19th November 2023
Abstract
Highly enantioretentive alcoholysis of epoxides is an important way to synthesize enantiopure β-alkoxy alcohols, which are irreplaceable intermediates demanded by biomedicines, fine chemicals and other industries. In this report, we exploit a series of Zr-based metal–organic frameworks (Zr-MOFs) as the catalysts to achieve high activity and enantioretentivity in the alcoholysis of styrene oxide via modulating their assembly fashions. It is explored that hcp-UiO-66 not only exhibits a ∼10 fold improved catalytic activity than both hxl-CAU-26 and fcc-UiO-66 of varied assemblies but also maintains superior product enantioretentivity. Theoretic calculations together with experimental proof discloses the origin of distinct catalytic activity caused by different assembly fashions. This assembly modulation strategy offers a potential protocol for seeking high-performance catalysts among MOFs by virtue of their rich polymorphisms.
New conceptsAs an emerging type of heterogeneous catalyst, metal–organic frameworks (MOFs) provide multiple freedoms for regulating catalytic performances usually through metal node variations, linker functionalizations, pore structure adjustments and defect engineering. Absolutely different from the above strategies, we report the modulation of the assembly fashions of MOF-based catalysts to implement double excellence in catalytic activity and product selectivity. As demonstrated by the performances acquired in the enantioretentive alcoholysis of epoxides, hcp-UiO-66 with a hexagonal close-packed assembly fashion not only exhibits nearly 10-fold improved catalytic activity over the corresponding primitive hexagonal latticed hxl-CAU-26 and face-centered cubic fcc-UiO-66, but also holds the highest product ee values. The great performance disparity is disclosed to originate from the assembly-dependent amount and charge distribution of active sites among polymorphic MOFs. The assembly modulation strategy reported in this work enables additional possibilities for seeking more high-performance MOF- or COF-based porous catalysts with both high conversion efficiency and product selectivity. |
Introduction
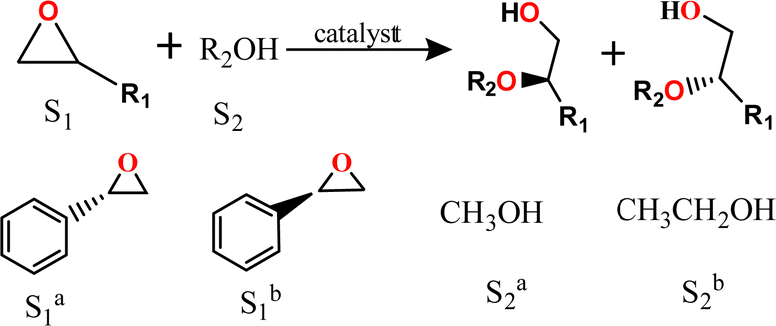
Chirality is one of the basic attributes of nature. Specifically, basic building blocks like amino acids, saccharides and ribonucleic acids in life are all enantiopure chiral substances and rigorously comply with a specific set of chirality selection rules when performing their biological functionalities.1–3 Thanks to vigorous advances of chiral science made in biomedicines,4 pesticides,5,6 fine chemicals,7 chiroptics,8 and so on,9 the increasing demand for various types of chiral substances in high enantiopurity has been an urgent task in chiral synthesis and asymmetric catalysis.10–13 For instance, the enantioretentive alcoholysis of epoxide substrates is an important way to synthesize chiral β-alkoxy alcohols with 1,2-disynthons introduced, which are multifunctional chiral intermediates irreplaceable in preparations of chiral polyurethane elastomers, anticancer eugenol β-amino alcohols and derivatives.14 However, owing to the poor nucleophilicity of alcohols, this reaction generally requires harsh conditions like high temperature, long reaction time and strong acid to get satisfactory conversions of epoxides.15,16 Unfortunately, the worrisome racemization of chiral products will be prone to occur under harsh reaction conditions, while additional enantiomer separations and purifications have to be conducted prior to further usage. As a result, the catalytic activation of epoxides in both high activity and enantioretentivity is highly expected in order to gain enantiopure target products as well as save posttreatment costs, but still represents a grand challenge.15–18As emerging crystalline porous materials, metal–organic frameworks (MOFs) have been widely recognized as efficient heterogeneous catalysts in versatile organic transformations, thanks to their abundant active sites, ordered pore structures, tuneable compositions and good thermal stabilities.19–23 Notably, MOF-based catalysts have enabled the regulation of many catalytic reactions via metal node variation,24,25 organic linker functionalization,26,27 pore size adjustment,28,29 and defect engineering.30,31 For example, by changing the metal node from Zr to Hf, Hf-NU-1000 [Hf6(OH)16(TBAPy)2, H4TBAPy = 1,3,6,8-tetrakis(p-benzoic acid)pyrene] showed higher conversion rate than the isoreticular Zr-NU-1000 in the catalytic activation of epoxides due to its weaker Lewis acidity.16 Moreover, defect engineering was recently proposed as a workable way for catalytic activity improvement.32,33 Prototypes such as MOF-808 [Zr6O4(CHOO)6(BTC)2, BTC = 1,3,5-benzenetricarboxylate] featuring abundant linker defects exhibited excellent conversion and regioselectivity towards the ring-opening reaction of styrene oxide;34 however, investigations about product enantioretentivity were rarely reported.
Beyond the aforementioned tricks in catalysis, in this work, we report the regulation of both catalytic activity and product enantioretentivity via modulating the assembly fashion among three typical Zr-MOFs. Those representatives are the primitive hexagonal latticed hxl-CAU-26 [Zr6O4(μ3-OH)4(BDC)3(CH3COO)6, BDC = 1,4-benzenedicarboxylate], hexagonal close-packed hcp-UiO-66 [Zr12O8(μ3-OH)8(μ2-OH)6(BDC)9], and face-centered cubic fcc-UiO-66 [Zr6O4(μ3-OH)4(BDC)6]. Note that the acetate modulator in hxl-CAU-26 only serves as a coordination auxiliary and does not involve the corresponding assembly fashion. Depending on the assembly fashions, those Zr-MOFs show distinct catalytic conversions as well as product enantioretentivities in the alcoholysis of epoxides.
Results and discussion
Three types of Zr-MOFs with varied assembly fashion are selected and constructed according to previous reports with minor modifications (the synthesis details are available in the ESI†).35–38 Among them, hxl-CAU-26 MOF is composed of a 6-coordinated Zr6O4(μ3-OH)4 node and BDC linker via adopting the hxl network within the crystallographic a–b plane (Fig. 1a). Along the c-direction, as-formed 2D lamellas are further assembled into a 3D framework via the eclipsed AA stacking fashion (Fig. 1b). For the sake of charge balance, there are six additional acetate modulators occupied at the bottom and top of each metal node separately, thereby giving the formula of [Zr6O4(μ3-OH)4(BDC)3(CH3COO)6] for hxl-CAU-26. The hcp-UiO-66 MOF consists of Zr6O4(μ3-OH)4 dimer [Zr12O8(μ3-OH)8(μ2-OH)6] bridged by six μ2-OH as the metal node (Fig. 1c), in which the 18-coordinated [Zr12O8(μ3-OH)8(μ2-OH)6] nodes linked by ditopical BDC linkers are assembled into a 3D hcp framework characteristic of the staggered AB stacking fashion (Fig. 1d). Moreover, the classical fcc-UiO-66 MOF is also constructed by connecting the 12-coordinated [Zr6O4(μ3-OH)4] metal node with BDC linkers, which is characteristic of a 3D fcc topology with the ABC assembly fashion (Fig. 1e and f).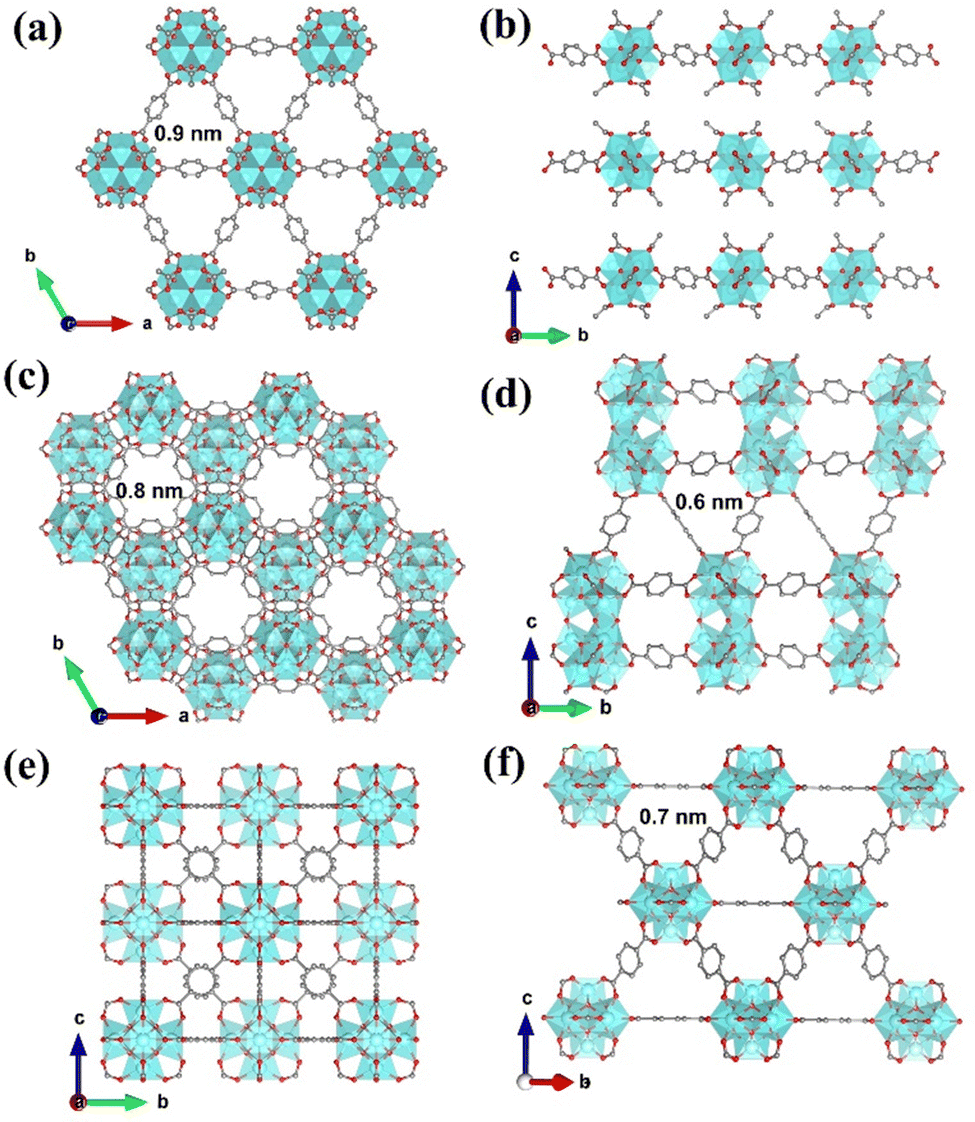 | ||
| Fig. 1 (a) Crystallographic structure of hxl-CAU-26 projected within the a–b plane. (b) Crystallographic structure of hxl-CAU-26 assembled along the c-direction. (c) Crystallographic structure of hcp-UiO-66 projected within the a–b plane. (d) Crystallographic structure of hcp-UiO-66 assembled along the b–c plane. (e) Crystallographic structure of fcc-UiO-66 projected within the a–b plane. (f) Crystallographic structure of fcc-UiO-66 assembled along the c-direction. | ||
According to the scanning electron microscope (SEM) image, one can see the successful construction of lamellar-shaped hxl-CAU-26 MOF with an averaged lateral size of 1.1 μm (Fig. 2a and Fig. S1a, ESI†). Its experimental powder X-ray diffraction (PXRD) pattern well correlates with the simulated one, with the first three diffraction peaks assigned to the (001), (100), and (110) facets in sequence (Fig. 2b). The hcp-UiO-66 MOF is also characteristic of the hexagonal plate morphology and shaped in a larger lateral size of 2.9 μm than hxl-CAU-26 (Fig. 2c and Fig. S1b, ESI†). In addition, the sharp diffraction peaks of hcp-UiO-66 also match with the simulation result, indicating its high crystallinity free of impurity phase (Fig. 2d). Differently, the fcc-UiO-66 MOF shows a characteristic (111)-exposed octahedral morphology with an averaged particle size of 5.8 μm (Fig. 2e and Fig. S1c, ESI†). Its high extinction PXRD pattern featured with the main (111) and (200) peaks further confirms the successful construction of the fcc-UiO-66 phase (Fig. 2f). Transmission electron microscopy (TEM) imaging and electron diffraction (ED) are further carried out on three types of MOFs, which also show consistent results with the corresponding structure simulations (Fig. S3–S5, ESI†).
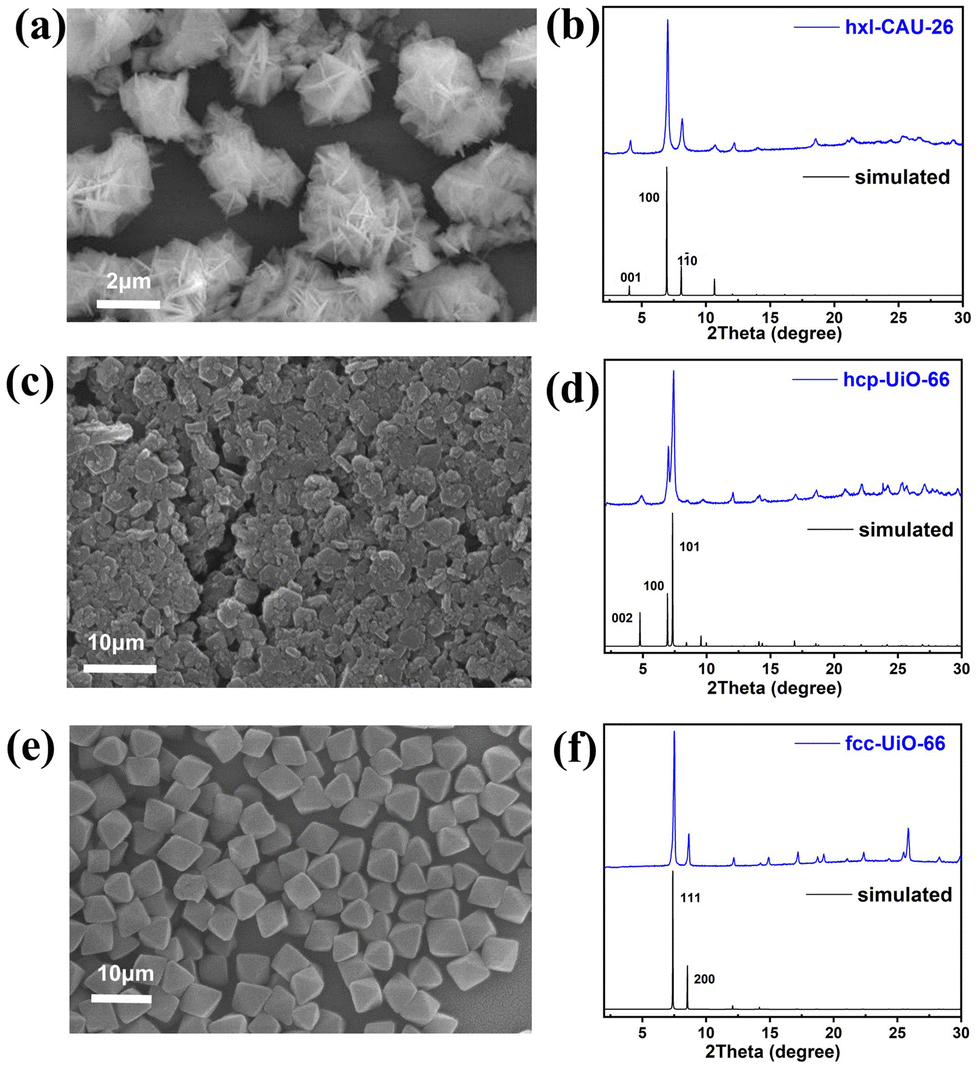 | ||
| Fig. 2 (a) SEM image and (b) PXRD pattern of hxl-CAU-26, (c) SEM image and (d) PXRD pattern of hcp-UiO-66, (e) SEM image and (f) PXRD pattern of fcc-UiO-66. | ||
Besides, Fourier transform infrared (FT-IR) spectra demonstrate the effective removal of residual H2BDC ligands inside the corresponding MOF pores (Fig. S2, ESI†), which are accessible for further evaluation.
The N2 adsorption and desorption of three types of Zr-MOFs (hxl-CAU-26, hcp-UiO-66, and fcc-UiO-66) are recorded at 77 K. As shown in Fig. S6 (ESI†), type I adsorption isotherms are discerned for hxl-CAU-26, hcp-UiO-66 and fcc-UiO-66 with the Brunauer–Emmett–Teller (BET) specific surface areas of 674.1 m2 g−1, 327.1 m2 g−1 and 876.5 m2 g−1, respectively. In addition to populations of crystallographic micropores (Fig. 1), pore size distribution (PSD) curves of hcp-UiO-66 and fcc-UiO-66 present 1.3–1.4 nm pore populations in Fig. 3a, indicating the existence of defective pores raised from missing linkers. Thermogravimetric analyses (TGA) also confirmed linker defects in both hcp-UiO-66 and fcc-UiO-66, according to their much lower BDC linker proportions than the theoretical ones (Fig. S7, ESI†). Moreover, the quantitative defect number of hcp-UiO-66 and fcc-UiO-66 is estimated to be 8.6 per [Zr12O8(μ3-OH)8(μ2-OH)6] node and 5.4 per [Zr6O4(μ3-OH)4] node, respectively (Table S1, ESI†). As a comparison, hxl-CAU-26 presents an inherent 6 linker defects per node based on its crystallographic formula of Zr6O4(BDC)3(CH3COO)6, as verified by the coincident BDC loss between experimental and theoretical ones (Table S1, ESI†). The above defect evaluations among MOFs are further validated by electron paramagnetic resonance (EPR) spectroscopy. As shown in Fig. 3b, the three types of Zr-MOFs all display strong isotropic EPR signals with the same g-factor of 2.003, a characteristic indicator of defect-induced oxygen vacancies.39 In line with the TGA results, the EPR signal of hcp-UiO-66 gives a spin count up to 6.41 × 1017 spins g−1, which is considerably higher than that of both hxl-CAU-26 (3.53 × 1017 spins g−1) and fcc-UiO-66 (3.23 × 1017 spins g−1).
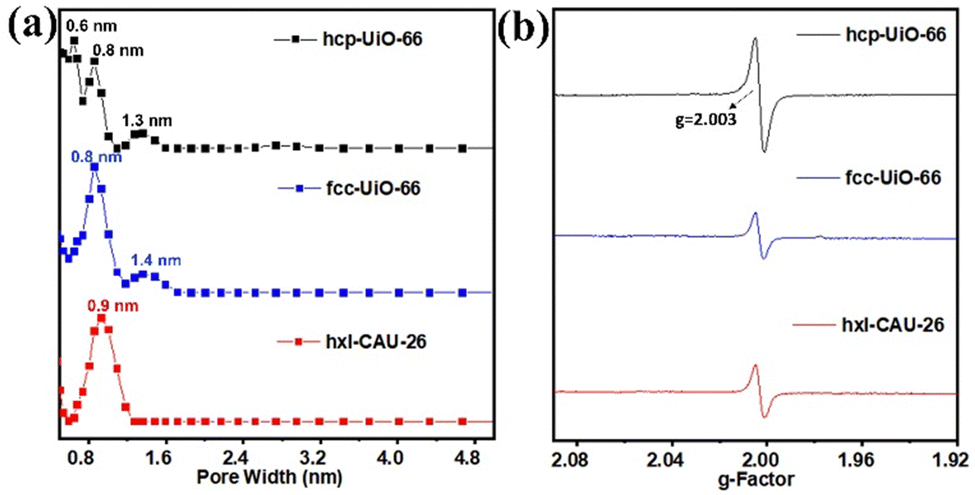 | ||
| Fig. 3 (a) Pore size distribution of hcp-UiO-66, fcc-UiO-66 and hxl-CAU-26 according to the N2-DFT method. (b) EPR spectra of hcp-UiO-66, fcc-UiO-66 and hxl-CAU-26. | ||
Bearing defect-induced metal sites, the above Zr-MOFs with varied assembly fashion are tested as Lewis acid catalysts in the alcoholysis of epoxides. As outlined in Table 1, hcp-UiO-66 MOF achieved nearly 100% conversion in the methanolysis of (S)-styrene oxide at 30 °C and within only 4 h (Entry 1). Benefitting from the very mild reaction temperature and short reaction time, 96.2% enantiomeric excess (ee) of 2-methoxy-2-phenylethan-1-ol product is maintained in comparison to the 97% ee of the initial substrate.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Sub.1 | Sub.2 | Catalyst | T/°C | t/h | Con./% | ee/% |
| The reaction conditions of enantioretentive alcoholysis: 2 mg catalyst, 28.5 μL of substrate 1 and 5 mL of substrate 2 were mixed into a 15 mL glass flask to react at a certain temperature for a certain time. | |||||||
| 1 | S1a | S2a | hcp | 30 | 4 | 99.9 | 96.2 |
| 2 | S1a | S2a | fcc | 80 | 48 | 93.6 | 91.3 |
| 3 | S1a | S2a | hxl | 80 | 48 | 96.4 | 93.5 |
| 4 | S1b | S2a | hcp | 30 | 4 | 94.0 | 95.8 |
| 5 | S1b | S2a | fcc | 80 | 48 | 91.8 | 84.8 |
| 6 | S1b | S2a | hxl | 80 | 48 | 92.5 | 87.8 |
| 7 | S1a | S2b | hcp | 60 | 48 | 97.0 | 94.8 |
| 8 | S1a | S2b | fcc | 100 | 72 | 84.6 | 91.1 |
| 9 | S1a | S2b | hxl | 100 | 72 | 93.3 | 91.9 |
| 10 | S1b | S2b | hcp | 60 | 48 | 96.3 | 96.1 |
| 11 | S1b | S2b | fcc | 100 | 72 | 90.3 | 85.1 |
| 12 | S1b | S2b | hxl | 100 | 72 | 91.1 | 86.8 |
In sharp contrast, fcc-UiO-66 and hxl-CAU-26 present only ∼10% conversions and the homogeneous ZrCl4 fails to show any catalytic activity under the otherwise identical reaction conditions (Table S2, ESI†). Accordingly, harsher reaction conditions (80 °C for 48 h) are required to get high substrate conversions (entries 2 and 3). Unfortunately, the product ee values consequently decrease to 91.3% and 93.5% for fcc-UiO-66 and hxl-CAU-26, respectively, caused by accelerated racemizations under harsh conditions. Similarly, the performances taken by using (R)-styrene oxide as the substrate also give inferior ee values for fcc-UiO-66 and hxl-CAU-26 compared with that of hcp-UiO-66 (entries 4–6). To further demonstrate both superior conversion activity and enantioretentivity of hcp-UiO-66, ethanol of weaker nucleophilicity is used in replacement of methanol for the addition of styrene oxide (entries 7–12). One can see that hcp-UiO-66 gives conversion over 95% and product ee of 94.8% at 60 °C for 48 h. The comparable substrate conversions are gained by fcc-UiO-66 and hxl-CAU-26 at raised temperature (100 °C) and prolonged reaction time (72 h), but at the cost of reduced product ee values.
The best performances achieved by hcp-UiO-66 are intuitively associated most with its highest defect amount among the three types of Zr-MOFs, as disclosed by both TGA and EPR characterizations (Fig. 3b and Table S1, ESI†). Similarly, the superior performance of hxl-CAU-26 over fcc-UiO-66 is also ascribed to the higher defect amount in the former. Besides, the inherent catalytic activity (i.e. Lewis acidity) of each metal site may also vary among Zr-MOFs of varied assembly fashion. Hence, the N-methylacridone (NMA)-based fluorescent probe is used to distinguish the Lewis acidity disparity of metal sites among MOFs.34,40–42 Initially, the free NMA probe shows a blue emission in CH3CN with the maximum peak wavelength (λmax) centered at 433.0 nm (Fig. S13, ESI†). Upon binding to metal sites among MOFs, the emission of NMA undergoes a redshift with a positive relationship to the binding energy (ΔE) between NMA and MOFs. As shown in Fig. 4a, the initial λmax changes significantly from 433.0 nm to 455.2 nm, 457.4 nm and 463.8 nm after binding to fcc-UiO-66, hxl-CAU-26 and hcp-UiO-66, respectively. The largest ΔE of 0.836 eV is therefore obtained for hcp-UiO-66, meaning the highest catalytic activity of the corresponding metal site. Furthermore, we calculate the turnover frequency based on each defect site (TOF per defect) for three Zr-MOFs of varied assembly fashions (Table S2, ESI†). Remarkably, the highest TOF per defect (up to 10.1 h−1) is offered by hcp-UiO-66 while fcc-UiO-66 and hxl-CAU-26 provided only 1.0 h−1 and 1.2 h−1 TOF per defect, respectively. Meanwhile, a consistent correlation between the TOF per defect with ΔE of each type of MOF is found in Fig. 4b. These results clearly uncover the assembly fashion-varied catalytical activity, especially between the hcp one and the others.
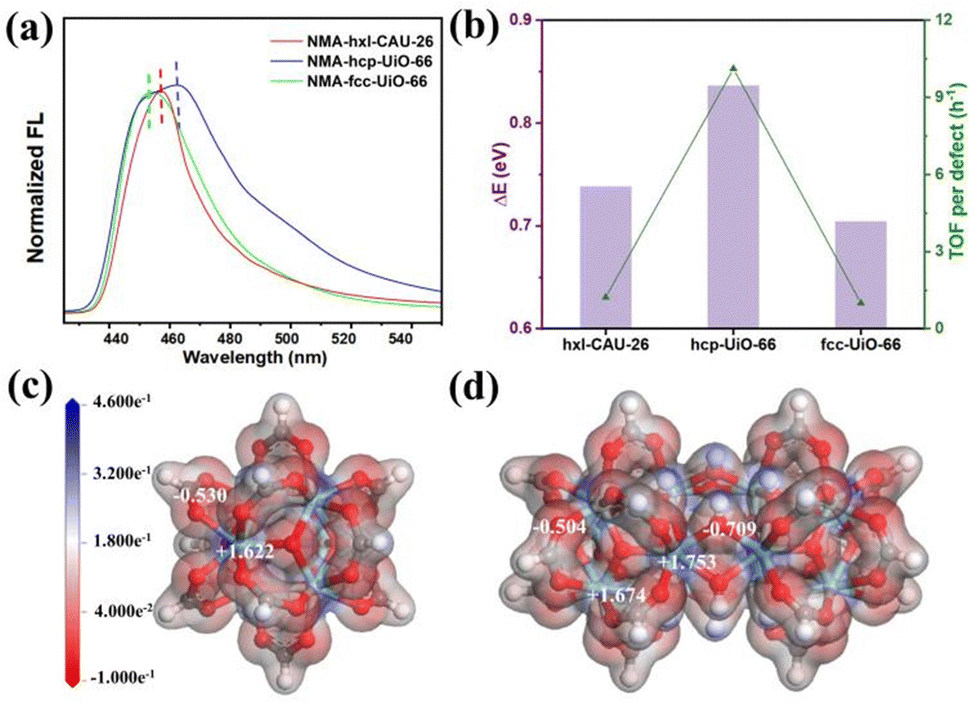 | ||
| Fig. 4 (a) Fluorescence spectra of NMA binding to hxl-CAU-26, hcp-UiO-66 and fcc-UiO-66 in CH3CN. (b) The correlation between ΔE and TOF per defect among hxl-CAU-26, hcp-UiO-66 and fcc-UiO-66. The charge density distribution profiles of Zr6O4(μ3-OH)4 in both hxl-CAU-26 and fcc-UiO-66 node (c) and (d) Zr12O8(μ3-OH)8(μ2-OH)6 in hcp-UiO-66. | ||
Scrutiny on the example of hcp-UiO-66 figures out an additional shoulder peak (455.0 nm) close to the peak position of fcc-UiO-66, an indication of two different Zr sites in the hcp assembly scaffold. To get an in-depth understanding, we then calculate the charge density distribution of the Zr sites among three assembly types. As both hxl-CAU-26 and fcc-UiO-66 share the same node, there are only Zr6O4(μ3-OH)4 (Fig. 4c) and Zr12O8(μ3-OH)8(μ2-OH)6 (Fig. 4d) nodes taken as theoretical moderns for consideration. As displayed in Fig. 4c, each Zr site in Zr6O4(μ3-OH)4 for both hxl-CAU-26 and fcc-UiO-66 is identically coordinated with 4 carboxylate oxygens and 4 μ3-OH, which presents a positive charge of +1.622 according to the Mulliken population analysis. In comparison, two distinct types of Zr site are clearly found for Zr12O8(μ3-OH)8(μ2-OH)6 in hcp-UiO-66 (Fig. 4d). Except for one same coordination as Zr6O4(μ3-OH)4, the other type of Zr site appears as replacing 2 carboxylate oxygens with 2 bridged μ2-OH. Impressively, the latter Zr site obviously shows a higher Mulliken charge up to +1.753 due to the stronger electronegativity of μ2-OH than the carboxylate group, which is on account of the congenitally higher activity of hcp-UiO-66 over both hxl and fcc ones.
Conclusions
In summary, three types of Zr-MOFs including hxl-CAU-26, hcp-UiO-66 and fcc-UiO-66 with varied assembly fashion are adopted to regulate the catalytic performances in the alcoholysis of epoxides. Taking advantage of a much higher proportion of linker defect and inherently stronger activation capability, hcp-UiO-66 not only exhibits nearly 10-fold TOF per node compared to both hxl-CAU-26 and fcc-UiO-66 but also maintains superior product enantioretentivity. The assembly-dependent performances found in this work enlighten an alternative way, namely tuning the polymorphism of MOFs, for driving more organic transformations in both high conversion efficiency and product selectivity.Experimental
Experimental details and supplementary figures can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (22103055, J. G., 21905195, M. T. Z., 92056204, 21890381 and 21721002, Z. Y. T.), Science and Technology Plans of Tianjin (21ZYJDJC00050, J. G.), National College Students Innovation and Entrepreneurship Training Program (202210058019, J. G.), National Key Research and Development Program of China (2021YFA1200302, Z. Y. T.), Strategic Priority Research Program of Chinese Academy of Sciences (XDB36000000, Z. Y. T.) and Wenzhou Key Laboratory of Biomaterials and Engineering (WIUCASSWCL21005, J. G.). We would like to thank the Analytical & Testing Center of Tiangong University for the help with SEM imaging.Notes and references
- W. Gong, Z. Chen, J. Dong, Y. Liu and Y. Cui, Chem. Rev., 2022, 122, 9078–9144 CrossRef CAS PubMed.
- J. Liang, A. Hao, P. Xing and Y. Zhao, ACS Nano, 2021, 15, 5322–5332 CrossRef CAS PubMed.
- J. Guo, Y. Zhang, Y. Zhu, C. Long, M. Zhao, M. He, X. Zhang, J. Lv, B. Han and Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 6873–6877 CrossRef CAS PubMed.
- F. Wang, X. Yue, Q. Ding, H. Lin, C. Xu and S. Li, Nanoscale, 2023, 15, 2541–2552 RSC.
- S. Gómez Graña, J. Pérez Juste and P. Hervés, Adv. Colloid Interface Sci., 2021, 288, 102338 CrossRef PubMed.
- X. Zhang, Z. Wang, X. Huang, Q. Huang, Y. Wen, B. Li, M. Holmes, J. Shi and X. Zou, Chem. Eng. J., 2023, 451, 138928 CrossRef CAS.
- P. Barbaro, V. Dal Santo and F. Liguori, Dalton Trans., 2010, 39, 8391–8402 RSC.
- N. H. Cho, A. Guerrero Martínez, J. Ma, S. Bals, N. A. Kotov, L. M. Liz Marzán and K. T. Nam, Nat. Biomed. Eng, 2023, 1, 88–106 Search PubMed.
- L. Zhang, H. X. Wang, S. Li and M. Liu, Chem. Soc. Rev., 2020, 49, 9095–9120 RSC.
- Q. Han, B. Qi, W. Ren, C. He, J. Niu and C. Duan, Nat. Commun., 2015, 6, 10007 CrossRef PubMed.
- H. Liu, H. Li, Y. He, P. Cheng, Y. Q. Zhang, B. Feng, H. Li, K. Wu and L. Chen, Nat. Commun., 2023, 14, 2100 CrossRef CAS PubMed.
- N. Antil, N. Akhtar, R. Newar, W. Begum, A. Kumar, M. Chauhan and K. Manna, ACS Catal., 2021, 11, 10450–10459 CrossRef CAS.
- J. Guo, Y. Wan, Y. Zhu, M. Zhao and Z. Tang, Nano Res., 2020, 14, 2037–2052 CrossRef.
- C. Teixeira, R. B. Pereira, N. F. S. Pinto, C. M. M. Coelho, M. J. G. Fernandes, A. G. Fortes, M. S. T. Goncalves and D. M. Pereira, Int. J. Mol. Sci., 2022, 23, 3759 CrossRef CAS PubMed.
- K. Berijani and A. Morsali, J. Catal., 2019, 378, 28–35 CrossRef CAS.
- H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- Y. Gu, B. A. Anjali, S. Yoon, Y. Choe, Y. G. Chung and D. W. Park, J. Mater. Chem. A, 2022, 10, 10051–10061 RSC.
- X. Chen, Z. Qiao, B. Hou, H. Jiang, W. Gong, J. Dong, H. Li, Y. Cui and Y. Liu, Nano Res., 2020, 14, 466–472 CrossRef.
- Y. Zhao, S. Qi, Z. Niu, Y. Peng, C. Shan, G. Verma, L. Wojtas, Z. Zhang, B. Zhang, Y. Feng, Y. S. Chen and S. Ma, J. Am. Chem. Soc., 2019, 141, 14443–14450 CrossRef CAS PubMed.
- O. Yang, X. Gao, G. Qi, Y. Wang, W. Dong, Z. Tian, J. Zhao, D. Li and Q. Zhang, ACS Appl. Energy Mater., 2022, 6, 334–341 CrossRef.
- J. Guo, C. Yang and Y. Zhao, Acc. Chem. Res., 2022, 55, 1160–1170 CrossRef CAS PubMed.
- C. Yang, J. Yu, L. Zhai, S. Jia, T. Yang, W. Xiong and Q. Zhang, Chem. Eng. J., 2023, 467, 143511 CrossRef CAS.
- J. Guo, Y. Qin, Y. Zhu, X. Zhang, C. Long, M. Zhao and Z. Tang, Chem. Soc. Rev., 2021, 50, 5366–5396 RSC.
- H. Wang, M. Gu, X. Huang, A. Gao, X. Liu, P. Sun and X. Zhang, J. Mater. Chem. A, 2023, 11, 7239–7245 RSC.
- A. H. Valekar, M. Lee, J. W. Yoon, J. Kwak, D. Y. Hong, K. R. Oh, G. Y. Cha, Y. Kwon, J. Jung, J. Chang and Y. Hwang, ACS Catal., 2020, 10, 3720–3732 CrossRef CAS.
- T. Y. Zhou, B. Auer, S. J. Lee and S. G. Telfer, J. Am. Chem. Soc., 2019, 141, 1577–1582 CrossRef CAS PubMed.
- C. Tan, X. Han, Z. Li, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16229–16236 CrossRef CAS PubMed.
- Y. H. Hu, C. X. Liu, J. C. Wang, X. H. Ren, X. Kan and Y. B. Dong, Inorg. Chem., 2019, 58, 4722–4730 CrossRef CAS PubMed.
- J. Li, P. M. Bhatt, J. Li, M. Eddaoudi and Y. Liu, Adv. Mater., 2020, 32, 2002563 CrossRef CAS PubMed.
- Q. Fu, D. Liu, W. Niu, S. Zhang, R. Chen, Y. Wang, P. Zhao, H. Jiang, Y. Zhao, L. Yang, L. Yan, H. Wang and X. Zhao, Fuel, 2022, 327, 125085 CrossRef CAS.
- X. Feng, H. S. Jena, C. Krishnaraj, K. Leus, G. Wang, H. Chen, C. Jia and P. Van Der Voort, ACS Appl. Mater. Interfaces, 2021, 13, 60715–60735 CrossRef CAS PubMed.
- Z. Niu, W. Zhang, P. C. Lan, B. Aguila and S. Ma, Angew. Chem., Int. Ed., 2019, 58, 7420–7424 CrossRef CAS PubMed.
- X. Rong, H. J. Wang, X. L. Lu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CrossRef CAS PubMed.
- P. Ji, X. Feng, P. Oliveres, Z. Li, A. Murakami, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 14878–14888 CrossRef CAS PubMed.
- L. Zhou, S. Wang, Y. Chen and C. Serre, Microporous Mesoporous Mater., 2019, 290, 109674 CrossRef CAS.
- S. Leubner, H. Zhao, N. Van Velthoven, M. Henrion, H. Reinsch, D. E. De Vos, U. Kolb and N. Stock, Angew. Chem., Int. Ed., 2019, 58, 10995–11000 CrossRef CAS PubMed.
- Y. Wu, X. Wang, K. O. Kirlikovali, X. Gong, A. Atilgan, K. Ma, N. M. Schweitzer, N. C. Gianneschi, Z. Li, X. Zhang and O. K. Farha, Angew. Chem., Int. Ed., 2022, 61, e202117528 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- R. Xu, Q. Ji, P. Zhao, M. Jian, C. Xiang, C. Hu, G. Zhang, C. Tang, R. Liu, X. Zhang and J. Qu, J. Mater. Chem. A, 2020, 8, 7870–7879 RSC.
- Y. Quan, G. Lan, W. Shi, Z. Xu, Y. Fan, E. You, X. Jiang, C. Wang and W. Lin, Angew. Chem., Int. Ed., 2021, 60, 3115–3120 CrossRef CAS PubMed.
- K. Ohkubo, S. C. Menon, A. Orita, J. Otera and S. Fukuzumi, J. Org. Chem., 2003, 68, 4720–4726 CrossRef CAS PubMed.
- P. Ji, T. Drake, A. Murakami, P. Oliveres, J. H. Skone and W. Lin, J. Am. Chem. Soc., 2018, 140, 10553–10561 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00419h |
| This journal is © The Royal Society of Chemistry 2024 |