
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rotationally-driven dynamic solid phase sodium bisulfite conversion disc for forensic epigenetic sample preparation†
R.
Turiello
 *a,
R. L.
Nouwairi
a,
J.
Keller
a,
L. L.
Cunha
a,
L. M.
Dignan
a and
J. P.
Landers
abc
*a,
R. L.
Nouwairi
a,
J.
Keller
a,
L. L.
Cunha
a,
L. M.
Dignan
a and
J. P.
Landers
abc
aDepartment of Chemistry, University of Virginia, Charlottesville, VA, USA. E-mail: rat3a@virginia.edu
bDepartment of Mechanical Engineering, University of Virginia, Charlottesville, VA, USA
cDepartment of Pathology, University of Virginia, Charlottesville, VA, USA
First published on 27th November 2023
Abstract
The approaches to forensic human identification (HID) are largely comparative in nature, relying upon the comparison of short tandem repeat profiles to known reference materials and/or database profiles. However, many profiles are generated from evidence materials that either do not have a reference material for comparison or do not produce a database hit. As an alternative to individualizing analysis for HID, researchers of forensic DNA have demonstrated that the human epigenome can provide a wealth of information. However, epigenetic analysis requires sodium ![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif) i
i![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) ulfite
ulfite ![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) onversion (BSC), a sample preparation method that is time-consuming, labor-intensive, prone to contamination, and characterized by DNA loss and fragmentation. To provide an alternative method for BSC that is more amenable to integration with the forensic DNA workflow, we describe a rotationally-driven, microfluidic method for dynamic solid phase-BSC (dSP-BSC) that streamlines the sample preparation process in an automated format, capable of preparing up to four samples in parallel. The method permitted decreased incubation intervals by ∼36% and was assessed for relative DNA recovery and conversion efficiency and compared to gold-standard and enzymatic approaches.
onversion (BSC), a sample preparation method that is time-consuming, labor-intensive, prone to contamination, and characterized by DNA loss and fragmentation. To provide an alternative method for BSC that is more amenable to integration with the forensic DNA workflow, we describe a rotationally-driven, microfluidic method for dynamic solid phase-BSC (dSP-BSC) that streamlines the sample preparation process in an automated format, capable of preparing up to four samples in parallel. The method permitted decreased incubation intervals by ∼36% and was assessed for relative DNA recovery and conversion efficiency and compared to gold-standard and enzymatic approaches.
Introduction
Current approaches to human identification (HID) of unknown persons remain largely comparative in nature, whereby short tandem repeat (STR) profiles from unknown evidence samples are compared with known reference materials/database profiles.1 Alternatively, unidentified human remains are morphologically categorized by visual interpretation by an anthropologist, as compared to discrete, published standards.2 Despite the statistical success of producing a match via STR analysis, many genetic profiles are generated from crime scenes, human remains, and sexual assault and evidence collection kits (SAECKs) that do not have a genetic reference material for comparison and do not produce a database hit. Further, the precision with which any trait is discerned via anthropological assessment for identification has been determined to be dependent upon the completeness of the remains and the anthropologist's prior experience.3 For these types of cases, the human epigenome has been suggested as a reservoir of information for sex typing,4 monozygotic twin individualization,5 body fluid identification,6 behavioral traits,7 and DNA phenotyping (FDP) by estimation of human chronological age.8 In particular, over the past 15 years more than 300 research studies and review articles have been published suggesting the utility of epigenetic methylation status at specified genetic loci for approximation of human age.9 Studies have demonstrated predictive success within 0.94 years10 from forensically-relevant body fluids including, but not limited to, blood,11–16 saliva,17–19 semen,20,21 and teeth.22 However, despite great research success and the forensic community's high regard for DNA-based testing, epigenetic age prediction has not been adapted into the forensic DNA analysis workflow or even used as a routine investigative technique by law enforcement personnel.If adopted, the forensic epigenetic workflow would require an additional step during sample preparation, referred to as sodium iulfite onversion (BSC), a method that has remained largely steadfast in its approach since its inception. Through a series of chemical modifications, the BSC process preferentially deaminates all unmethylated cytosines in the DNA transcript to yield uracil residues, leaving those cytosines containing a methyl group (e.g., 5-methylcytosines) intact and distinguishable for downstream analysis by methylation-specific real-time polymerase chain reaction (RT-PCR) or sequencing. Unfortunately, these techniques are characterized by extensive DNA loss; in fact, a 2015 study by Leontiou and co-workers comparing four of the most widely used BSC kits determined that the recovery from 200 ng of input DNA averaged as low as 33.2% and only as high as 55%. Further, these methods require time-consuming, labor-intensive workflows with a high propensity for contamination due to the multitude of open-tube pipetting steps. We conclude that adaptation of the epigenetic analysis workflow by the forensic community has stalled given the current requirement for large amounts of input DNA and the constraint that implementation of the associated laborious processes are not optimal for integration into the existing forensic DNA workflow.
We describe a microfluidic solution for forensic epigenetic sample preparation that leverages centrifugal force to enable rapid, efficient conversion of forensically-relevant DNA input masses in an automated microCD (μCD) format. Shortened conversion intervals are possible with the use of reduced reagent and sample volumes in chambers with an enhanced surface-area-to-volume ratio when compared with the conventional, in-tube BSC method; diffusion theories associated with miniaturization dictate that a system 1/10th of the original reaction chamber size will result in 100-fold reduction in time, thus minimizing the need for long incubations.23,24 Few examples of miniaturized bisulfite conversion methods exist in the literature. One such technology originating from the Wang Laboratory at Johns Hopkins University leverages a polydimethylsiloxane (PDMS) cartridge consisting of a linear array of chambers containing BSC buffer droplets that are combined and traversed through via magnetic actuation of DNA-containing silica beads.25,26 The primary benefit of this BSC method is automation and the parallel processing of up to three samples on one device; however, there is no exploration of reduced incubation intervals or measurement of conversion efficiency besides what can be gleaned from amplification results.26 A system was also developed by Yoon et al., wherein a conversion module is coupled with a detection platform employing isothermal solid-phase amplification; while the integration of components is impressive, the system is tested with high concentrations of input DNA not relevant to some applications.27 The proposed work varies from those in literature on a number of fronts, but namely we sought to develop a method more amenable to low template DNA. Additionally, our method employs centrifugal force as the mechanism for fluid movement, which is advantageous for forensics applications for three primary reasons. First, this ensures that only a single mechanism is required for propulsion, eliminating the need for bulky external hardware (e.g., syringe pumps, electronics, tubing, etc.) that hinder portability and take up valuable bench space. Second, the mechanism permits automation in a fully closed system to mitigate contamination risk. Third, the forces controlling fluid movement through channels and into reaction chambers for precise chemistries are easily controlled by simply adjusting rotational speed, an aspect that may be coded for automation via a corresponding graphical user interface (GUI). With regard to automation specifically, the μCD approach is fully programmable via custom, external systems capable of heating, imparting rotational and magnetic forces at specified frequencies, and laser valving to open and close fluidic channels. For this application, the use of a silica dynamic solid phase (dSP) enables magnetically-actuated, bead-based conversion; together with careful consideration of fluidic architecture and valving strategy, this permits the completion of several sequential unit operations on-board. Conversion discs were designed to accommodate approximately 1/10th of the fluid volumes required by conventional BSC methods and with a view of multiplexing in mind: that is, each μCD is capable of converting up to four samples per disc.
Microfluidic integration was assessed with standards and multiple downstream analytical processes, including RT-PCR, high resolution melting (HRM), and electrophoresis. Early phase goals of this project included testing the chemistry at the microfluidic scale, adjusting the parameters of the reaction steps associated with DNA loss, optimizing microfluidic architecture, and comparing μCD converted eluates with those originating from an in-tube, gold-standard method for conversion. For proof-of-concept, assay characterization was completed with primers targeting FHL2, a locus associated with age determination. Results suggest the increased surface-area-to-volume ratio at the microscale enabled reduction of incubation intervals, thereby decreasing the total assay time, with some evidence of increased DNA recovery and comparable conversion efficiency to a gold-standard method. Finally, we compared this method to an alternative, enzymatic method for cytosine conversion.
Experimental
Sample materials
Preliminary testing of the in-tube and μCD dSP-BSC methods was accomplished with the human methylated & non-methylated DNA set (Zymo Research, Irvine, CA, USA) at an initial concentration of 250 ng μL−1 to assess relative DNA recovery and BSC efficiency without potential variability resulting from DNA extraction. Universal methylation human DNA standard (Zymo Research, Irvine, CA, USA) at a starting concentration of 20 ng μL−1 was used as a positive control for amplification and pyrosequencing; here, the positive control is fully methylated at all cytosine positions and bisulfite converted by the manufacturer. Negative controls were included during the BSC process, whereby human sample was substituted for nuclease free water and otherwise handled as if containing human genetic material. No template controls consisting of nuclease free water in place of the BSC eluate were included in all amplification and HRM detection modes.In-tube dSP-BSC
For comparison with a gold-standard method, the dSP-BSC process was completed with the EZ-96 DNA Methylation-Lightning MagPrep (Zymo Research, Irvine, CA, USA) kit, according to the manufacturer recommended protocol, adapted for lower sample throughput (e.g., replacing the 96-well plate format with individual tubes and a magnet stand). For in-tube microfluidic reactions, volumes corresponding to the microdevice chamber capacities were used. Here, 2.5 μL DNA standard was added to 12.5 μL of lightning conversion reagent (Zymo Research, Irvine, CA, USA) in a 0.2 mL tube; the 15 μL reaction mixture was heated on the Veriti thermal cycler (Thermo Fisher Scientific, Waltham, MA, USA) at 95 °C for 1 min and 54 °C 45 min for sulphonation and hydrolytic deamination. In separate 1.5 mL tubes, 40 μL of M-binding buffer and 10 μL MagBinding beads were combined with the partially-converted DNA. Samples were mixed by vortexing, incubated at room temperature for 1 min, and placed on a magnetic stand before the supernatant was removed and discarded. The beads were then resuspended in 40 μL M-Wash buffer, mixed by vortexing, and placed on the magnet stand for supernatant removal. Beads were then mixed with 40 μL L-desulphonation buffer, mixed by vortexing, and incubated at room temperature for 20 min following desulphonation, a second wash step was completed, as before, and the tubes were subsequently placed on a dry bath set to 55 °C for 1 min to remove residual M-Wash buffer. Finally, the beads were resuspended in 25 μL of M-elution buffer, heated to 55 °C for 4 min and placed back on the magnetic stand. The BSC eluate was separated from the bead fraction by pipette and added to a 0.2 mL tube, which was then retained and stored at −20 °C until further analysis. For downstream detection via RT-PCR and HRM, a 5 μL volume of the BSC eluate was used, corresponding to a final PCR concentration of 5 ng μL−1, except for the bead volume optimization study, wherein the final PCR concentration was 2 ng μL−1. All in-tube BSC conversions were completed in technical replicates of 3 and PCR/HRM was also run in replicates of 3.Real-time polymerase chain reaction and high resolution melting
Amplification and detection of BSC eluates and corresponding controls was accomplished using the ZymoTaq™ DNA polymerase (Zymo Research, Irvine, CA, USA) chemistry. Detection was made possible with the inclusion of an intercalating LAMP fluorescent dye (New England Biolabs, Ipswich, MA, USA). Forward and reverse primers for the FHL2 region were designed according to specifications published previously.28 For conservation of reagents, half-reactions totaling 25 μL were used, including 12.5 μL 2× reaction buffer (1×), 0.25 μL dNTP mix (0.25 mM), 0.625 μL of 10 μM forward and reverse primers (0.4 μM) (Integrated DNA Technologies, Coralville, Iowa, USA), 0.2 μL ZymoTaq™ DNA polymerase (2 U/50 μL), 1.25 μL LAMP fluorescent dye (2.5 μM), 4.55 μL PCR-grade water, and 5 μL of BSC eluate, positive control, or nuclease-free water. All samples were run in triplicate on the QuantStudio 5 Real-Time PCR system with detection in the FAM channel (Thermo Fisher Scientific, Waltham, MA, USA). Thermal conditions included initial denaturation (95 °C, 600 s), 45 cycles of denaturation (95 °C, 30 s), annealing (50 °C, 45 s), and extension (72 °C, 60 s), and a final extension step (72 °C, 420 s). For data analysis, eluates and controls were considered positive if they crossed the instrument-defined threshold, producing a Ct value. HRM was accomplished immediately following amplification on the QuantStudio 5 system and included thermal conditions whereby the reaction was denatured at 95 °C for 1 s, subsequently cooled to 50 °C and held for 20 s, before being incrementally heated to 95 °C at a rate of 0.1 °C s−1, with data acquisition occurring at each interval. The Tm of each sample was determined via the instrument's own algorithm. For visual clarity, some RT-PCR and HRM plots were recreated in excel using raw fluorescence values extracted from the QuantStudio 5 system. To show the threshold line, baseline subtraction was calculated from cycles 3 through 15 and the threshold was plotted at three times the standard deviation of the mean baseline, as before.29Operation of mechatronic systems
Spin systems to impart centrifugal force, enable laser-based valving to open and close fluidic channels, perform magnetic mixing, and on-disc heating were all designed in-house, as described previously.30–32 These systems are controlled by 8-core microcontrollers (Propeller P8X32A-M44; Propeller, Inc., Rockland, CA, USA) and corresponding, custom programs written in Spin and run from an external laptop. Rotational fluid propulsion and laser-based valving was accomplished with the power, time, and Z-height adjustable (PrTZAL) system.32 Here, valves were opened to permit flow into a new fluidic layer and into the corresponding chamber using laser power settings of 500 mW for a actuation time of 500 ms, and positioned 15 mm above the surface of the disc,33 Similarly, fluidic channels were closed by the same 638 nm laser diode to prevent backflow into the system using power, time, and z-height settings of 700 mW, 2500 ms, and 26 mm, respectively.32 Here, automation is enabled through use of the GUI code, instructing the PrTZAL system when to stop rotation and position the disc under the valve for each valving event. A separate dynamic solid-phase extraction (dSPE) platform was used to impart external magnetic control over the silica solid phase for efficient mixing of both DNA for capture and reagents for effective conversion, using settings optimized by Dignan et al.30 On-disc heating was accomplished with a dual-clamped Peltier system.34Microdevice design and fabrication
Iterative and final μCD prototyping was accomplished with AutoCAD software (Autodesk, Inc., Mill Valley, CA, USA). Designs were laser ablated into thermoplastic substrates and corresponding adhesives via a CO2 laser (VLS 3.50, Universal Laser Systems, Scottsdale, AZ, USA). The core device contains five primary poly(ethylene terephthalate) (PeT) layers (Film Source, Inc., Maryland Heights, MO, USA); whereby primary fluidic layers (layers 2 and 4) are encapsulated with heat-sensitive adhesive (HSA) (EL-7970-39, Adhesives Research, Inc., Glen Rock, PA, USA). At the center of the μCD, a black PeT (bPeT) (Lumirror X30, Toray Industries, Inc., Chuo-ku, Tokyo, Japan) layer enables laser-based valving and provides a barrier between the two primary fluidic layers.32,33 Following alignment of the 5-layer device, layers were heat-bonded using a commercial-off-the-shelf laminator (UltraLam 250B, Akiles Products, Inc., Mira Loma, CA, USA). according to the “print, cut, laminate” method, described elsewhere.35 Multiple accessory pieces were added to the device via pressure-sensitive adhesive (PSA) transfer tape (MSX-7388, 3M, Saint Paul, MN, USA). Poly-(methyl methacrylate) (PMMA) (1.5 mm thickness, McMaster Carr, Elmhurst, IL, USA) capped with PeT was added to all chambers, not including the bisulfite conversion chamber, to increase chamber volume capacity. Polytetrafluoroethylene (PTFE) hydrophobic membranes (0.2 μm, Sterlitech, Auburn, WA, USA) were added to the vents of the bisulfite conversion and magnetic manipulation chambers to permit gas exchange during heated incubations on-board. Fluidic channels enabling flow from one chamber to another upon device rotation were designed to be approximately 100 μm deep and have widths between 400 and 500 μm.Fluidic dye studies and corresponding image analysis
For early optimization of fluidic architecture, blue and yellow aqueous dye solutions were used to visually represent sample reagents. Following each workflow step (e.g., sulphonation and deamination), scanned images of the μCD were captured using an Epson Perfection V100 Photo desktop scanner (Seiko Epson Corporation, Suwa, Nagano Prefecture, Japan). Characterization of fluidic loss during the initial heating steps of the reaction was completed with 0.1 M Allura red dye solution (Sigma-Aldrich, St. Louis, MO, USA) diluted in 1× Tris-EDTA buffer, pH 7.5 (Sigma-Aldrich, St. Louis, MO, USA). Fiji Image J Freeware was used to evaluate fluid loss via ‘The Crop-Threshold-and-Go’ method of analysis.36 Briefly, cropped chamber images from digital scans were analyzed via the ImageJ color thresholding module to overlay a mask denoting the region of interest (ROI) from any background and providing a number of pixels associated with that mask. To build the calibration curve and measure fluid loss pre- and post-heating, a total of 5 technical replicates were measured for each parameter.Microdevice dynamic solid phase sodium bisulfite conversion
The complete μCD dSP-BSC process can be followed in the dye study, described below. The reaction begins with reagent and sample loading, wherein C1 is loaded with 13 μL lightning conversion reagent and 2 μL of DNA sample. The neighboring C2 is loaded with a mixture of 40 μL bead binding buffer and 10 μL magnetic beads. Chambers 4 and 8 are loaded with 40 μL of wash buffer and C6 is loaded with 40 μL of desulphonation buffer, while C10 is loaded with 25 μL of elution buffer. V1 is closed and C1 is positioned within the dual-clamped heating system for the following temperature intervals: 95 °C for 1 min and 54 °C for 45 min to complete the denaturation, sulphonation, and deamination steps. Following incubation, V2 is opened and the disc is spun (2000 rpm, 30 s) to introduce the partially converted DNA to C2 for bead binding. V3 is closed and the mixture is magnetically agitated on the dSPE system for 1 min beads are subsequently pelleted (2000 rpm, 30 s), V4 is opened, and the disc is spun (1500 rpm, 30 s) to remove waste to C3, and V5 is closed. Wash #1 begins with the opening of V6 and disc rotation (1500 rpm, 15 s) to introduce wash buffer to C2. Following magnetic mixing (1 min), beads are pelleted once again (2000 rpm, 30 s), V7 is opened, and the disc is spun (1500 rpm, 30 s) to remove supernatant to C5 before V8 is closed. To begin the desulphonation step, V9 is opened and the disc is spun (1500 rpm, 15 s) to introduce desulphonation buffer from C6 to C2. The cocktail is magnetically mixed (1 min) and held at room temperature for 20 min to complete conversion. Desulphonation waste is removed following bead pelleting (2000 rpm, 30 s), the opening of V10, a spin step (1500 rpm, 30 s), and the closing of V11. The final wash occurs when V12 is opened and the disc is spun (1500 rpm, 15 s), introducing wash buffer into C2. The mixture is magnetically mixed (1 min), beads are pelleted (2000 rpm, 30 s), V13 is opened, the disc is spun again (1500 rpm, 30 s), and V14 is closed off to the upstream architecture. C2 is then placed between the dual-clamping Peltier system at a temperature of 55 °C for 5 min for wash buffer evaporation, prior to DNA elution. Elution is initiated when V15 is opened and the disc is rotated (1500 rpm, 30 s) to introduce elution buffer to C2 and the beads. V16 is closed and C2 is once again placed under the clamping system and heated to 55 °C, except for only 4 min Once the DNA has been released from the beads, they are once again pelleted (2000 rpm, 30 s), V17 is opened, and the disc is spun to move the eluate from C2 to C11 for pipette removal.Electrophoresis
Electrophoresis was completed with the 2100 Bioanalyzer Instrument (Agilent Technologies, Santa Clara, CA, USA). The Agilent DNA 1000 kit (Agilent) was used according to manufacturer recommendations, whereby 9 μL of gel dye matrix, 5 μL of DNA 1000 marker, 1 μL of DNA 1000 ladder, and 1 μL of converted DNA eluate was added to the requisite wells in the microfluidic chip. Analysis was completed with the 2100 Expert software (Agilent).Degradation study
Degradation associated with the on-disc sample preparation method was assessed with the Quantifiler Trio Quantification kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer recommendations and using the QuantStudio 5 Real-Time PCR system. Degradation indices were calculated by the HID Real-Time PCR Analysis software (Thermo Fisher Scientific, Waltham, MA, USA) and were based upon the Ct values of diluted standards for large and small autosomal targets from 50–0.005 ng μL−1 according to manufacturer recommendations. Non-methylated DNA standards were bisulfite converted using the on-disc μCD approach at a final concentration of 25 ng μL−1 in technical replicates of 3 and exactly 1 μL of converted eluate was used from each conversion replicate for evaluation of resultant degradation, equating to 1.25 ng μL−1 in each Quantifiler Trio reaction.Enzymatic methyl-Seq (EM) conversion
A total of 13 μL human non-methylated control DNA (Zymo Research, Irvine, CA, USA) was added to 117 μL 10 mM Tris-EDTA buffer (Sigma-Aldrich, St. Louis, MO, USA), pH 8.0, for DNA fragmentation at a final concentration of 25 ng μL−1. Shearing was completed using an S2 Ultrasonicator (Covaris, Woburn, MA, USA) with the 6 × 16 mm AFA Fiber microTubes (Covaris, Woburn, MA, USA) and settings associated with mean fragment sizes of 1.5 kilobases (kb) for a downstream application in RT-PCR and HRM, per manufacturer recommendations. The requisite volumes of sheared DNA were mixed with 10 mM Tris-EDTA buffer to a total volume of 28 μL to begin conversion and amplify converted product to a final DNA input amount of 100, 10, and 1 ng of total input DNA in technical replicates of 2. The NEBNext® enzymatic methyl-Seq conversion module (New England Biolabs, Ipswitch, MA, USA) was used for enzymatic conversion according to the manufacturer's protocol and with Hi-Di formamide (Applied Biosystems, Waltham, MA, USA) for denaturation and NEBNExt® sample purification beads (New England Biolabs, Ipswitch, MA, USA) for purification. Subsequent amplification and HRM of converted eluates was completed as described here previously for the FHL2 target in replicates of 3.Statistics and reproducibility
All statistical calculations related to significance testing were completed with GraphPad Prism software (San Jose, CA, USA). Ct and Tm values were described as the mean ± standard deviations for all technical BSC replicates and/or amplification replicates. All described t-tests are two-tailed, using unpaired comparison parameters, and with a significance (α) of 0.05 (e.g., 95% confidence interval). Any analysis of variance (ANOVA) used a one-way framework and with a the same 95% confidence interval parameters.Results and discussion
The development and characterization of a rotationally-driven microfluidic device for the dynamic solid phase sodium bisulfite conversion (dSP-BSC) of differentially-methylated human DNA is described. Preliminary characterization of the chemical workflow was accomplished in-tube and all iterative changes to the method were tested by comparing resultant eluates to those produced from the manufacturer recommended protocol to a corresponding ‘in-tube microfluidic’ protocol using reduced reagent volumes and incubation parameters. Likewise, BSC eluates produced following sample preparation via the ‘on-disc’ μCD approach were compared with those using the previously described in-tube approach. The selected target for early characterization is in the promoter region of FHL2, and is one associated with forensic human age prediction across multiple tissues.12,19,22,37 The primer sequences were adapted from Hamano et al.,28 wherein the assay was used for age prediction using PCR amplification and HRM and have thus been previously vetted for PCR bias, function, and relevance to human age approximations.Dynamic solid phase sodium bisulfite conversion workflow
The conventional, ‘gold-standard’ BSC workflow was developed according to the manufacturer's protocol but modified for in-tube sample preparation (Fig. 1a). During the initial incubation, samples are heated to facilitate complete denaturation and subsequent progression of unmethylated cytosines through two intermediate structures, including 5,6-dihydrocytosine-6-sulphate and 5,6-dihydrouracil-6-sulphonate via ammonium bisulfite; this phase is referred to collectively as sulphonation and hydrolytic deamination (Fig. 1b). Following a bead wash and immobilization step, desulphonation occurs, forming uracil residues via incubation in a sodium hydroxide solution. Elution of the chemically converted DNA from the dynamic solid phase is completed following another bead wash (Fig. 1b). Following the reaction, only unmethylated cytosines are converted to uracil; methylated cytosines remain intact, as the addition of a methyl group to the ring contributes to stabilization of the structure and a lack on conversion due steric hindrance and electrostatic repulsion.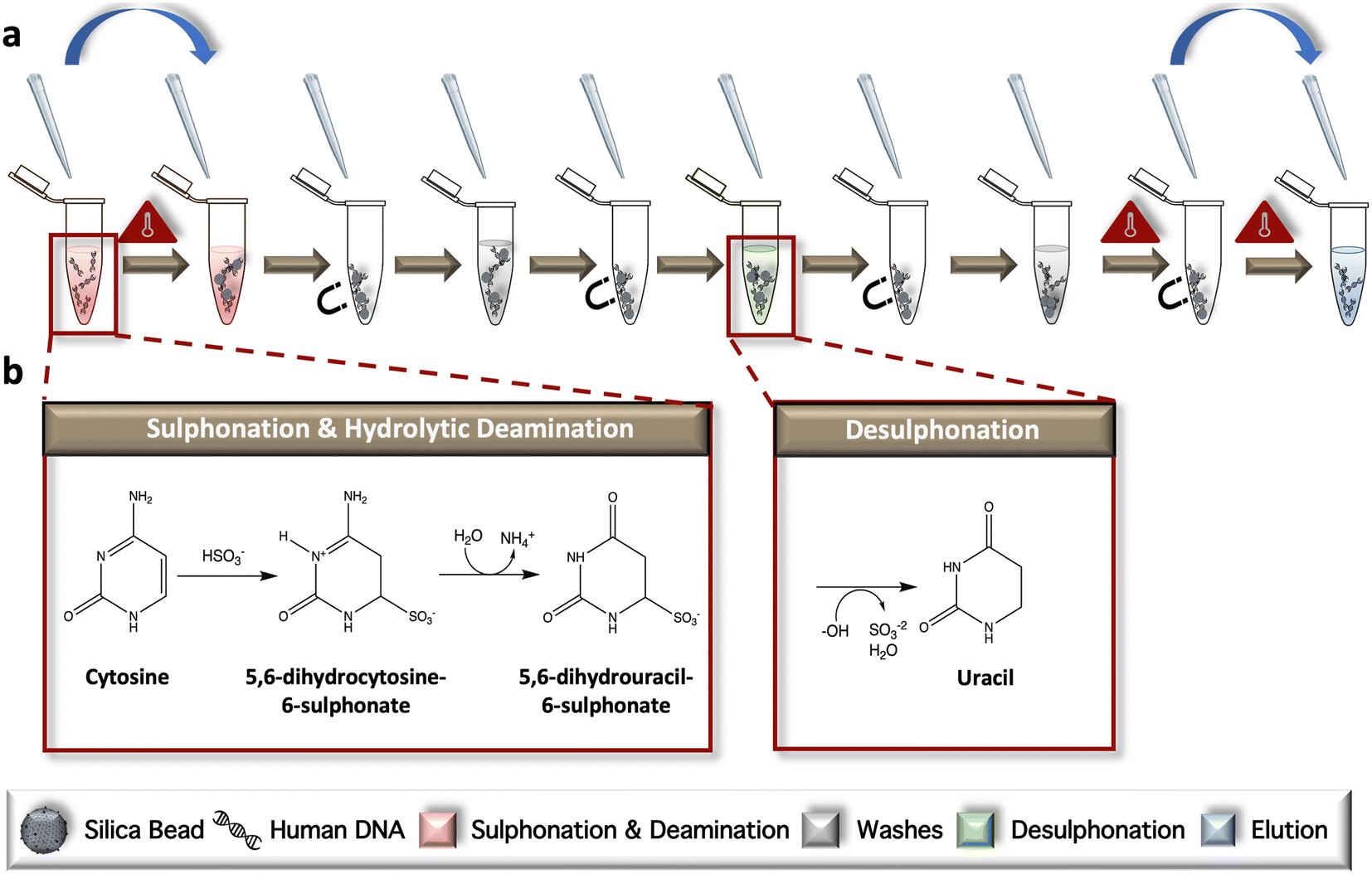 | ||
| Fig. 1 Adapted dSP-BSC workflow. a and b, The in-tube gold-standard method for bead-based conversion; sulphonation and deamination of unmethylated cytosine residues begins when the dsDNA is denatured and incubated with sodium bisulfite (HSO3−) at elevated temperatures. Genomic material with partially converted bases is affixed to a silica dSP and washed prior to final desulphonation. A second wash step is completed, and the beads are heated in an open tube to evaporate residual ethanol before the DNA is eluted and removed for downstream analysis. Blue arrows indicate tube transfers. | ||
Downstream analysis strategy by RT-PCR and HRM
To assess the analytical performance of the upstream sample preparation method, multiple techniques were used to measure relative DNA recovery and conversion efficiency. Relative DNA recovery was evaluated via RT-PCR, whereby resultant cycle threshold (Ct) values were compared. Because these values are representative of starting concentration, it follows that samples prepared with optimal BSC conditions for DNA preservation would produce more rapid amplification (e.g., lower Ct values). Here, the ZymoTaq™ DNA polymerase chemistry was used, as it was specifically designed for the amplification of bisulfite-treated DNA; however, the protocol was modified for reagent conservation and to enable real-time detection by utilizing only half-reactions and adding an intercalating Syto 9 dye, respectively. For verification of this detection method, methylated and non-methylated DNA standards were bisulfite converted in triplicate along with BSC negative controls and using the manufacturer's adapted protocol described above (Fig. 1). Resultant eluates were successfully amplified along with methylated positive controls (previously converted by the manufacturer) and no template controls (Fig. 2a); non-methylated standards produced Ct values of ∼33.9 (±0.60) and methylated standards and converted positive controls produced values of ∼38.99 (±0.54) and ∼38.33 (±0.13), respectively. Noticeably, there was no statistical difference detected between the methylated standard converted in-house and with the modified in-tube approach and the positive control previously modified by the manufacturer (unpaired t-test, α = 0.05, p-value = 0.7446). While this may not signify that the modified ‘gold-standard’ method performs comparably in terms of conversion, since this is a fully methylated standard and conversion is not taking place, this does indicate that the methods are comparable in recovery (e.g., degradation). In addition, all samples were amplified with an initial concentration of 5 ng μL−1 per reaction; however, there is a reproducible shift in Ct units between the non-methylated and methylated samples. This shift may be explained by PCR bias, whereby the GC content of the methylated template is higher than that of the non-methylated sample post-conversion, leading to a comparatively diminished amplification product.38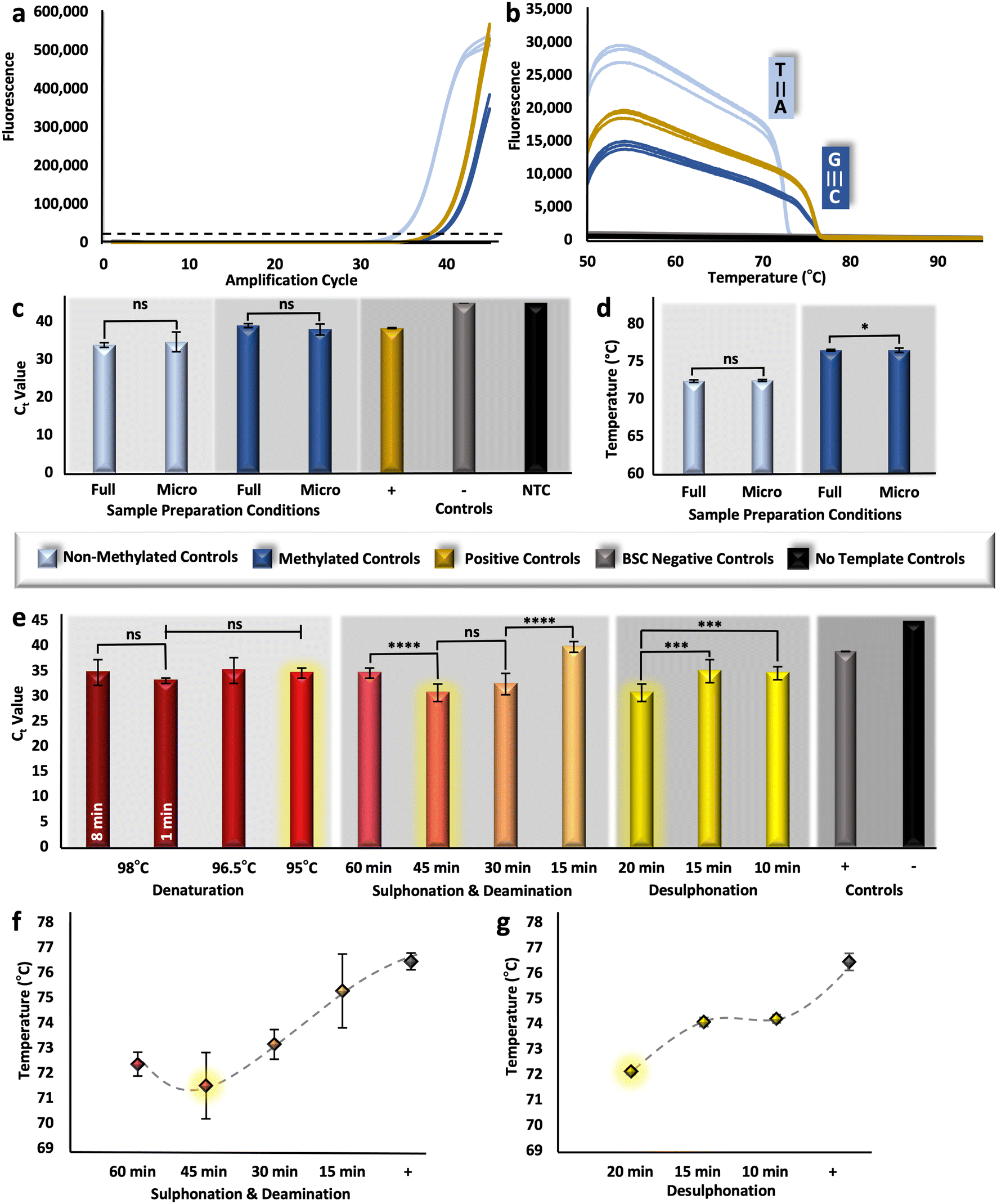 | ||
| Fig. 2 Assessment of DNA recovery and conversion efficiency. a, RT-PCR and b, HRM results, originating from methylation standards, bisulfite converted with the gold-standard approach. c and d, RT-PCR and HRM results, comparing methylation standards converted with either the manufacturer recommended ‘full’ volume or the decreased ‘micro’ volumes. e, Ct values corresponding to reduced incubation parameters for phases of the conversion reaction with non-methylated control DNA. f and g, Tm values corresponding to reduced incubation parameters. Selected incubation intervals are highlighted in yellow (e–g). | ||
Likewise, relative conversion efficiency was demonstrated with HRM analysis, whereby the Tm of non-methylated and methylated control samples post-conversion was determined, and the corresponding differences were associated with a shift in GC content. Assuming 99–100% conversion efficiency with the gold-standard method, as is alleged, would dictate that all unmethylated cytosines are converted to uracil and then to thymine following PCR, thus, these transcripts should consistently exhibit a much lower Tm than their methylated counterparts. However, if BSC conditions are such that conversion efficiency becomes diminished, the Tm of unmethylated amplicons will undoubtedly shift upward, approaching that of the methylated sequences with higher GC content. As a baseline, non-methylated and methylated amplicons melted at temperatures of ∼72.39 °C (±0.16 °C) and ∼76.51 °C (±0.11 °C), respectively (Fig. 2b), indicating that, post conversion, non-methylated standards will have a lowered GC content compared with that of methylated standards, due to the overall reduction in hydrogen bonds in the template. For additional confirmation of the HRM method, methylated positive controls, previously converted by the manufacturer, also showed reproducible melt temperatures at ∼76.08 (±0.23 °C). Moving forward, if a statistically significant difference is detected for those non-methylated transcripts that have been bisulfite converted, it may be assumed that conversion efficiency has be altered. However, it is important to note here that HRM is only a measure of relative conversion efficiency and cannot be used to calculate the precise percentage (0–100%) typically associated with this metric.
In-tube optimization of the microfluidic method
The conventional, gold-standard dSP-BSC process is a multi-step workflow requiring several sequential tube transfers, vortexing steps, incubations (both heated and at room temperature), and magnetic manipulations. Given the complexity, in-tube studies were completed prior to microdevice adaptation to isolate each variable for optimal performance at the microfluidic scale. First, samples were prepared with decreased BSC reagent volumes, approximately 1/10th of the manufacturer recommended amount; however, the concentration of silica beads remained consistent, as a reduction in the volume of beads resulted in diminished DNA recovery (Fig. S1†). Ct values originating from samples with decreasing volumes of silica bead solutions from 10 μL to 5 μL and 1 μL show statistical differences overall (one-way ANOVA, α = 0.05, p-value = 0.0002), with the lowest Ct values demonstrated with preparation using 10 μL volumes (32.09 ± 1.76) (Fig. S1†). Likewise, the elution volume was kept consistent to ensure a large enough volume for downstream testing. Regarding remaining BSC reagents, unpaired t-tests comparing Ct values originating from samples bisulfite converted with full and microfluidic volumes show no statistical differences for non-methylated and methylated control samples (α = 0.05, p-values = 0.3803 and 0.1016, respectively), indicating similar recovery (Fig. 2c). Likewise, non-methylated control samples, for which all cytosines would ostensibly be converted to uracil, produced statistically similar HRM values (unpaired t-test, α = 0.05, p-value = 0.248) and were consistent with the known Tm for that locus (Fig. 2d), indicating comparable conversion efficiency. Dissimilarly, a comparison of the Tm values for the methylated controls converted with different conditions showed statistical differences (unpaired t-test, α = 0.05, p-value = 0.0265); however, the difference between means was only calculated to be ∼0.266 °C (±0.11 °C) (Fig. 2d). These results were considered acceptable and further in-tube optimization to decrease dwell temperatures and intervals was completed with microfluidic volumes.Three incubation parameters were optimized at the microfluidic scale to increase adaptability of the protocol to our microfluidic system and reduce total analytical time, including 1) denaturation, 2) sulphonation and deamination, and 3) desulphonation. Conventional denaturation parameters necessitated an 8 min incubation at 98 °C; this parameter was reduced first from 8 min to 1 min with no difference in estimated recovery (unpaired t-test, α = 0.05, p-value = 0.0972). Dwell temperature was subsequently decreased from 98 °C to 96.5 °C and 95 °C for 1 min; likewise, one-way ANOVA results indicated no statistical differences between eluates produced with decreasing dwell temperature overall (α = 0.05, p-value = 0.0511) (Fig. 2e). For successive studies, samples were converted at 95 °C for 1 min, parameters much more amenable to microfluidic integration. For optimization of the next incubation step, sulphonation and deamination intervals were reduced from 60 min to 45, 30, and 15 minutes; interestingly, samples prepared via the conventional protocol (e.g., 60 min incubation) showed higher Ct values (34.60 ± 0.98) than those incubated for only 45 min (30.67 ± 1.75), indicating recovery was enhanced by reducing the incubation time (Fig. 2e). This trend was reversed when samples were only incubated for 30 or 15 min, likely as a result of incomplete conversion and corresponding primer mismatch during amplification. In fact, Fig. 2f shows the corresponding Tm values, providing evidence of incomplete conversion as incubation time decreased lower than a 45 min interval. Also evident from this figure is a statistical difference between Tm values undergoing sulphonation and deamination for 60 or 45 min (α = 0.05, p-value = 0.027), with 45 min incubated samples providing lower Tm values, indicating potentially increased conversion efficiency. Notably, samples undergoing sulphonation and deamination for less than 45 minutes showed either reduced recovery (Fig. 2e) or conversion efficiency (Fig. 2f). Thus, to test the optimal conversion time of the final reaction interval, desulphonation, a 45 min sulphonation and deamination was used. For desulphonation, the ideal interval in terms of both recovery and conversion efficiency was determined to be the conventional one of 20 min, producing eluates with the lowest Ct (Fig. 2e) and Tm values (Fig. 2g). In summary, the BSC parameters have been adapted for microfluidic integration, with decreased reagent volumes (e.g., 1/10th of the standard workflow, not including silica beads), and reduced incubation intervals, with denaturation occurring at 95 °C for 1 min and sulphonation and deamination shortened to 45 min total. Moving forward, the on-disc workflow incorporated all optimized parameters discussed above; additionally, wash evaporation time was reduced from 20 min to 1 min, as no wash buffer was visibly detected with on-disc evaporation at 55 °C for the shortened interval, reducing the entire workflow by ∼36.61% compared to the gold-standard method (Table S1†).
Microdevice design
The rotationally-driven μCD was designed for multiplexed analysis of up to four samples in parallel (Fig. 3a and b). Each domain includes all the necessary architectural features to support the sequential unit operations associated with the dSP-BSC workflow, wherein all of the architecture situated toward the center of rotation from the magnetic manipulation chamber houses the aqueous reagents, and the chambers closer to the edge of the disc accommodate reaction waste and the final BSC eluate (Fig. 3c). Initial conversion steps, including denaturation and sulphonation and deamination, are completed in the bisulfite conversion chamber. Following these steps, the partially converted material and aqueous buffer is spun into the magnetic manipulation chamber featuring the dynamic solid phase and a chaotropic solution to promote DNA-silica bead interactions. Note that the concave-shaped magnetic manipulation chamber was designed to retain magnetic beads during waste removal and was previously optimized elsewhere (Fig. 3c).30,31 Both the bisulfite conversion and magnetic manipulation chambers undergo heated incubations that may cause thermal pumping and subsequent fluid loss; thus, each of these chambers feature a hydrophobic membrane composed of polytetrafluoroethylene (PTFE) on the vent and a ‘closed’ loading arm channel. The device makes use of sacrificial valves to enable sequential unit operations, making each device single-use and preventing the potential for contamination and device failure from repeated use. The valving strategy is depicted in the schematic shown in Fig. 3d. Briefly, this approach was adapted from one described by Garcia-Cordero et al.33 and makes use of an optically-dense intermediate layer at the center of the disc that is thermally ablated by an external laser to form a pinhole, permitting fluid to flow from layer 2 to layer 4. To subsequently close channels and prevent backflow, the laser is positioned upstream from the opened valve, and laser parameters, including output power, contact time, and height from the surface of the disc, are altered to thermally deform and occlude flow. This method was developed in-house32 and the precise parameters for both valve opening and channel or ‘valve’ closing are detailed in the methods section.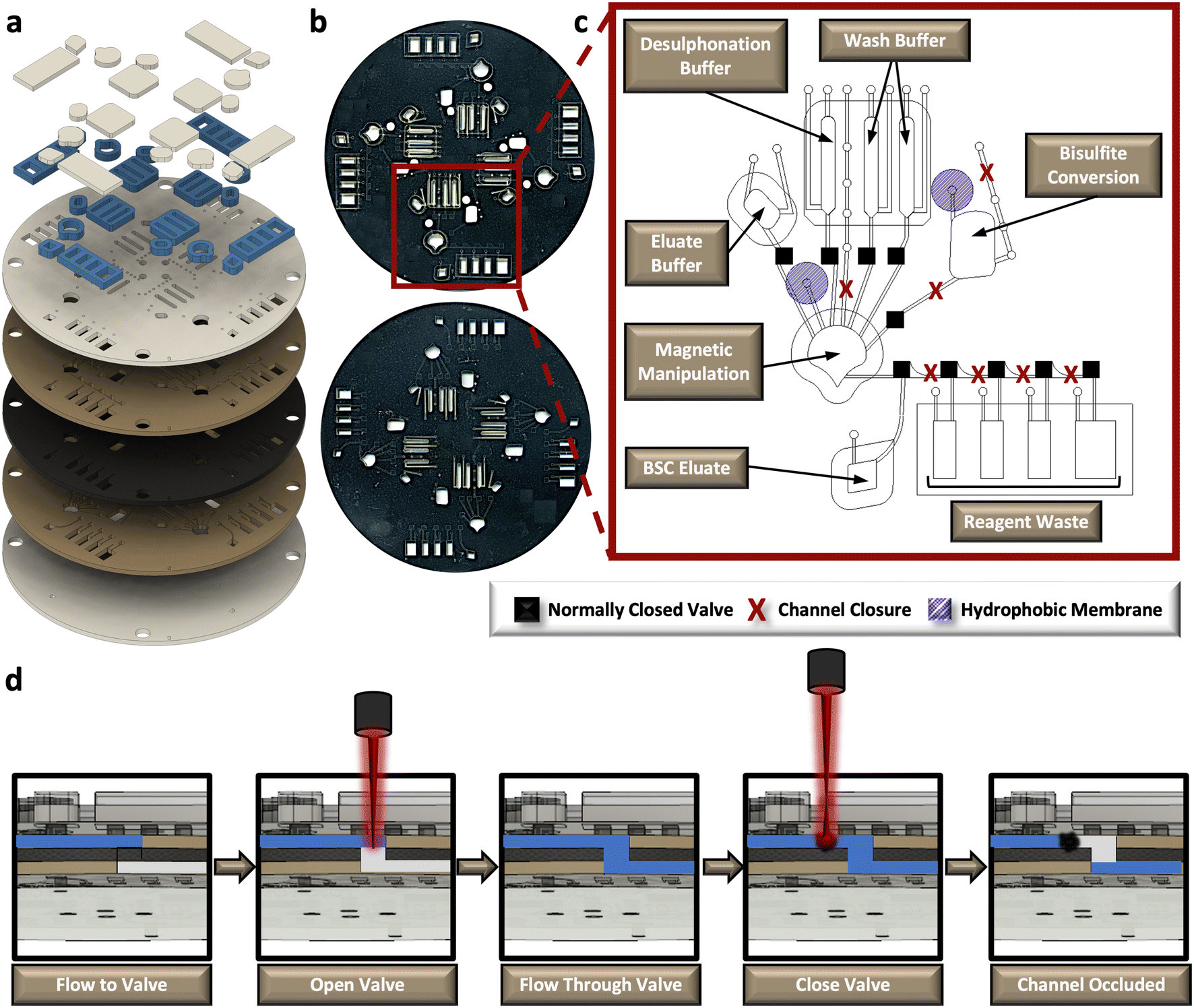 | ||
| Fig. 3 Rotationally-driven microfluidic device for automated sodium bisulfite conversion. a, Exploded rendering of the core, 5-layer polymeric disc and corresponding accessory pieces that increase reagent volume capacities. b, Digital scans of the front and back of a fabricated μCD. c, One labeled domain from the 4-plex μCD depicting the positions of reagent chambers, fluidic channels, pressure vents, sacrificial valve locations, reagent loading ports, and hydrophobic membrane patches. d, Schematic representation of the sacrificial valving process to enable sequential unit operations. | ||
Fluidic control testing and characterization
Reliability of the microfluidic BSC method is based upon the reproducibility of the fully-integrated μCD. To assess the architectural features and their ability to complete unit operations during discrete reactions, fluidic dye studies were completed. Fig. 4a shows the progress of one representative dye study as it progresses through each of the BSC steps, including sulphonation and deamination, bead binding, wash steps, desulphonation, and the final DNA elution. Alternating blue and yellow dye solutions were moved throughout each domain of a 4-plex disc through the requisite channels and chambers successfully, indicating fluidic control and reproducibility. A schematic detailing the placement and radial position of valves on each domain of the microdevice is depicted in Fig. S2.†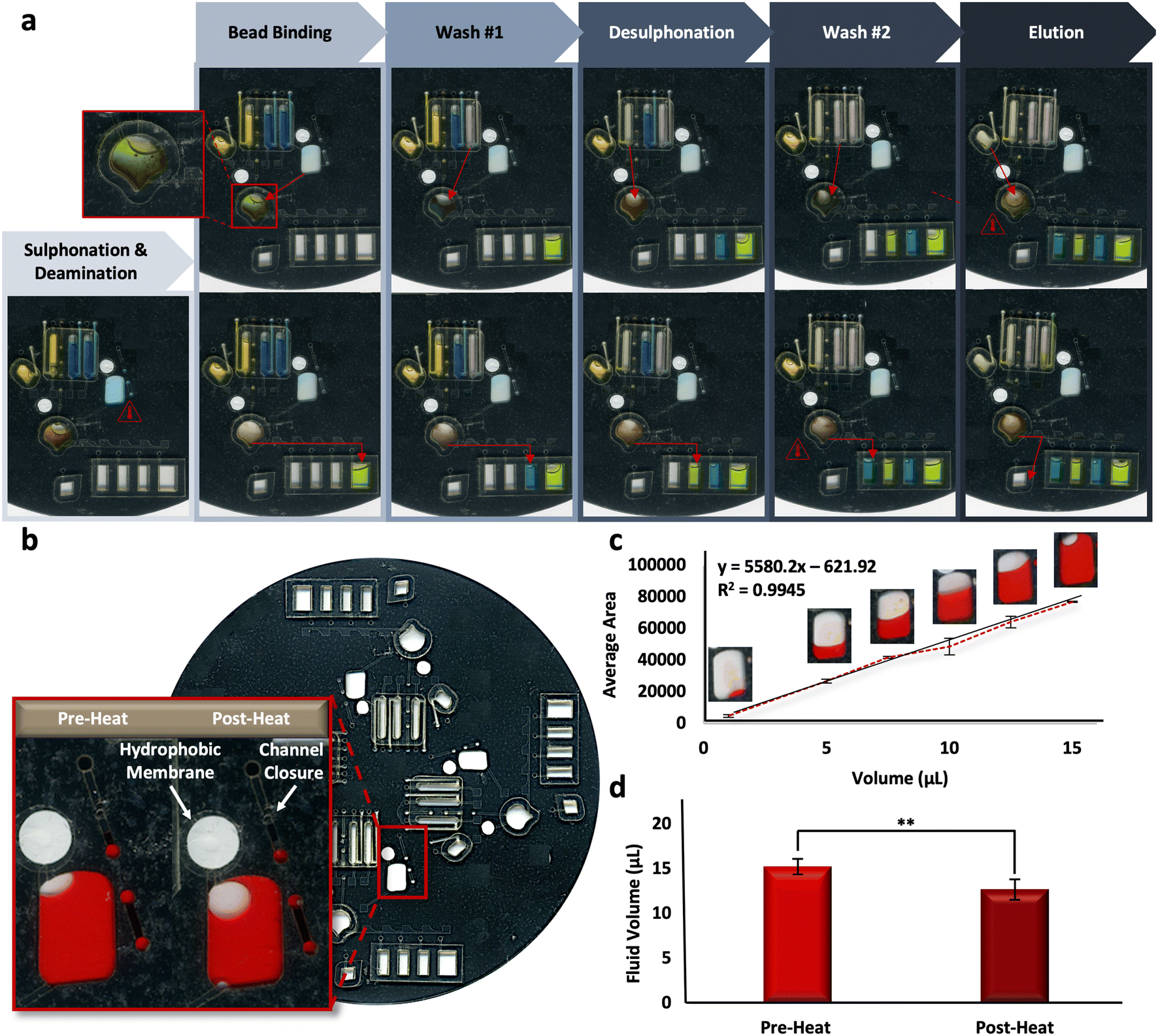 | ||
| Fig. 4 Fluidic dye studies. a, Digital scans showing one representative dye study. b, Digital scans of the bisulfite conversion chamber pre- and post-heating associated with the initial incubation steps (e.g., denaturation, followed by sulphonation and deamination). c, Calibration curve corresponding to the average pixel areas of digital scans of bisulfite conversion chambers loaded with Allura red dye. d, Quantification of fluid loss pre- and post-heating protocol. | ||
Complete adaptation to the microdevice requires that all incubations be completed on disc. Following the shortening of reaction intervals described above, it stands that the longest on-disc heating interval occurs during the sulphonation and deamination step (54 °C for 45 min), preceded by a brief denaturation in the same chamber (95 °C for 1 min). Upon visual inspection, it appeared some fluid loss was reproducibly occurring during this step (Fig. 4b). A dye study was completed to quantify this loss per a previously described protocol, known as ‘The Crop-Threshold-and-Go’ method of image segmentation and analysis.36 Here, a calibration curve correlating average pixel area and fluid volume was constructed from digital scans of bisulfite conversion chambers loaded with Allura Red dye at regular intervals, including 1, 5, 7.5, 10, 12.5, and 15 μL (R2 = 0.9945, y = 55.80.2x − 621.92) (Fig. 4c). Subsequent scans were taken of the conversion chambers pre- and post-heating, and the corresponding volumes were extrapolated from image analysis and according to their placement along the standard curve. Results indicated that ∼83% of the fluid was retained following this heated incubation step, with pre- and post-heat volumes approximated to be 15.28 ± 0.95 and 12.73 ± 1.28, respectively (Fig. 4d). We can speculate that fluid is being lost to the intermediate layers surrounding the chamber, given all outlets are closed to the external environment. In particular, the chamber vent incorporates a hydrophobic PTFE membrane to prevent fluid loss and the loading port channel is thermally occluded (e.g., ‘closed’) prior to heating (Fig. 4b, inset). Thus, we cannot confirm whether the “leaking” leads to DNA loss from the chamber or simply concentration of the DNA into a reduced aliquot of fluid.
Microdevice testing with methylation standards
To compare the performance of the conventional ‘gold-standard’ method and the optimized on-disc method, non-methylated controls were converted at equivalent concentrations and subsequently assessed for relative DNA recovery and conversion efficiency. Post-BSC, the total amount of DNA in each corresponding amplification reaction totaled 100, 10, and 1 ng (e.g., 4 ng μL−1, 400 pg μL−1, and 40 pg μL−1, respectively); unpaired t-tests of resultant Ct values were not statistically different at each concentration (α = 0.05, p-values = 0.6083, 0.0804, 0.4596, respectively), indicating similar recovery between the gold-standard and on-disc methods (Fig. 5a). Equivalent concentrations of non-methylated standards were also prepared in-tube using the microfluidic volumes and incubation parameters. Relative recovery results indicate similar recovery across conditions for samples prepared with DNA input amounts of 10 and 1 ng total (unpaired t-tests, α = 0.05, p-values = 0.5368 and 0.3693, respectively); however, the in-tube microfluidic method demonstrated markedly increased recovery compared to the on-disc method at 100 ng total (unpaired t-test, α = 0.05, p-value = <0.0001), with average Ct values of 28.89 ± 0.51 and 36.23 ± 2.89, respectively (Fig. S3†). This may indicate the potential of the microfluidic method at concentrations higher than 4 ng μL−1 with optimal microdevice performance. At this point, the microdevice provides a faster, automated BSC alternative that performs comparably in terms of DNA recovery, and with only ∼1/10th of the reagent volumes, theoretically decreasing cost at scale.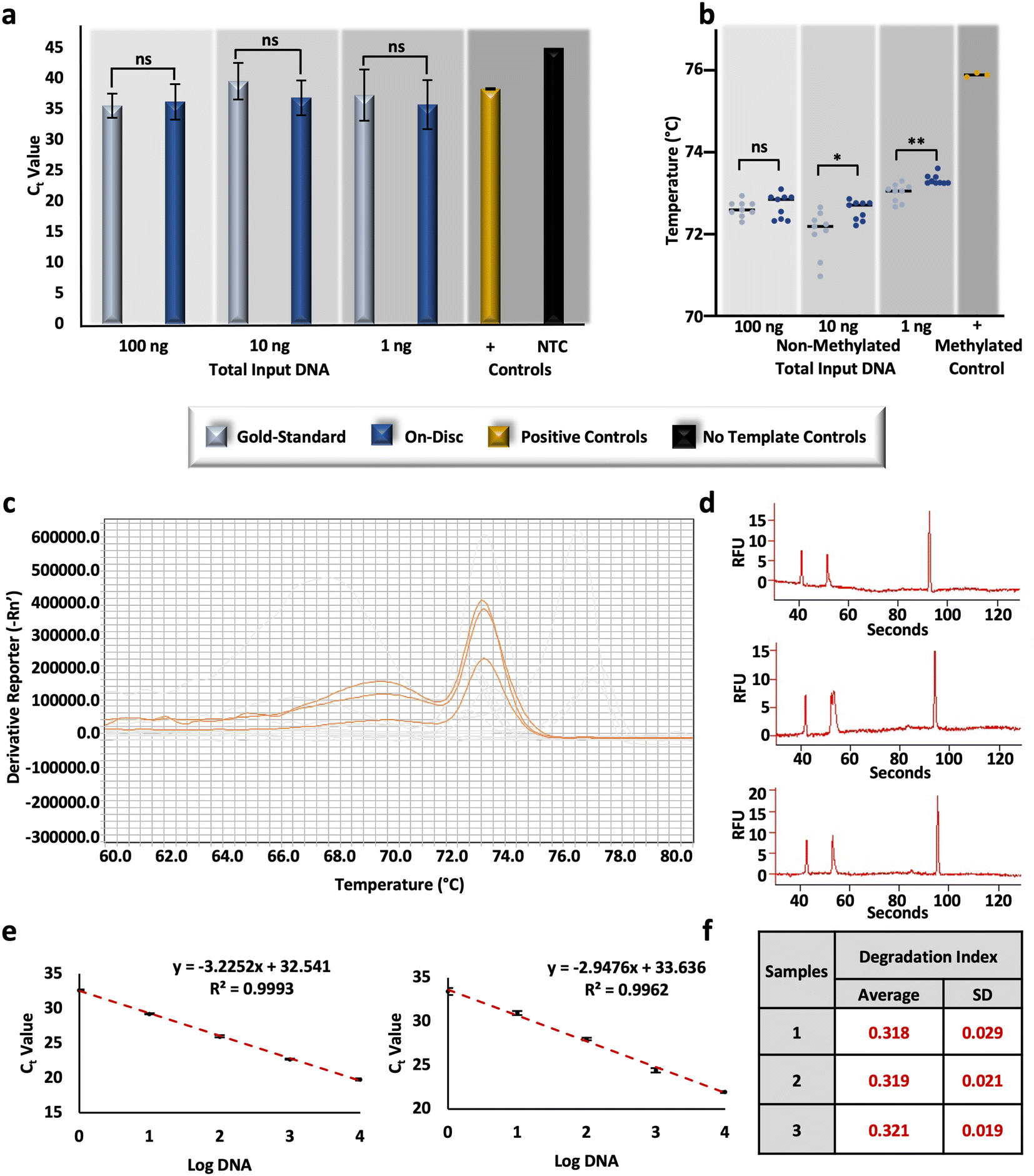 | ||
| Fig. 5 Characterization of comparative DNA recovery and conversion efficiency on the microdevice. a and b, RT-PCR and HRM results, respectively, originating from non-methylated DNA standards, bisulfite converted gold-standard approach and microfluidic ‘on-disc’ approaches. c, Melt curve results depicting the profiles from samples prepared on-disc with a total DNA input of 100 ng. d, Electropherograms resulting from the samples prepared on-disc at 100 ng total, post RT-PCR and HRM. An electrophoretic artifact, or ‘split-peak’ is observed in the second (middle) replicate. e, Standard curves constructed for large (left) and small (right) autosomal targets. f, Degradation indices calculated for three replicates prepared via the on-disc method. | ||
As before, relative conversion efficiency was assessed with HRM following the RT-PCR reaction. While no statistical difference was determined at the higher concentration (unpaired t-test, α = 0.05, p-value = 0.5477), differences were observed with the lower DNA input amounts, including 10 ng (unpaired t-test, α = 0.05, p-value = 0.0152), and 1 ng (unpaired t-test, α = 0.05, p-value = 0.0014). However, the differences between Tm values were negligible overall; on average, differences between 10 ng eluates ranged 0.55 °C ± 0.20 °C and 1 ng eluates were only different by 0.32 °C ± 0.08 °C (Fig. 5b). Comparing these results to the same concentrations of standards prepared using the in-tube microfluidic method trends reverse with no statistical difference at the lowest concentration (unpaired t-test, α = 0.05, p-value = 0.0001) and noticeable differences at the higher DNA input amounts of 100 and 10 ng total (unpaired t-tests, α = 0.05, p-values = <0.0001 and 0.0001, respectively) (Fig. S3†). At 100 ng total, the in-tube microfluidic method exhibits lower Tm values of 0.68 °C ± 0.14 °C compared to its on-disc counterpart, once again indicating the potential of the microfluidic scheme, if fully optimized to reduce fluid loss. Overall, the standard deviations, or spread, of Tm values was the lowest with the automated, on-disc method when comparing all conditions and concentrations, speaking to the reproducibility of this mode. Interestingly, a comparison of each method across concentrations, reveals a statistical difference between them (two-way ANOVA, α = 0.05, p-value = <0.0001). While this may seem peculiar, studies here are completed in a stochastic regime so as to be forensically-relevant (e.g., 4, 0.4, and 0.04 ng μL−1), where starting DNA template is known to impact the resulting Tm.39 This same trend is demonstrated later in this work, with conversion by an alternative commercial method in this same concentration range. Diving deeper into the variation across BSC preparation conditions and the estimation of conversion efficiency, at the highest DNA input amount (e.g., 100 ng total in the PCR reaction), an additional peak was reproducibly observed with HRM (Fig. 5c). Generally, multiple melt curves suggest nonspecific amplification; however, the NTCs did not indicate contamination and the additional ‘peak’ exhibited low amplitude and appeared broad and unresolved (Fig. 5c). This brings up a shortcoming of HRM, whereby an assumption is made that DNA melting is a 2-stage process resulting in only the detection of amplicons in their double- and single-stranded states. In reality, there may often be an intermediate state wherein the G/C rich portions of the amplicon maintain a double-stranded configuration and A/T rich regions disassociate first.40 To confirm this phenomenon with the FHL2 amplicons at the highest concentrations and with on-disc BSC eluates, resultant amplicons were separated via microchip electrophoresis. Results indicate the presence of only one amplicon at 133 base pairs (bp), as anticipated,28 and suggest a multi-stage melt may occur at higher concentrations with this particular target (Fig. 5d). This may also account for the variation observed here between Tm values across all sample preparation conditions at 100 ng total.
To evaluate the potential for DNA degradation resulting from conversion-related fragmentation, the Quantifiler Trio DNA Quantification kit was used. This kit is typically used in forensic DNA analysis workflows to quantify DNA, test for the contribution of male genetic material, and assess the quality of forensic samples that are often subject to environmental influences that lead to nucleic acid degradation.41,42 Degradation indices are automatically calculated by the associated software and based upon Ct values of diluted standards for large and small autosomal targets. Here, R2 values were high (>0.99) (Fig. 5e) and the associated internal PCR control (IPC) amplified as expected, indicating the amplification reaction was not affected by any inhibitors and efficiency was as expected.43 Calculated degradation indices from non-methylated DNA standards converted on-disc via the μCD method are <1, indicating that the DNA is not degraded or inhibited.43 Additionally, indices are fairly consistent between conversion replicates and indicate consistency with regard to degradation (Fig. 5f). These results are both relevant to forensic use of the workflow and confirm that degradation via the μCD method should not interfere with interpretation at this concentration (∼1.25 ng μL−1).
Comparison to an enzymatic method for cytosine deamination
In response to the aforementioned issues associated with gold-standard sodium bisulfite conversion, namely DNA fragmentation and loss, alternative methods for the conversion of cytosines for epigenetic analysis have been developed commercially.44–47 One such commercialized method forgoes chemical conversion and relies upon an apolipoprotein B mRNA-editing enzyme, catalytic peptide (APOBEC) for the deamination of cytosine to uracil, leaving modified cytosines (e.g., 5-methylcytosines and 5-hydroxymethylcytosines) intact via enzymatic modification by a ten-eleven translocation 2 (TET2) enzyme and oxidation enhancer.48,49 To compare the results from μCD dSP-BSC (Fig. 5a and b) with this alternative method for conversion, non-methylated control DNA was enzymatically converted at equivalent amounts of input DNA, as before. Fig. 6a and b show the results from duplicate enzymatic conversion reactions in terms of relative DNA recovery and conversion efficiency, respectively. Generally, relative recovery results were inconsistent in comparison with the μCD method, indicating that the microdevice method showed greater DNA recovery at total DNA input amounts of 100 ng (μCD mean Ct values 5.42 ± 2.13 lower) and lower recovery at 10 ng total (μCD mean Ct values 3.37 ± 1.26 higher) (Fig. 6a). While recovery at 1 ng total, perhaps the most forensically-relevant range, was found to show no statistical differences between average Ct values (unpaired t-test, α = 0.05, p-value = 0.4174). However, this is likely the result of stochastic differences across all samples processed within this concentration range; a direct comparison of units reveals a lower mean Ct difference of 1.28 ± 0.06 for samples prepared via the microdevice, indicating overall higher recovery (Fig. 6a). When comparing Tm values associated with the enzymatic approach, temperatures are statistically different across concentrations (one-way ANOVA, α = 0.05, p-value = <0.0001), potentially indicating that DNA input amount influences conversion efficiency (Fig. 6b). However, in estimations of conversion efficiency via HRM, the enzymatic method outperformed the microfluidic approach by a mean temperature difference of 1.02 ± 0.11 °C; these differences were also found to be statistically significant (unpaired t-test, α = 0.05, p-value = <0.0001). In total, preliminary results comparing the μCD and enzymatic approaches indicate that performance is likely dependent upon DNA input amount with regard to recovery and slightly improved in terms of conversion efficiency with the enzymatic method. Of course, the data set and analytical range tested here is relatively small and further testing is required for a true comparison. With regard to manual intervention and time at the bench, the enzymatic approach required DNA pre-processing (e.g., shearing), 11 more reagents and associated manual handling steps/tube transfers, and 6 additional hours of processing time compared to the μCD method.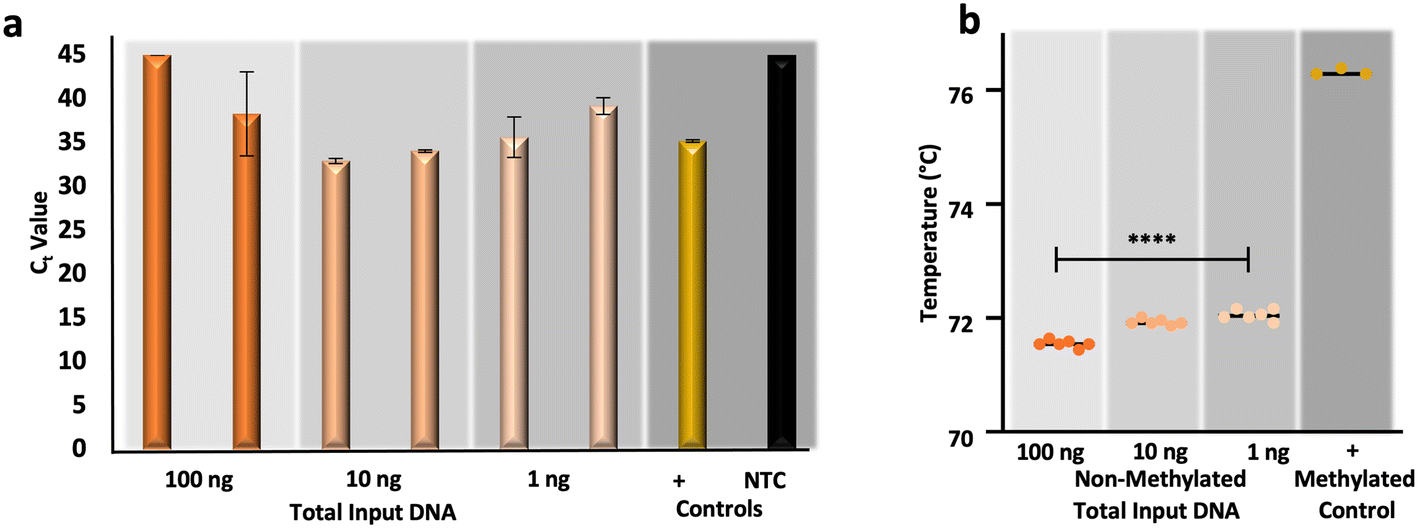 | ||
| Fig. 6 Preliminary data from an enzymatic approach to cytosine conversion. a, RT-PCR and b, HRM results, respectively, originating from enzymatically converted non-methylated DNA standards. | ||
Conclusions
The chemical modification of cytosine residues to uracil via sodium bisulfite conversion has remained largely steadfast since its conception several decades ago50 and is widely accepted to be associated with DNA degradation and loss.51 For the preponderance of epigenetic applications, this loss may be compensated for by using samples known to contain higher concentrations of nucleic acids and/or by incorporating upstream enrichment techniques to increase DNA concentration from a large volume of sample. Unfortunately, forensic casework samples are known to have limited DNA contributions that are often fragmented for a number of reasons, including limited sample deposits, environmental exposure, or sample partitioning for individualizing identification efforts, to name a few. Thus, applications in forensic epigenetics, wherein DNA is subject to an additional, deleterious sample preparation process post-extraction, may result in complete loss of the sample and are therefore not ideal for integration with the forensic workflow in their current form. Additionally, the adage of another ‘open-tube’ process with several labor-intensive pipetting steps increases time at the bench, the risk for contamination, and opportunities for errors by the analyst.We describe a microfluidic solution for forensic epigenetic sample preparation that decreases contamination risks and the potential for interoperability issues that are often associated with manual handling. By leveraging decreased, microfluidic volumes, the described method enables reduced incubation times by ∼36% compared to the gold-standard and modified in-tube approaches, and preliminary results indicate increased recovery compared to a gold-standard method. The μCD itself incorporates centrifugal force and sacrificial, laser-based valving for fluidic control and the performance of discrete unit operations, permitting automation, reproducibility, and a small overall footprint for preparation of up to four samples in parallel. The fully-integrated device does exhibit some fluid loss through uptake to the surrounding material during the longest incubation step (e.g., sulphonation and deamination) that may be associated with loss of sensitivity compared to an in-tube microfluidic approach; yet, when comparing controls converted with both gold-standard and on-disc approaches at multiple concentrations, there are no statistical differences in recovery and only negligible differences in conversion efficiency. Likewise, samples prepared via the μCD show no evidence of DNA degradation or inhibition from residual reagents (e.g., ethanol) in the converted eluate, as indicated by a commercial kit intended for forensic characterization of these particular factors. Finally, in a limited comparison of the μCD method and an alternative, enzymatic approach for cytosine conversion, the results were largely stochastic, but indicate that DNA input concentration may be a key factor of performance. Additionally, the enzymatic method necessitated shearing the DNA up front for successful conversion and required a 300× increase in time at the bench, several manual handling steps, and 11 more reagents when compared with the μCD approach. In summary, this work demonstrates progress toward a microfluidic sodium bisulfite conversion method that is more amenable to integration with the forensic DNA workflow but will benefit from further quantitative validation and characterization in the future. Looking to the future, there are a variety of methylation-based applications that would benefit from an automated and miniaturized sample preparation workflow across multiple sectors, as predictive biomarkers have been identified for cancer,52 neurodegenerative53 and psychiatric54 disorders, and therapeutic outcomes,55 to name a few.
Author contributions
R. T. and J. P. L. contributed to idea conception. R. T. and L. M. D. contributed to microfluidic design. R. T. contributed DNA recovery and conversion efficiency estimations. R. T. and R. L. N. contributed to figure development, electrophoresis results, and enzymatic conversion. R. T. and J. K. contributed to fluidic dye studies and design validation. R. T. and L. L. C. contributed to degradation study.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the National Institute of Justice (NIJ), U.S. Department of Justice (DOJ) for the Graduate Research Fellowship support (2020-R2-CX-0030), the Forensic Sciences Foundation, Inc. for the Jan S. Bashinski Criminalistics Graduate Thesis Grant, the University of Virginia (UVA) Graduate School of Arts and Sciences Council for the Research Grant, and the Achievement Rewards for College Scientists (ARCS) Foundation for Fellowship support. We would also like to recognize the International Society for Forensic Genetics (ISFG), the International Society for Applied Biological Sciences (ISABS) and the University of Virginia for their acknowledgement and support of this work.Notes and references
- J. M. Butler, Advanced Topics in Forensic DNA Typing: Methodology, Elsevier/Academic Press, Walthan, MA, 2012 Search PubMed.
- W. M. Bass, Human Osteology: A Laboratory and Field Manual, Special publication no. 2 of the Missouri Archaeological Society, Missouri Archaeological Society, Columbia, Mo, 5th edn, 2005 Search PubMed.
- J. M. Wiersema, Evolution of Forensic Anthropological Methods of Identification, Acad. Forensic. Pathol., 2016, 6(3), 361–369, DOI:10.23907/2016.038.
- E. Naito, K. Dewa, H. Yamanouchi, S. Takagi and R. Kominami, Sex Determination Using the Hypomethylation of a Human Macro-Satellite DXZ4 in Female Cells, Nucleic Acids Res., 1993, 21(10), 2533–2534, DOI:10.1093/nar/21.10.2533.
- C. Li, S. Zhang, T. Que, L. Li and S. Zhao, Identical but Not the Same: The Value of DNA Methylation Profiling in Forensic Discrimination within Monozygotic Twins, Forensic Sci. Int. Genet. Suppl. Ser., 2011, 3(1), e337–e338, DOI:10.1016/j.fsigss.2011.09.031.
- D. Frumkin, A. Wasserstrom, B. Budowle and A. Davidson, DNA Methylation-Based Forensic Tissue Identification, Forensic Sci. Int.: Genet., 2011, 5(5), 517–524, DOI:10.1016/j.fsigen.2010.12.001.
- A. Vidaki and M. Kayser, From Forensic Epigenetics to Forensic Epigenomics: Broadening DNA Investigative Intelligence, Genome Biol., 2017, 18(1), 238, DOI:10.1186/s13059-017-1373-1.
- S. Bocklandt, W. Lin, M. E. Sehl, F. J. Sánchez, J. S. Sinsheimer, S. Horvath and E. Vilain, Epigenetic Predictor of Age, PLoS One, 2011, 6(6), e14821, DOI:10.1371/journal.pone.0014821.
- C. Maulani and E. I. Auerkari, Age Estimation Using DNA Methylation Technique in Forensics: A Systematic Review, Egypt. J. Forensic Sci., 2020, 10(1), 38, DOI:10.1186/s41935-020-00214-2.
- A. Freire-Aradas, C. Phillips and M. V. Lareu, Forensic Individual Age Estimation with DNA: From Initial Approaches to Methylation Tests, Forensic Sci. Rev., 2017, 29(2), 121–144 CAS.
- R. Zbieć-Piekarska, M. Spólnicka, T. Kupiec, Ż. Makowska, A. Spas, A. Parys-Proszek, K. Kucharczyk, R. Płoski and W. Branicki, Examination of DNA Methylation Status of the ELOVL2 Marker May Be Useful for Human Age Prediction in Forensic Science, Forensic Sci. Int.: Genet., 2015, 14, 161–167, DOI:10.1016/j.fsigen.2014.10.002.
- R. Zbieć-Piekarska, M. Spólnicka, T. Kupiec, A. Parys-Proszek, Ż. Makowska, A. Pałeczka, K. Kucharczyk, R. Płoski and W. Branicki, Development of a Forensically Useful Age Prediction Method Based on DNA Methylation Analysis, Forensic Sci. Int.: Genet., 2015, 17, 173–179, DOI:10.1016/j.fsigen.2015.05.001.
- A. Freire-Aradas, C. Phillips, A. Mosquera-Miguel, L. Girón-Santamaría, A. Gómez-Tato, M. Casares de Cal, J. Álvarez-Dios, J. Ansede-Bermejo, M. Torres-Español, P. M. Schneider, E. Pośpiech, W. Branicki, Á. Carracedo and M. V. Lareu, Development of a Methylation Marker Set for Forensic Age Estimation Using Analysis of Public Methylation Data and the Agena Bioscience EpiTYPER System, Forensic Sci. Int.: Genet., 2016, 24, 65–74, DOI:10.1016/j.fsigen.2016.06.005.
- J.-L. Park, J. H. Kim, E. Seo, D. H. Bae, S.-Y. Kim, H.-C. Lee, K.-M. Woo and Y. S. Kim, Identification and Evaluation of Age-Correlated DNA Methylation Markers for Forensic Use, Forensic Sci. Int.: Genet., 2016, 23, 64–70, DOI:10.1016/j.fsigen.2016.03.005.
- J. Naue, H. C. J. Hoefsloot, O. R. F. Mook, L. Rijlaarsdam-Hoekstra, M. C. H. van der Zwalm, P. Henneman, A. D. Kloosterman and P. J. Verschure, Chronological Age Prediction Based on DNA Methylation: Massive Parallel Sequencing and Random Forest Regression, Forensic Sci. Int.: Genet., 2017, 31, 19–28, DOI:10.1016/j.fsigen.2017.07.015.
- L. Feng, F. Peng, S. Li, L. Jiang, H. Sun, A. Ji, C. Zeng, C. Li and F. Liu, Systematic Feature Selection Improves Accuracy of Methylation-Based Forensic Age Estimation in Han Chinese Males, Forensic Sci. Int.: Genet., 2018, 35, 38–45, DOI:10.1016/j.fsigen.2018.03.009.
- S. R. Hong, S.-E. Jung, E. H. Lee, K.-J. Shin, W. I. Yang and H. Y. Lee, DNA Methylation-Based Age Prediction from Saliva: High Age Predictability by Combination of 7 CpG Markers, Forensic Sci. Int.: Genet., 2017, 29, 118–125, DOI:10.1016/j.fsigen.2017.04.006.
- S. R. Hong, K.-J. Shin, S.-E. Jung, E. H. Lee and H. Y. Lee, Platform-Independent Models for Age Prediction Using DNA Methylation Data, Forensic Sci. Int.: Genet., 2019, 38, 39–47, DOI:10.1016/j.fsigen.2018.10.005.
- S.-E. Jung, S. M. Lim, S. R. Hong, E. H. Lee, K.-J. Shin and H. Y. Lee, DNA Methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 Genes for Age Prediction from Blood, Saliva, and Buccal Swab Samples, Forensic Sci. Int.: Genet., 2019, 38, 1–8, DOI:10.1016/j.fsigen.2018.09.010.
- H. Y. Lee, S.-E. Jung, Y. N. Oh, A. Choi, W. I. Yang and K.-J. Shin, Epigenetic Age Signatures in the Forensically Relevant Body Fluid of Semen: A Preliminary Study, Forensic Sci. Int.: Genet., 2015, 19, 28–34, DOI:10.1016/j.fsigen.2015.05.014.
- L. Li, F. Song, Y. Huang, H. Zhu and Y. Hou, Age-Associated DNA Methylation Determination of Semen by Pyrosequencing in Chinese Han Population, Forensic Sci. Int. Genet. Suppl. Ser., 2017, 6, e99–e100, DOI:10.1016/j.fsigss.2017.09.042.
- C. Giuliani, E. Cilli, M. G. Bacalini, C. Pirazzini, M. Sazzini, G. Gruppioni, C. Franceschi, P. Garagnani and D. Luiselli, Inferring Chronological Age from DNA Methylation Patterns of Human Teeth: Inferring Age From Dna Methylation Of Human Teeth, Am. J. Phys. Anthropol., 2016, 159(4), 585–595, DOI:10.1002/ajpa.22921.
- N.-T. Nguyen, S. T. Wereley and S. A. M. Shaegh, Fundamentals and Applications of Microfluidics, Artech House integrated microsystems series, Artech House, Norwood, Massachusetts, 3rd edn, 2019 Search PubMed.
- D. R. Reyes, D. Iossifidis, P.-A. Auroux and A. Manz, Micro Total Analysis Systems. 1. Introduction, Theory, and Technology, Anal. Chem., 2002, 74(12), 2623–2636, DOI:10.1021/ac0202435.
- D. J. Shin, A. Stark and T. H. Wang, Droplet Bisulfite Conversion Platform for Epigenetic Cancer Biomarker Detection, in 2013 Transducers & Eurosensors XXVII: The 17th International Conference on Solid-State Sensors, Actuators and Microsystems (Transducers & Eurosensors XXVII), IEEE, Barcelona, Spain, 2013, pp. 2181–2184, DOI:10.1109/Transducers.2013.6627235.
- A. Stark, D. J. Shin, T. Pisanic, K. Hsieh and T.-H. Wang, A Parallelized Microfluidic DNA Bisulfite Conversion Module for Streamlined Methylation Analysis, Biomed. Microdevices, 2016, 18(1), 5, DOI:10.1007/s10544-015-0029-8.
- J. Yoon, M. K. Park, T. Y. Lee, Y.-J. Yoon and Y. Shin, LoMA-B: A Simple and Versatile Lab-on-a-Chip System Based on Single-Channel Bisulfite Conversion for DNA Methylation Analysis, Lab Chip, 2015, 15(17), 3530–3539, 10.1039/C5LC00458F.
- Y. Hamano, S. Manabe, C. Morimoto, S. Fujimoto, M. Ozeki and K. Tamaki, Forensic Age Prediction for Dead or Living Samples by Use of Methylation-Sensitive High Resolution Melting, Leg. Med., 2016, 21, 5–10, DOI:10.1016/j.legalmed.2016.05.001.
- R. L. Nouwairi, L. L. Cunha, R. Turiello, O. Scott, J. Hickey, S. Thomson, S. Knowles, J. D. Chapman and J. P. Landers, Ultra-Rapid Real-Time Microfluidic RT-PCR Instrument for Nucleic Acid Analysis, Lab Chip, 2022, 22, 3424–3435, 10.1039/D2LC00495J.
- L. M. Dignan, M. S. Woolf, C. J. Tomley, A. Q. Nauman and J. P. Landers, Multiplexed Centrifugal Microfluidic System for Dynamic Solid-Phase Purification of Polynucleic Acids Direct from Buccal Swabs, Anal. Chem., 2021, 93(19), 7300–7309, DOI:10.1021/acs.analchem.1c00842.
- R. Turiello, L. M. Dignan, B. Thompson, M. Poulter, J. Hickey, J. Chapman and J. P. Landers, Centrifugal Microfluidic Method for Enrichment and Enzymatic Extraction of Severe Acute Respiratory Syndrome Coronavirus 2 RNA, Anal. Chem., 2022, 94(7), 3287–3295, DOI:10.1021/acs.analchem.1c05215.
- M. S. Woolf, L. M. Dignan, H. M. Lewis, C. J. Tomley, A. Q. Nauman and J. P. Landers, Optically-Controlled Closable Microvalves for Polymeric Centrifugal Microfluidic Devices, Lab Chip, 2020, 20(8), 1426–1440, 10.1039/C9LC01187K.
- J. L. Garcia-Cordero, D. Kurzbuch, F. Benito-Lopez, D. Diamond, L. P. Lee and A. J. Ricco, Optically Addressable Single-Use Microfluidic Valves by Laser Printer Lithography, Lab Chip, 2010, 10(20), 2680, 10.1039/c004980h.
- J. A. DuVall, D. Le Roux, A.-C. Tsuei, B. L. Thompson, C. Birch, J. Li, D. A. Nelson, D. L. Mills, M. M. Ewing, R. S. McLaren, D. R. Storts, B. E. Root and J. P. Landers, A Rotationally-Driven Polyethylene Terephthalate Microdevice with Integrated Reagent Mixing for Multiplexed PCR Amplification of DNA, Anal. Methods, 2016, 8(40), 7331–7340, 10.1039/C6AY01984F.
- B. L. Thompson, Y. Ouyang, G. R. M. Duarte, E. Carrilho, S. T. Krauss and J. P. Landers, Inexpensive, Rapid Prototyping of Microfluidic Devices Using Overhead Transparencies and a Laser Print, Cut and Laminate Fabrication Method, Nat. Protoc., 2015, 10(6), 875–886, DOI:10.1038/nprot.2015.051.
- M. S. Woolf, L. M. Dignan, A. T. Scott and J. P. Landers, Digital Postprocessing and Image Segmentation for Objective Analysis of Colorimetric Reactions, Nat. Protoc., 2021, 16(1), 218–238, DOI:10.1038/s41596-020-00413-0.
- S. Cho, S.-E. Jung, S. R. Hong, E. H. Lee, J. H. Lee, S. D. Lee and H. Y. Lee, Independent Validation of DNA-Based Approaches for Age Prediction in Blood, Forensic Sci. Int.: Genet., 2017, 29, 250–256, DOI:10.1016/j.fsigen.2017.04.020.
- H. G. Hernández, M. Y. Tse, S. C. Pang, H. Arboleda and D. A. Forero, Optimizing Methodologies for PCR-Based DNA Methylation Analysis, BioTechniques, 2013, 55(4), 181–197, DOI:10.2144/000114087.
- Applied Biosystems, A Guide to High Resolution Melting (HRM) Analysis, 2010, https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms_070283.pdf Search PubMed.
- N. Downey, Z. Dwight and C. Wittwer, Explaining Multiple Peaks in qPCR Melt Curve Analysis, https://www.idtdna.com/pages/education/decoded/article/interpreting-melt-curves-an-indicator-not-a-diagnosis Search PubMed.
- N. Gouveia, P. Brito, A. Serra, F. Balsa, L. Andrade, M. São Bento, P. Cunha, V. Bogas, V. Lopes and M. J. Porto, Validation of Quantifiler ® Trio DNA Quantification Kit in Forensic Samples, Forensic Sci. Int. Genet. Suppl. Ser., 2015, 5, e24–e25, DOI:10.1016/j.fsigss.2015.09.010.
- S. Vernarecci, E. Ottaviani, A. Agostino, E. Mei, L. Calandro and P. Montagna, Quantifiler ® Trio Kit and Forensic Samples Management: A Matter of Degradation, Forensic Sci. Int.: Genet., 2015, 16, 77–85, DOI:10.1016/j.fsigen.2014.12.005.
- A. Lackey, How To Evaluate Forensic DNA Quality With Quantifiler Trio DNA Quantification Kit, ThermoFisher Scientific, https://www.thermofisher.com/blog/behindthebench/how-to-evaluate-forensic-dna-quality-with-quantifiler-trio-dna-quantification-kit/ Search PubMed.
- M. Yu, G. C. Hon, K. E. Szulwach, C.-X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen, B. Park, J.-H. Min, P. Jin, B. Ren and C. He, Base-Resolution Analysis of 5-Hydroxymethylcytosine in the Mammalian Genome, Cell, 2012, 149(6), 1368–1380, DOI:10.1016/j.cell.2012.04.027.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Quantitative Sequencing of 5-Methylcytosine and 5-Hydroxymethylcytosine at Single-Base Resolution, Science, 2012, 336(6083), 934–937, DOI:10.1126/science.1220671.
- L. Zhang, K. E. Szulwach, G. C. Hon, C.-X. Song, B. Park, M. Yu, X. Lu, Q. Dai, X. Wang, C. R. Street, H. Tan, J.-H. Min, B. Ren, P. Jin and C. He, Tet-Mediated Covalent Labelling of 5-Methylcytosine for Its Genome-Wide Detection and Sequencing, Nat. Commun., 2013, 4(1), 1517, DOI:10.1038/ncomms2527.
- E. K. Schutsky, J. E. DeNizio, P. Hu, M. Y. Liu, C. S. Nabel, E. B. Fabyanic, Y. Hwang, F. D. Bushman, H. Wu and R. M. Kohli, Nondestructive, Base-Resolution Sequencing of 5-Hydroxymethylcytosine Using a DNA Deaminase, Nat. Biotechnol., 2018, 36(11), 1083–1090, DOI:10.1038/nbt.4204.
- R. Vaisvila, V. K. C. Ponnaluri, Z. Sun, B. W. Langhorst, L. Saleh, S. Guan, N. Dai, M. A. Campbell, B. S. Sexton, K. Marks, M. Samaranayake, J. C. Samuelson, H. E. Church, E. Tamanaha, I. R. Corrêa, S. Pradhan, E. T. Dimalanta, T. C. Evans, L. Williams and T. B. Davis, Enzymatic Methyl Sequencing Detects DNA Methylation at Single-Base Resolution from Picograms of DNA, Genome Res., 2021, 31(7), 1280–1289, DOI:10.1101/gr.266551.120.
- L. Williams, Y. Bei, H. E. Church, N. Dai, E. T. Dimalanta, L. M. Ettwiller, T. C. Evans, Jr., B. W. Langhorst, J. G. Borgaro, S. Guan, K. Marks, J. F. Menin, N. M. Nichols, V. K. C. Ponnaluri, L. Saleh, M. Samaranayake, B. S. Sexton, Z. Sun, E. Tamanaha, R. Vaisvilla, E. Yigit and T. B. Davis, Enzymatic Methyl-Seq: The Next Generation of Methylome Analysis, New England Biolabs, https://www.neb.com/tools-and-resources/feature-articles/enzymatic-methyl-seq-the-next-generation-of-methylome-analysis Search PubMed.
- R. Shapiro, R. Servis and M. Welcher, Reactions of Uracil and Cytosine Derivatives with Sodium Bisulfite. A Specific Deamination Method, J. Am. Chem. Soc., 1970, 92(2), 422–424, DOI:10.1021/ja00705a626.
- C. A. Leontiou, M. D. Hadjidaniel, P. Mina, P. Antoniou, M. Ioannides and P. C. Patsalis, Bisulfite Conversion of DNA: Performance Comparison of Different Kits and Methylation Quantitation of Epigenetic Biomarkers That Have the Potential to Be Used in Non-Invasive Prenatal Testing, PLoS One, 2015, 10(8), e0135058, DOI:10.1371/journal.pone.0135058.
- W. J. Locke, D. Guanzon, C. Ma, Y. J. Liew, K. R. Duesing, K. Y. C. Fung and J. P. Ross, DNA Methylation Cancer Biomarkers: Translation to the Clinic, Front. Genet., 2019, 10, 1150, DOI:10.3389/fgene.2019.01150.
- The Australian Imaging Biomarkers and Lifestyle study, The Alzheimer's Disease Neuroimaging Initiative, M. F. Nabais, S. M. Laws, T. Lin, C. L. Vallerga, N. J. Armstrong, I. P. Blair, J. B. Kwok, K. A. Mather, G. D. Mellick, P. S. Sachdev, L. Wallace, A. K. Henders, R. A. J. Zwamborn, P. J. Hop, K. Lunnon, E. Pishva, J. A. Y. Roubroeks, H. Soininen, M. Tsolaki, P. Mecocci, S. Lovestone, I. Kłoszewska, B. Vellas, S. Furlong, F. C. Garton, R. D. Henderson, S. Mathers, P. A. McCombe, M. Needham, S. T. Ngo, G. Nicholson, R. Pamphlett, D. B. Rowe, F. J. Steyn, K. L. Williams, T. J. Anderson, S. R. Bentley, J. Dalrymple-Alford, J. Fowder, J. Gratten, G. Halliday, I. B. Hickie, M. Kennedy, S. J. G. Lewis, G. W. Montgomery, J. Pearson, T. L. Pitcher, P. Silburn, F. Zhang, P. M. Visscher, J. Yang, A. J. Stevenson, R. F. Hillary, R. E. Marioni, S. E. Harris, I. J. Deary, A. R. Jones, A. Shatunov, A. Iacoangeli, W. Van Rheenen, L. H. Van Den Berg, P. J. Shaw, C. E. Shaw, K. E. Morrison, A. Al-Chalabi, J. H. Veldink, E. Hannon, J. Mill, N. R. Wray and A. F. McRae, Meta-Analysis of Genome-Wide DNA Methylation Identifies Shared Associations across Neurodegenerative Disorders, Genome Biol., 2021, 22(1), 90, DOI:10.1186/s13059-021-02275-5.
- Z. Shirvani-Farsani, Z. Maloum, Z. Bagheri-Hosseinabadi, N. Vilor-Tejedor and I. Sadeghi, DNA Methylation Signature as a Biomarker of Major Neuropsychiatric Disorders, J. Psychiatr. Res., 2021, 141, 34–49, DOI:10.1016/j.jpsychires.2021.06.013.
- S. Romero-Garcia, H. Prado-Garcia and A. Carlos-Reyes, Role of DNA Methylation in the Resistance to Therapy in Solid Tumors, Front. Oncol., 2020, 10, 1152, DOI:10.3389/fonc.2020.01152.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00867c |
| This journal is © The Royal Society of Chemistry 2024 |