
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterplay between coordination sphere engineering and properties of nickel diketonate–diamine complexes as vapor phase precursors for the growth of NiO thin films†
Mattia
Benedet
ab,
Davide
Barreca
 *b,
Ettore
Fois
c,
Roberta
Seraglia
b,
Gloria
Tabacchi
*c,
Marco
Roverso
ab,
Gioele
Pagot
d,
Cristiano
Invernizzi
c,
Alberto
Gasparotto
ab,
Alexandra A.
Heidecker
e,
Alexander
Pöthig
e,
Emanuela
Callone
f,
Sandra
Dirè
f,
Sara
Bogialli
ab,
Vito
Di Noto
d and
Chiara
Maccato
ab
*b,
Ettore
Fois
c,
Roberta
Seraglia
b,
Gloria
Tabacchi
*c,
Marco
Roverso
ab,
Gioele
Pagot
d,
Cristiano
Invernizzi
c,
Alberto
Gasparotto
ab,
Alexandra A.
Heidecker
e,
Alexander
Pöthig
e,
Emanuela
Callone
f,
Sandra
Dirè
f,
Sara
Bogialli
ab,
Vito
Di Noto
d and
Chiara
Maccato
ab
aDepartment of Chemical Sciences – Padova University and INSTM, Via Marzolo 1, 35131 Padova, Italy
bCNR-ICMATE and INSTM – Department of Chemical Sciences – Padova University, Via Marzolo 1, and Corso Stati Uniti 4, 35127 Padova, Italy. E-mail: davide.barreca@unipd.it
cDepartment of Science and High Technology – Insubria University and INSTM, Via Valleggio 11, 22100 Como, Italy. E-mail: gloria.tabacchi@uninsubria.it
dSection of Chemistry for the Technology (ChemTech), Department of Industrial Engineering – Padova University and INSTM, Via Marzolo 9, 35131 Padova, Italy
eCatalysis Research Center & Department of Chemistry – Technische Universität München, Lichtenbergstr. 4, 85747 Garching, Germany
f“Klaus Müller” Magnetic Resonance Laboratory, Department of Industrial Engineering – Trento University, Via Sommarive 9, 38123 Trento, Italy
First published on 24th May 2023
Abstract
NiO-based films and nanostructured materials have received increasing attention for a variety of technological applications. Among the possible strategies for their fabrication, atomic layer deposition (ALD) and chemical vapor deposition (CVD), featuring manifold advantages of technological interest, represent appealing molecule-to-material routes for which a rational precursor design is a critical step. In this context, the present study is focused on the coordination sphere engineering of three heteroleptic Ni(II) β-diketonate–diamine adducts of general formula [NiL2TMEDA] [L = 1,1,1-trifluoro-2,4-pentanedionate (tfa), 2,2-dimethyl-6,6,7,7,8,8,8-heptafluoro-3,5-octanedionate (fod) or 2,2,6,6-tetramethyl-3,5-heptanedionate (thd), and TMEDA = N,N,N′,N′-tetramethylethylenediamine]. Controlled variations in the diketonate structure are pursued to investigate the influence of steric hindrance and fluorination degree on the chemico-physical characteristics of the compounds. A multi-technique investigation supported by density functional calculations highlights that all complexes are air-insensitive and monomeric and that their thermal properties and fragmentation patterns are directly dependent on functional groups in the diketonate ligands. Preliminary thermal CVD experiments demonstrate the precursors’ suitability for the obtainment of NiO films endowed with flat and homogeneous surfaces, paving the way to future implementation for CVD end-uses.
Introduction
Thin films and nanostructures based on nickel(II) oxide, a multi-functional and transparent p-type semiconductor (EG ≈ 3.7 eV) with high chemical stability,1–5 have been extensively studied due to their manifold of attractive properties for a large variety of applications.1,6 In addition to its use in light emitting diodes (LEDs)7 and electrochromic8 and solar cell devices,9 NiO has been investigated as a (photo)-10,11 and electrocatalyst12–15 for various processes of technological interest. Among the fabrication routes to supported NiO thin films/nanoarchitectures,11,16–21 atomic layer deposition (ALD)22–27 and chemical vapor deposition (CVD)1,6,9,12–14,28,29 stand out as preferred choices thanks to a plethora of benefits, encompassing conformal step coverage even on complex 3D substrates, potential upscaling, and availability of many degrees of freedom to tailor material physicochemical properties.29–32 Since such advantages are eased by chemical surface reactions of a molecular precursor and a co-reactant, the precursor's characteristics play a key role in determining the resulting system features.12,30,33,34 In this regard, it is important to recall that, different from the case of CVD, in ALD the precursor and the co-reactant are never present simultaneously in the reactor, but are inserted through separate sequential pulses, during which each of them reacts with the surface in a self-limiting way.35,36 Hence, a different reaction mechanism is expected for the same precursor under CVD vs. ALD conditions. Nonetheless, both processes require molecular sources, ensuring good volatility, clean decomposition, and tailored reactivity.To date, a number of Ni precursors for CVD/ALD applications have been developed and tested. They include cyclopentadienyl Ni(II) derivatives,2,4,5,7,10,13 bis(2,4-pentanedionato)Ni(II) [Ni(acac)2],14,22 and bis(2,2,6,6-tetramethyl-3,5-heptanedionato)Ni(II) [Ni(thd)2].24,25 Nevertheless, the latter two compounds suffer from relatively poor gas phase stability23 and present drawbacks associated with the formation of oligomers,37 the presence of coordinated water molecules, and the high melting points (≥220 °C).6,22,34,37,38 Other studies have focused on Ni ketoiminate,12,23,39 guanidinate,40 propanolate9,26 butanolate,41 and imino-carbonylenolato derivatives,28 as well as bis(dimethylglyoximato)23 and bis(N-propyl-3-methylsalicylaldimine) complexes.6 These two compounds, nonetheless, required high volatilization temperatures, and/or led to film contamination.
The addition of a Lewis base is a well-established strategy to suppress the tendency of the compounds to oligomerize and the coordination of water molecules to the metal center.37 In this regard, research works have been focused on β-diketonate derivatives containing either TMEDA (N,N,N′,N′-tetramethylethylenediamine)1,27 or PDA (1,3-diaminopropane).42,43 Yet, some of these compounds required high vaporization temperatures,1 and there is still a dearth of CVD/ALD Ni precursors combining high volatility, improved shelf-stability, and clean decomposition pathways.12 To this aim, an amenable approach involves the use of fluorinated ligands to suppress intermolecular interactions,31,33 which has led to the preparation of [Ni(hfa)2TMEDA] and [Ni(tta)2TMEDA] (hfa = 1,1,1,5,5,5-hexafluoro-2,4-pentanedionate; tta = 2-thenoyltrifluoroacetonate).29,34
In the course of our studies, we have focused on a detailed chemico-physical investigation of first-row transition metal(II) hfa–TMEDA adducts of manganese,44,45 iron,46,47 cobalt,30,48 copper,49–51 and zinc.52,53 The favorable properties of these monomeric metal complexes make them promising molecular precursors for the vapor phase preparation of the corresponding oxides. In addition, as demonstrated for Mn,44,45 Fe,46,47 and Co,30,31 the compound features and reactivity depend on both the metal center's nature and the variations in the diketonate structure from hfa to tfa (1,1,1-trifluoro-2,4-pentanedionate). Based on such results, the present study is devoted to the coordination sphere engineering of Ni(II) β-diketonate-TMEDA compounds by pursuing variations of the ligand fluorination degree and steric hindrance. In particular, three heteroleptic adducts, [Ni(tfa)2TMEDA] (1), [Ni(fod)2TMEDA] (2) (fod = 2,2-dimethyl-6,6,7,7,8,8,8-heptafluoro-3,5-octanedionate), and [Ni(thd)2TMEDA] (3), were synthesized and characterized by a joint multi-technique approach, focusing on their structure/property interplay. It is worth observing that although the preparation of 1 and 3 has already been reported,37,38 only some data on their structure and thermal behavior are available so far. CVD validation of 1 yielded Ni films, requiring an ex situ high-temperature annealing process for the conversion into NiO,37 whereas the use of 3 as a CVD/ALD precursor has not yet been attempted. Furthermore, to the best of our knowledge, 2 was never reported, and a theoretical–experimental characterization of all three compounds is completely missing so far.
Herein, we have undertaken for the first time a comprehensive multi-technique investigation to examine the interplay between the diketonate hindrance/fluorination degree and the volatility, stability, and gas-phase fragmentation of the target Ni complexes. Particular attention has been paid to the rational analysis of differences and analogies in the compounds’ structure, bonding, and reactivity. Finally, preliminary data on the compound functional validation in thermal CVD experiments are also presented and discussed, along with structural, compositional, and morphological analyses of the fabricated nickel(II) oxide films.
Experimental
Precursors’ synthesis
All manipulations were carried out on open benches and at ambient pressure. NiCl2·6H2O (98%, Thermo Scientific), Htfa (98%, Sigma-Aldrich), Hfod (98%, Aldrich), Hthd (98%, abcr), and TMEDA (99%, Strem Chemicals) were used as received. The syntheses were performed starting from a NiCl2·6H2O solution in deionized water [2.76 g (11.6 mmol) in 80 mL; (I)].The obtained compound batches could be stored under ambient conditions and readily handled in air without any degradation.
Precursors’ characterization
Elemental analyses were performed on a Fisons Carlo Erba EA1108 instrument. X-ray diffraction data were collected on a Bruker D8 Venture system equipped with a Bruker Photon II detector, a Helios mirror optic monochromator and a Mo IμS microsource (λ = 0.71073 Å). Crystal structures were solved and refined using the Bruker Software Suite APEX4 and implemented programs (SHELXT and SHELXL-2019/1 in conjunction with SHELXLE).54–56 Additional details are provided in the ESI.† CCDC reference numbers: 2245881 [Ni(tfa)2TMEDA] (1) and 2245880 [Ni(thd)2TMEDA] (3).†1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 400WB spectrometer (400.13 MHz) equipped with a BBO 5 mm probe, under the following conditions: 1H: π/6 3.5 μs, recycle delay 1 s, and 16 scans. 13C analyses were carried out with inverse gated decoupling (π/6 2 μs, recycle delay 1 s, and 16k scans). The compounds were diluted in both CDCl3 and acetone-d6. Optical absorption spectra were recorded by using a Varian Cary 50 spectrophotometer (spectral bandwidth = 1 nm) on 1.50 × 10−5 M ethanol solutions of the target complexes, using quartz cuvettes (optical path = 1 cm). Electron ionization mass spectrometry (EI-MS) analyses were carried out using a gas chromatograph (Trace 1300, Thermo Scientific) coupled with a TSQ 8000 Evo triple quadrupole mass spectrometer (Thermo Scientific) equipped with a 70 eV EI source. Thermogravimetric (TG) analyses were performed in air and nitrogen at ambient pressure using a NETZSCH TG 209F1 Libra instrument (sample weight ≈ 4.5 mg; heating rate = 10 °C min−1). High-resolution isothermal studies were performed in air by heating each sample at a fixed temperature (ramp rate = 10 °C min−1), and measuring weight loss as a function of time. Melting and boiling points were determined by differential scanning calorimetry (DSC) analyses, performed with a Netzsch Phoenix DSC 204 instrument (heating rate = 10 °C min−1; ≈6 mg of each compound). Analogous results were obtained for fresh and aged batches, thus enabling one to rule out detrimental degradation processes.Computational methods
Complexes 1–3 were modelled by density functional theory (DFT), using the ωb97XD approximation57 for exchange and correlation functionals and the Gaussian 09 code.58 For Ni, Stuttgart–Dresden ECP pseudopotential and a basis set were used.59 For all the other atoms, the chosen basis set was D95 V(d).60 This combination has already enabled one to properly reproduce physico-chemical properties of other β-diketonate-TMEDA compounds.52,53 The computed optimized geometries of 1–3 all showed positive (harmonic) frequencies and triplet spin multiplicity. All the reported energy differences included the zero-point-energy correction. The compound optical absorption spectra were calculated by time dependent (TD)-DFT at the CAM-B3LYP/D95++(2d,p) theory level;61 TD-DFT excitations (40 in all cases) were calculated using a polarizable continuum model for the ethanol solvent,62 and plotted using a Gaussian broadening (2 nm) to obtain the simulated spectra.Thin film fabrication and characterization
CVD of NiO films from the synthesized precursors was performed in a home-made cold wall reactor endowed with a resistively heated susceptor, adopting the same operating conditions apart from the vaporization temperature [75 °C, 1; 80 °C, 2; 120 °C, 3]. In each experiment, the used complex was heated in an external reservoir and transported into the reactor by a 100 sccm O2 flow. An additional oxygen flow (rate = 100 sccm) was independently introduced into the deposition chamber (total pressure = 10.0 mbar). Growth processes were performed for 2 h at 400 °C on pre-cleaned31 Si(100) substrates (MEMC®; 1 × 1 cm2). The films were characterized by X-ray diffraction (XRD), atomic force microscopy (AFM), field emission-scanning electron microscopy (FE-SEM), energy dispersive X-ray spectroscopy (EDXS), and X-ray photoelectron spectroscopy (see § S8.1 and S8.2† for details).Results & discussion
Synthesis and characterization of the [NiL2TMEDA] compounds
The eventual applicability of metal complexes as CVD molecular precursors is directly dependent on the possibility of obtaining them by a straightforward and potentially scalable route, followed by an easy purification.12 In the present work, Ni(II) β-diketonate-diamine complexes were synthesized through the reaction of metal chloride with two equivalents of diketonate ligands in alkaline solutions, followed by the introduction of one equivalent of TMEDA (Scheme 1). The synthesis can be easily conducted at room temperature under ordinary laboratory conditions and does not require exceedingly long reaction times or the separation of the bis-diketonate [NiL2] adduct, at variance with previous studies.37,38 The resulting [NiL2TMEDA] complexes, obtained in high yields, are non-hygroscopic and can be easily handled in air. The compounds are insoluble in water and relatively soluble in different solvents, such as acetone, acetonitrile, dichloromethane, chloroform, and alcohols.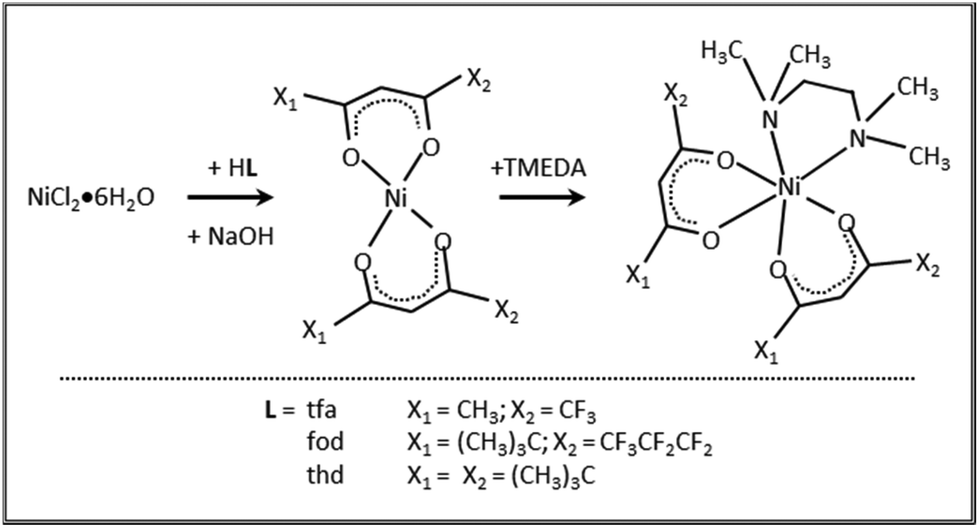 | ||
| Scheme 1 Synthetic pathway towards 1–3. | ||
Both 1 and 3 crystal structures, previously reported at different temperatures,37,38 showed positional disorder, which has been modelled only in part. Therefore, in this study we remeasured the two crystal structures at the same temperature in an attempt to better model the disorder. 1 and 3 were crystallized by evaporation of chlorinated solvents, and analysed by single crystal X-ray diffraction. In contrast, all efforts to obtain crystals of 2 suitable for X-ray analysis turned out to be unsuccessful (see Table S1†). Fig. 1 displays the solid state structures of 1 and 3, along with the minimum energy structures of 1, 2 and 3 obtained by theoretical modelling (see the ESI, § S3 and Fig. S2–S4†). Experimental and calculated structural data are presented in Table 1 and Table S2.† In each case, the Ni(II) center, surrounded by two β-diketonates and a TMEDA moiety, is six-fold coordinated in a distorted octahedral environment. In accordance with other transition metal β-diketonates and ketoiminates,31,32,45,63,641 and 3 exhibit shorter Ni–O bonds than Ni–N bonds. In addition, in the case of 1 (3), the two Ni–O bonds in trans to TMEDA are slightly elongated (shortened). These trends are well reproduced by DFT-computed values. As for 1 and 3, even for 2, the Ni–O bonds are all shorter than the Ni–N ones, and the Ni–O distances in trans to TMEDA are slightly shorter than the other ones (2.028 Å vs. 2.039 Å, respectively). A detailed data inspection indicates that the Ni–O bonds become shorter upon going from 1 to 2 and 3 – i.e., non-fluorinated 3 is characterized by the shortest Ni–O bonds. This finding is further supported by IR spectra (Fig. S5–S7†) and vibrational analysis (Tables S3–S5†). In fact, Ni–O stretching frequencies were 578, 618, and 638 cm−1 for 1, 2, and 3, respectively. Such a result, indicating that the Ni–O bonds are stronger in 3 with respect to those in 1 and 2, may suggest that, in the fragmentation process, the loss of a β-diketonate might be more unfavorable for 3, bearing non-fluorinated ligands.
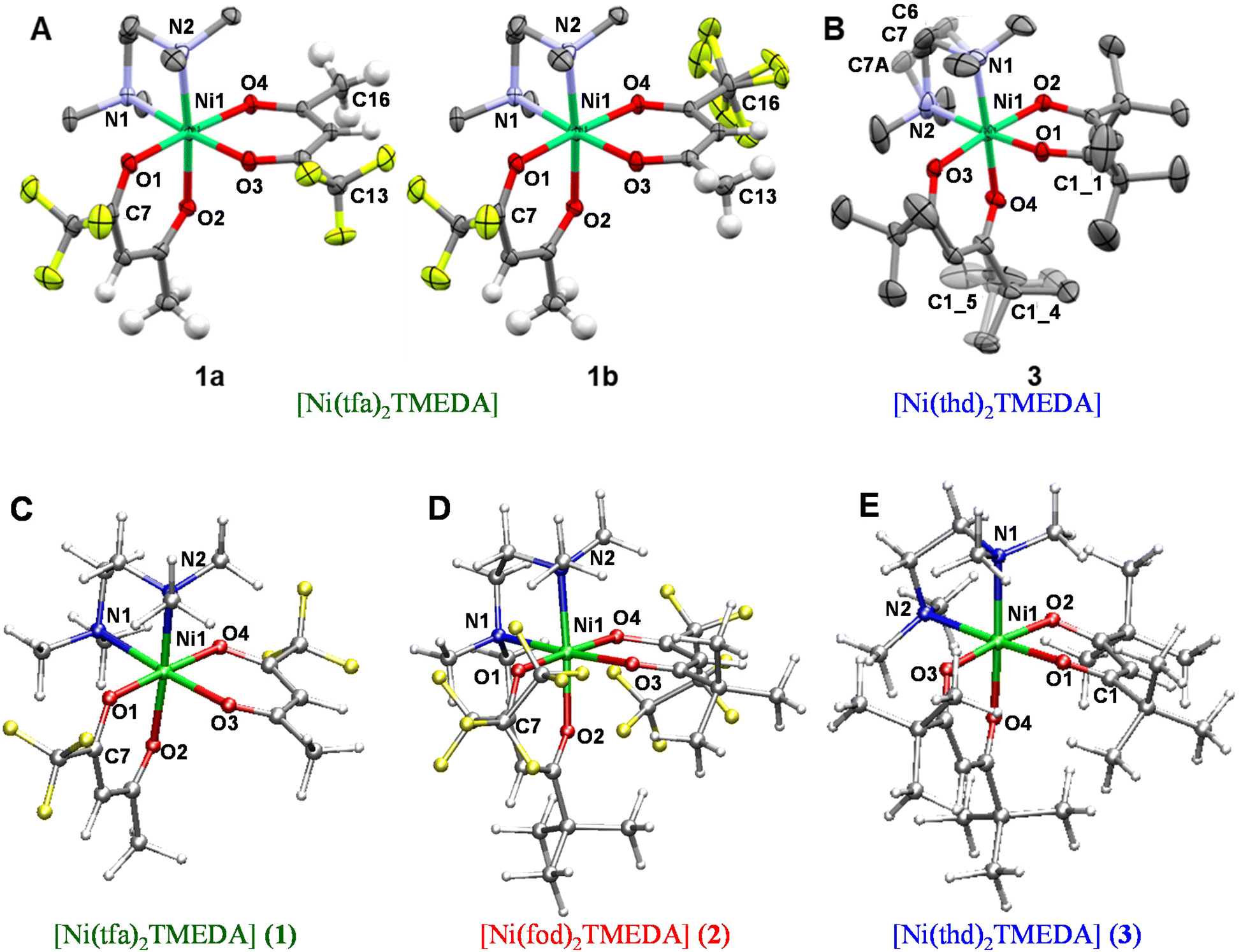 | ||
Fig. 1 (A and B) Solid-state molecular structures of compounds 1 and 3, respectively (selected hydrogen atoms are omitted for clarity). Ellipsoids are depicted at the 50% probability level. (A) Compound 1 showing two determined disordered isomers depending on the orientation of the –CF3 group of one tfa ligand (1a: –CF3@C13 vs.1b: –CF3@C16). For the 1b isomer, additional rotational disorder was detected and modelled. The ratio 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1b with the observed disorders is 13.7:86.3. (B) Molecular structure of compound 3 with modelled positional disorder in the TMEDA backbone and one of the tert-butyl groups of the thd ligand. (C–E) DFT-calculated minimum energy structures for 1, 2, and 3, respectively. :1b with the observed disorders is 13.7:86.3. (B) Molecular structure of compound 3 with modelled positional disorder in the TMEDA backbone and one of the tert-butyl groups of the thd ligand. (C–E) DFT-calculated minimum energy structures for 1, 2, and 3, respectively. | ||
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| Experimental | Calculated | Calculated | Experimental | Calculated | ||
| Bond length (Å) | Ni(1)–O(1) | 2.0200(12) | 2.028 | 2.039 | 1.9912(13) | 2.016 |
| Ni(1)–O(2) | 2.0484(12) | 2.045 | 2.028 | 2.0217(13) | 2.026 | |
| Ni(1)–O(3) | 2.0498(12) | 2.045 | 2.028 | 2.0216(14) | 2.026 | |
| Ni(1)–O(4) | 2.0361(11) | 2.028 | 2.039 | 2.0039(14) | 2.016 | |
| Ni(1)–N(1) | 2.1450(14) | 2.169 | 2.171 | 2.1683(19) | 2.182 | |
| Ni(1)–N(2) | 2.1617(14) | 2.169 | 2.171 | 2.1522(18) | 2.182 | |
| Bond angle (°) | O(1)–Ni(1)–O(4) | 176.79(5) | 177.9 | 178.1 | 93.45(5) | 93.3 |
| O(1)–Ni(1)–O(2) | 89.43(5) | 89.2 | 88.9 | 89.66(5) | 88.9 | |
| O(2)–Ni(1)–O(3) | 91.68(5) | 90.5 | 90.0 | 175.05(6) | 174.8 | |
| O(3)–Ni(1)–O(4) | 89.18(5) | 89.2 | 88.9 | 89.17(6) | 88.9 | |
| N(1)–Ni(1)–N(2) | 84.68(6) | 84.8 | 84.6 | 84.16(9) | 84.0 | |
| O(1)–Ni(1)–O(3) | 88.09(5) | 89.3 | 89.8 | 87.84(6) | 87.6 | |
| O(1)–Ni(1)–N(1) | 94.38(5) | 92.6 | 92.4 | 90.50(7) | 91.4 | |
| O(1)–Ni(1)–N(2) | 87.63(5) | 88.9 | 89.1 | 174.66(7) | 174.7 | |
| Ni(1)–O(1)–C(7/1) | 124.58(11) | 124.2 | 124.1 | 126.29(12) | 124.6 | |
Compound 1 was found to crystallize in the P21/c monoclinic space group, as reported by Bendt et al.37 Interestingly, in the previously reported crystal structure, a disorder in the position of one –CF3 group was observed, differing in the orientation of one tfa ligand (Fig. 1, 1avs. 1b). In this work, this observation was qualitatively reproduced, but a different ratio of the isomers was obtained (13.7% vs. 86.3%; see also the caption for Fig. 1, against 5.1% vs. 94.9% in the original report65). This finding suggests that, in solution, both isomers 1a and 1b are present, possibly in different ratios. In addition, crystallization does not seem to favour one isomer, rendering them energetically similar tectons. The DFT-computed minimum energy geometry of 1a (see the ESI, § S3, Fig. S2B†) is slightly less stable than the one for 1b (0.78 kcal mol−1), providing a possible molecular-level origin of the lower abundance of 1a with respect to 1b deduced by structural data. For 1b, an additional disorder can be observed, originating from one –CF3 group rotation. Hereby, the ratio between the main and the minor modelled split-layer components is 55.4% vs. 44.6%. Such a disorder was not detected in the calculated 1b structure. Geometry optimizations performed starting from the two crystallographic structures coalesced to a single –CF3 orientation (see Fig. 1C). This result suggested that the –CF3 rotational static disorder detected in the crystal may be due to intermolecular interactions.
3 was solved in the P21/n monoclinic space group,38 with slightly smaller cell parameters than those of the previously reported structure due to the lower measurement temperature [a = 10.0160(8) Å vs. 9.9651(7) Å, b = 18.9271(12) Å vs. 18.8124(15) Å, and c = 17.1938(16) Å vs. 17.0424(13) Å]. In the original room temperature structure, enhanced atomic displacement parameters (ADPs) were already observed for the ethylene bridge at the TMEDA ligand and the tert-butyl groups, and attributed to thermal or static disorder. We now could observe and explicitly refine the disorder in the redetermination at a low temperature, rendering it a static, rather than a thermally-induced, disorder, as originally discussed.38 The ethylene bridge crystallizes in two conformationally disordered positions with 69.7% of the main layer (C6, C7) vs. 30.3% (C6A, C7A) of the minor one (depicted in Fig. 1, greyed out). The ratio of the disorder due to rotating tert-butyl was found to be 63.5% vs. 36.5% between the main (C1_4) and the minor (C1_5) split layers. Accordingly, DFT calculations performed on 3 (see also § S3 and Fig. S4†) revealed that the optimized structure of the main component was slightly more stable (0.24 kcal mol−1) than that of the minor one, accounting for the co-presence of both ethylene bridge arrangements in the compound 3 crystals.
It is interesting to note that the Ni–N bond distances increase with the number of electron-donor tert-butyl groups on β-diketonate ligands. Such a bond-lengthening effect, which is maximum in compound 3, may likely arise from steric hindrance associated with the two tert-butyl groups on each thd ligand. Correspondingly, the stretching frequency associated with the Ni–N bonds is lower (461 cm−1) in 3. In compound 2, a balance between the increase of (electron-withdrawing) fluorine atoms and the steric hindrance of (electron-donor) tert-butyl groups gives rise to almost the same Ni–N stretching frequencies as in compound 1 (472 and 471 cm−1 in 2 and 1, respectively). Overall, these results may indicate that the tert-butyl-rich complex 3, bearing non-fluorinated ligands, might undergo an easier TMEDA release with respect to 1 and 2. An important common feature of 1–3 is that the Ni–N stretching frequencies are lower than Ni–O ones, indicating a weaker interaction between the Ni center and the TMEDA N atoms. This analysis could suggest that, upon thermal activation, the TMEDA detachment might be easier than β-diketonate loss.
To underpin this hypothesis, for compounds 1–3 the energetics correlated with the loss of TMEDA (ΔE1, eqn (1)) and of a β-diketonate ligand L (ΔE2, eqn (2)) were calculated according to the following pathways:
| [NiL2TMEDA] → [NiL2] + TMEDA | (1) |
| [NiL2TMEDA] → [NiL(TMEDA)]+ + L−. | (2) |
The calculated fragmentation energies, reported in Table 2, indicate that, for all complexes, the energetic cost of TMEDA removal is much lower than the loss of a β-diketonate ligand, in line with previous results on similar TMEDA-containing compounds.31,45 Also importantly, the detachment of TMEDA is significantly less energy demanding for compound 3 than for the fluorinated compounds, in line with all previous analyses. Overall, the discussed results suggest that TMEDA loss should be the first fragmentation step for all three complexes, and that such event should be the easiest in compound 3.
| Energies (kcal mol−1) | 1 | 2 | 3 |
|---|---|---|---|
| ΔE1 | 61.2 | 65.1 | 54.9 |
| ΔE2 | 142.9 | 143.9 | 146.6 |
| Δ(ΔE) | 81.7 | 78.8 | 91.7 |
To confirm the identity and spectroscopic purity of the synthesized complexes, NMR spectroscopy analyses were performed. 1H spectra of the ligands and complexes are reported in the ESI,† together with the suggested peak assignments (Fig. S8–S12 and Table S6†). As expected, due to the strong paramagnetic character of Ni(II) centers with a d8 configuration, only a few and broad resonances, spread over a wide chemical shift range, were observed.34 Regarding the 13C spectra (Fig. 2), the different ligand signals could be discriminated taking into account the correlations of positive/negative shifts with the bond distances from the paramagnetic center,66 also based on previous works on homologous compounds.31,32 In particular, TMEDA aliphatic carbon resonances, experiencing an upfield shift,31,32 were located in the −190 to −300 ppm region for all complexes. For 1, the resonances at −208, −258 and −308 ppm could be reasonably attributed to methyl and methylene groups. For 2, these resonances were located at −315, −273 and −215 ppm, whereas for 3 they appeared at −195, −257 and −281 ppm. The signals due to the three different carbonyl-bearing ligands could be identified in the positive chemical shift range up to 900 ppm. In the case of 1, the broad signal centred at ≈880 ppm could be assigned to the two tfa ligand carbonyls. The –CF3 group is represented by the quartet between 380 and 287 ppm, due to the large C–F J-coupling.67 Finally, the 449 ppm and the 163–160 ppm resonances could be assigned to the tfa methylene and methyl groups, respectively.31 Although the spectrum of 2 suffers from a lower signal-to-noise ratio, rendering unfeasible a precise assignment of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signals, the other ligands’ resonances could be recognized. Specifically, the three fod methyls exhibited the signal at 27 ppm, as in the pure reagent (Fig. S10b†), whereas the signals at 128 and 131 ppm were ascribed to quaternary and methylene carbons, and the resonances in the 330–400 ppm region were ascribed to the three fluorinated carbons.31 Finally, the spectrum of 3 shows a broad signal at 900 ppm, which is ascribed to the four thd carbonyls, in analogy with 1. The resonances at 339 and 298 ppm could be due to quaternary carbons, and the peak at 147 ppm is attributed to methylene groups. The signals of the thd methyls were almost unaffected by complexation (Fig. S11b†), maintaining the position at 30 ppm.
O signals, the other ligands’ resonances could be recognized. Specifically, the three fod methyls exhibited the signal at 27 ppm, as in the pure reagent (Fig. S10b†), whereas the signals at 128 and 131 ppm were ascribed to quaternary and methylene carbons, and the resonances in the 330–400 ppm region were ascribed to the three fluorinated carbons.31 Finally, the spectrum of 3 shows a broad signal at 900 ppm, which is ascribed to the four thd carbonyls, in analogy with 1. The resonances at 339 and 298 ppm could be due to quaternary carbons, and the peak at 147 ppm is attributed to methylene groups. The signals of the thd methyls were almost unaffected by complexation (Fig. S11b†), maintaining the position at 30 ppm.
 | ||
| Fig. 2 13C-NMR spectra of compounds 1–3, all as a sum of three 800 ppm wide spectra (the overlapped regions are highlighted). The intense CDCl3 solvent peak is marked by #. | ||
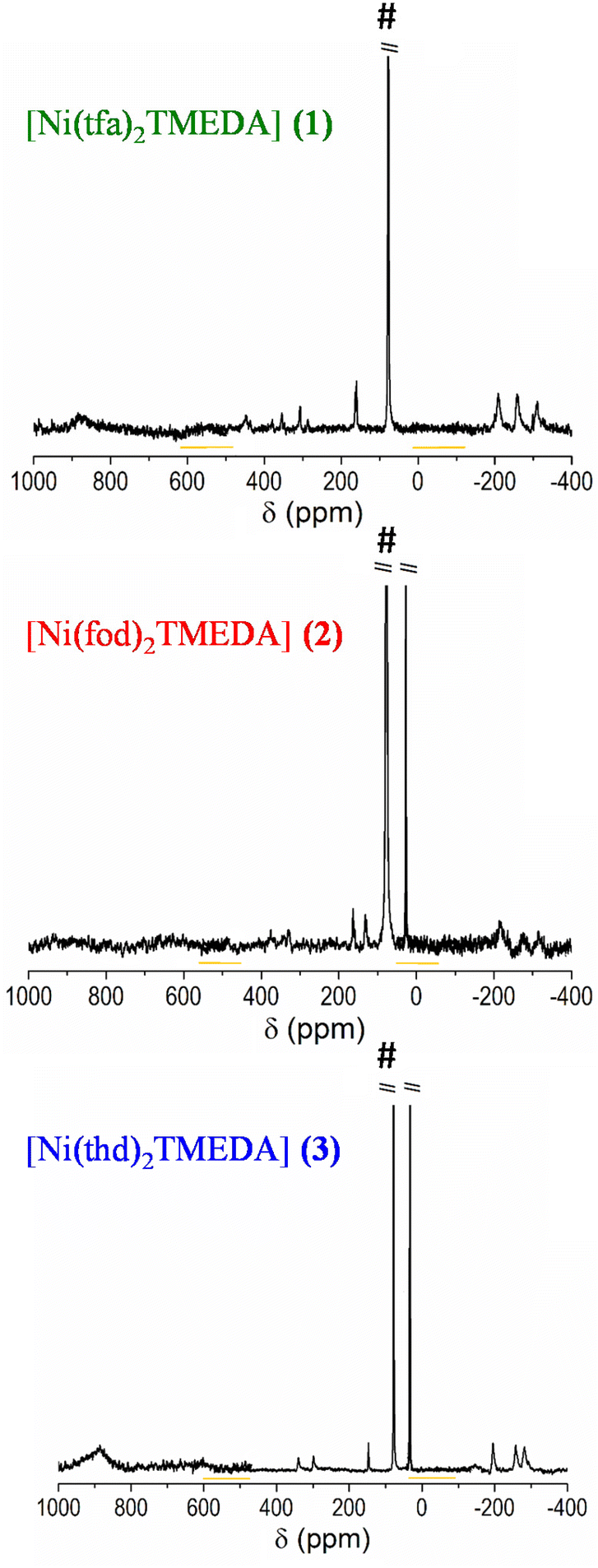
The optical spectra of 1–3 (Fig. 3) exhibited a very strong band in the near-UV region at ≈300 nm, suggesting that the three complexes might be potential precursors for UV photo-assisted CVD approaches.31 Indeed, the photo-induced dissociation of the complexes in the gas-phase may generate highly reactive radical species, thus enabling one to operate at lower growth temperatures, suitable for depositions on thermally sensitive substrates. In the experimental spectra, the main peak positions are found at 303, 307, and 312 nm for compounds 3, 1, and 2, respectively. TD-DFT calculations reproduced the same trend, yielding values of 271, 277 and 290 nm for 3, 1, and 2 respectively. This finding indicates that the absorption band is progressively shifted towards higher wavelengths by increasing the fluorine content in the diketonate ligand – i.e., the lowest wavelength value corresponds to the non-fluorinated derivative 3.
 | ||
| Fig. 3 Experimental and calculated optical absorption spectra of compounds 1, 2, and 3. | ||
For all compounds, a detailed analysis of computed electronic excitations enabled one to ascribe the main absorption band to ligand–ligand (L → L) transitions involving molecular spin orbitals predominantly localized on diketonate ligands (Fig. S13†). In particular, the transition could be classified as π → π*, as for the homologous Mn and Zn complexes.45,53
Supporting data on the reactivity and fragmentation behavior of the target compounds were obtained by EI-MS analyses. In this regard, it is worth highlighting that the identification of cation/radical-cation species and the underlying fragmentation pathway under EI conditions might provide useful indications on the potential behavior of the developed precursors in plasma assisted CVD processes.44,68
EI-MS spectra of 1, 2, and 3 (Fig. 4; see also Table 3) did not show the presence of the compound molecular cations, due to their complete fragmentation under EI conditions, i.e. interaction in the gas phase of neutral molecules with 70 eV accelerated electrons. Diketonate ligand loss was the most favored fragmentation for 1 (m/z = 327), and 2 (m/z = 469), as the signal due to the [M − L]+ species was the base peak in both spectra. In the case of 3, abundant ions for 3 are due to TMEDA release and the consequent loss of a C4H9 radical (m/z = 424 and 367). In 3, the absence of signals due to diketonate loss indicated that thd is strongly coordinated to the Ni center in the gas phase, at variance with tfa and fod in 1 and 2, respectively. For 2, other well detectable fragment ions resulted from the detachment of TMEDA (m/z = 648), followed by the loss of the C4H9 radical (m/z = 591), the loss of TMEDA from [M − L]+ (m/z = 353), and the formation of [N(CH3)2CH2]+ (m/z = 58), and of the C4H9 cation (m/z = 57). It is worth highlighting that for 2, at variance with 1 and previously reported [M(tfa)2TMEDA] complexes (M = Fe, Co),31,32 no ions arising from the fragmentation of the fluorinated moiety (CF2CF2CF3) were detected.
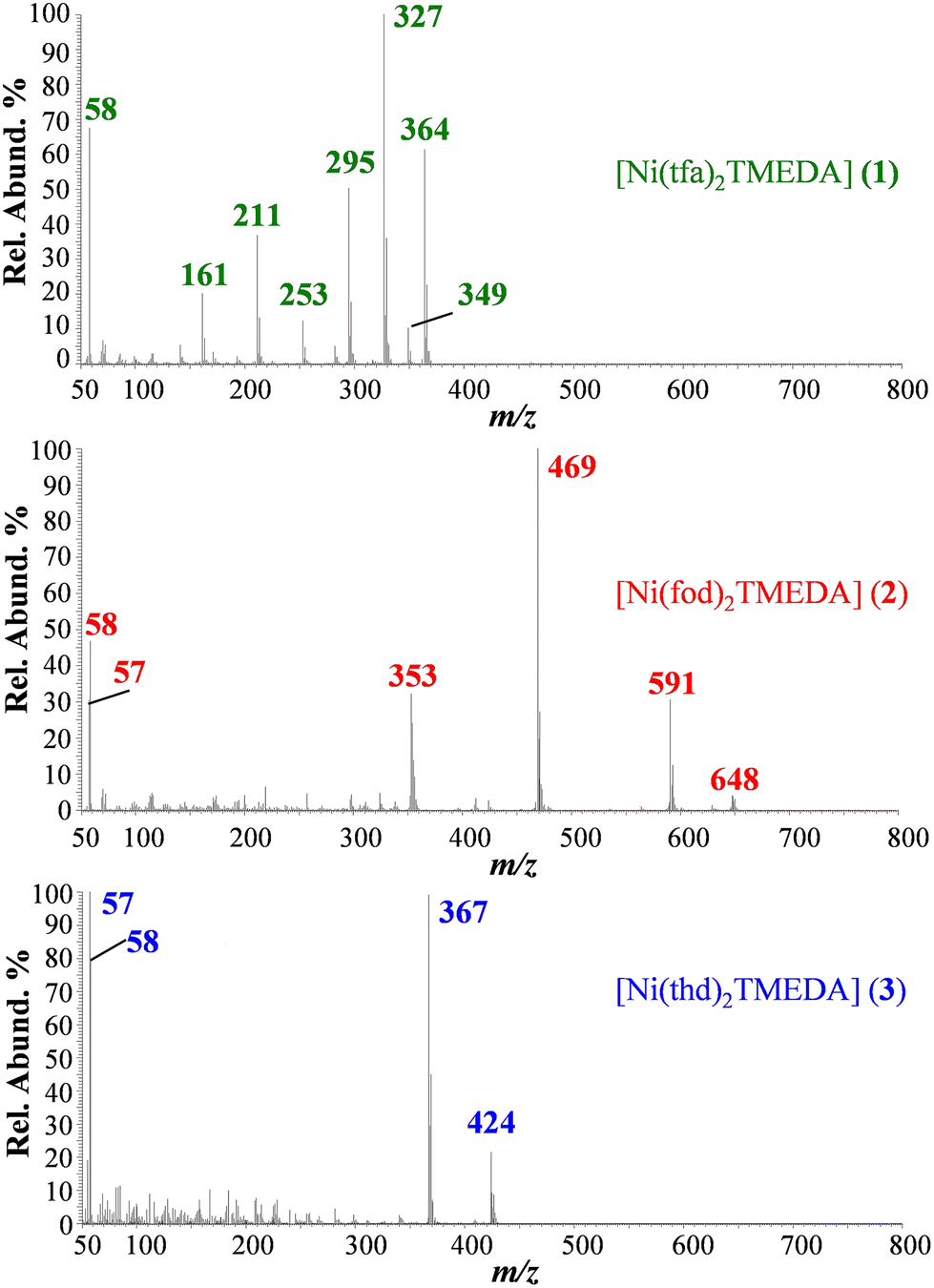 | ||
| Fig. 4 EI mass spectra of compounds 1–3. | ||
| Ionic species | 1 m/z (%) | 2 m/z (%) | 3 m/z (%) |
|---|---|---|---|
| M+˙ | 480 (0) | 764 (0) | 540 (0) |
| [M − TMEDA]+˙ | 364 (63) | 648 (8) | 424 (24) |
| [M − TMEDA − C4H9]+ | — | 591 (29) | 367 (99) |
| [M − TMEDA − CH3]+ | 349 (13) | — | — |
| [M − L]+ | 327 (100) | 469 (100) | — |
| [M − TMEDA − L]+ | 211 (38) | 353 (35) | — |
| [N(CH3)2CH2]+ |
58 (68) | 58 (48) | 58 (80) |
| [C4H9]+ | — | 57 (30) | 57 (100) |
| [M − TMEDA − CF3˙]+ | 295 (50) | — | — |
| [M − TMEDA − CF3 − COCH2]+˙ | 253 (18) | — | — |
| [M − TMEDA − L − CF2]+˙ | 161 (22) | — | — |
In order to evaluate the suitability of the target compounds as vapor phase molecular sources for NiO thin films/nanosystems, thermal analyses were undertaken. TGA curves (Fig. 5a) exhibited a distinct single-step mass loss, ultimately leading to zero weight for temperatures higher than 250 °C. This behaviour is an excellent starting point in view of compounds’ applications as CVD/ALD precursors, since it indicates the occurrence of a clean and quantitative vaporization free from any undesired side decomposition. Such results reveal the improved properties of the present compounds in comparison with a variety of Ni precursors proposed so far,1,6,9,12,26–28,34,40,43,69 showing more complex TGA curve shapes with a non-zero final weight and/or mass losses at higher temperatures. In particular, low values for the temperature of 50% mass loss (T50) are a useful marker of an improved volatility.40,69 The T50 data extrapolated from Fig. 5a (see Table 4) were found to be all lower than those of Ni ketoiminates,39 and, for 1 and 2, they were lower than those of iminoate and diketonate-PDA Ni(II) complexes.43 In particular, the T50 value for 1 was better than those of Ni(acac)2, Ni(acac)2TMEDA,43,69 and unsymmetrical Ni β-diketonate derivatives.1,42 The obtained T50 values increased in the order 1 < 2 < 3, suggesting that the compounds’ volatility followed the opposite trend. The volatilization onset (1% mass loss) displayed an analogous behaviour (Table 4), and the corresponding values were appreciably lower than those of variously substituted Ni ketoiminate compounds.12 The melting points of the present complexes were lower than those of Ni(II) ketoiminates,39 β-enamino ketoesters28 and dialkylaminoalkoxides,9 and, for 1 and 2, they were lower than those of many Ni(II) complexes.1,6,12,26,29,34,38–40,42,43 Isothermal analyses carried out at selected temperatures (see Fig. S14† and related observations) highlighted the long-term stability of the compounds and the occurrence of volatilization free from any undesired side decomposition. The fit of vaporization rates vs. temperature data using the Clausius–Clapeyron equation (Fig. 5b) enabled us to estimate the apparent molar vaporization enthalpy values (ΔHvap, Table 4), which are of importance for CVD/ALD end-uses, since lower ΔHvap accounts for a higher vapor pressure, and vice versa. For 1, ΔHvap was in line with those estimated for the homologous Fe and Co hfa/tfa derivatives.30–32,47 The actual ΔHvap values were also comparable to those reported for Ni ketoiminates12,39 and [bis(N-propyl-3-methyl-salicylaldimine)nickel(II)].6 For each compound, DSC curves (Fig. 5c and Table 4) revealed the presence of two endothermic peaks, due to melting (a) and boiling (b). As for T50 and volatilization onsets, the corresponding temperatures increased from 1 to 2, up to 3 (the least volatile compound).
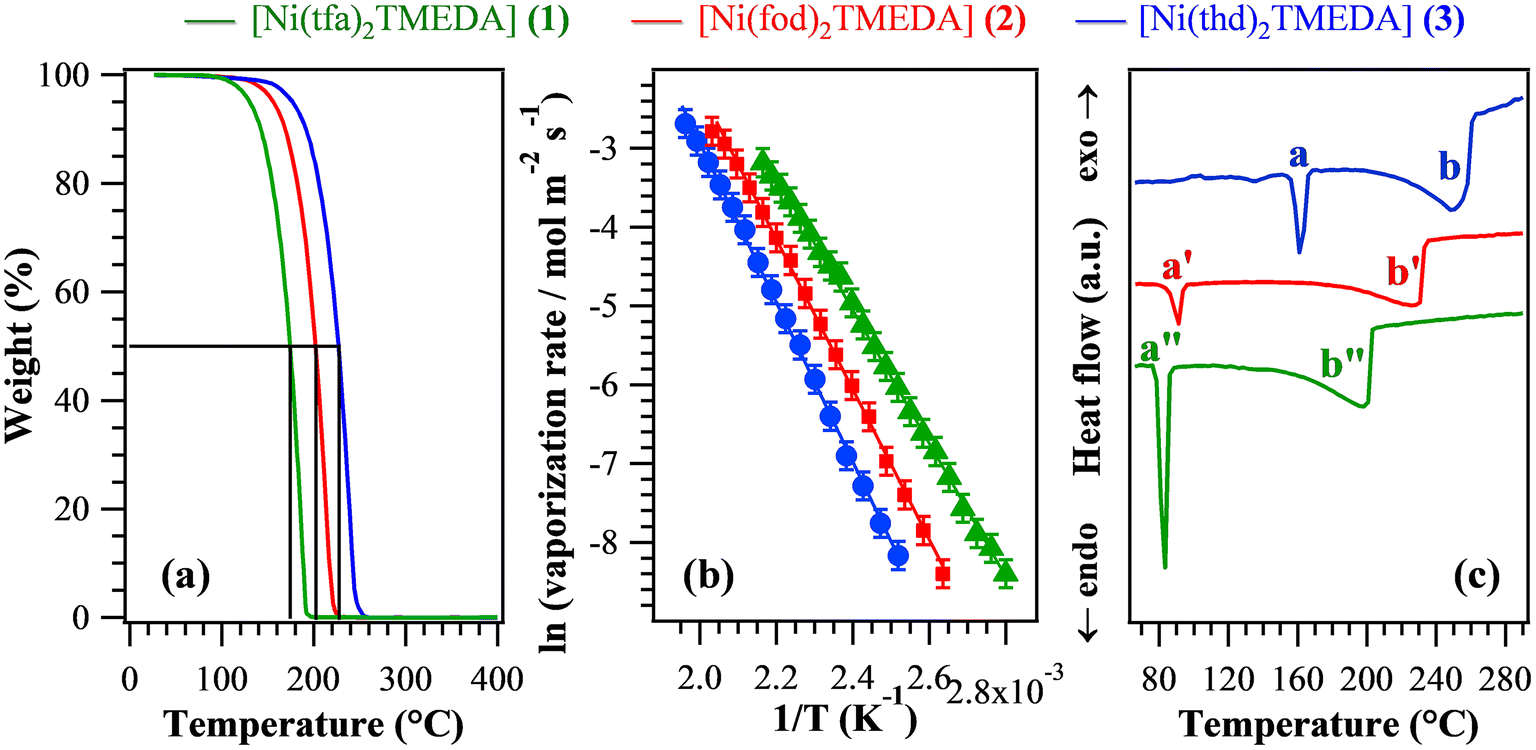 | ||
| Fig. 5 TGA curves (a), Arrhenius plot for the vaporization (b), and DSC traces (c) for compounds 1–3. For the present compounds, in the investigated temperature interval, vaporization rate values ranged between 2 × 10−4 and 8 × 10−2 mol m−2 s−1. | ||
| T 50 (°C) | Volatilization onseta (°C) | ΔHvapa (kcal mol−1) | Melting pointb (°C), a | Boiling pointb (°C), b | |
|---|---|---|---|---|---|
| The letters a and b refer to the peaks marked in Fig. 5c.a Determined from TGA analyses.b Determined from DSC analyses. | |||||
| 1 | 174 | 100 | (16.8 ± 0.1) | 83 | 198 |
| 2 | 202 | 116 | (18.9 ± 0.1) | 91 | 226 |
| 3 | 227 | 126 | (20.2 ± 0.1) | 162 | 247 |
Overall, the above discussed results indicate that although all complexes were endowed with a clean vaporization, their volatility was directly dependent on both the fluorination degree and the steric hindrance of the used diketonate ligands. In particular, complex 3, containing fluorine-free ligands bearing bulky tert-butyl groups on the terminal sides, turned out to be the least volatile, whereas the lighter tfa derivative 1 was the most volatile one. These outcomes are consistent with the fact that the introduction of fluorinated groups into the coordinated ligands can: (i) reduce intermolecular interactions;6 (ii) decrease the basic character of β-diketonate oxygen donor sites thanks to their electron-withdrawing action, increasing the compounds’ volatility.31,33,39
Although the above-discussed thermal analyses do not provide direct indications about the decomposition mechanism of complexes 1–3, a ligand detachment event – for example, a TMEDA loss – appears to be unlikely in TGA experiments due to the high energy cost of the process (at least 54.9 kcal mol−1, see Table 2). Nevertheless, the activation of the ligand detachment process may occur when the complexes interact with the heated substrate surface, under conditions typical of thermal CVD experiments.50,52,53
Precursors’ validation: thin film fabrication and analysis
Based on the promising chemico-physical properties of the synthesized Ni compounds, their suitability for the CVD of NiO was tested in preliminary experiments carried out on Si(100) substrates under dry O2 reaction atmospheres. The present growth and/or vaporization temperatures (see the Experimental section) were lower than those previously utilized for other Ni precursors {ketoiminates,12 [Ni(acac)2], as such14,22 or modified with various ligands,27 [Ni(thd)2],24,25 other β-diketonate-TMEDA derivatives,1,29,34 and bis(N-propyl-3-methylsalicylaldimine)nickel(II)6}.XRD analyses (Fig. 6a and Fig. S15†) revealed that only the specimen deposited from 3 gave rise to detectable diffraction peaks. The latter, located at 2θ ≈ 37.2° and 43.3°, corresponded respectively to the (111) and (200) crystallographic planes of NiO with the rocksalt structure (bunsenite, cubic; Fig. 6b),70 and their relative intensities matched those of the reference pattern. No other appreciable reflections could be observed, indicating the formation of phase-pure systems. The actual dislocation density (δ) and microstrain (ε) values (§ S8.2†) were higher than those of NiO films obtained by spray pyrolysis20 and sputtering followed by thermal oxidation.19 The absence of net diffraction peaks for the systems obtained from 1 and 2 (Fig. S15†) at the same temperature and in the same reaction environment was ascribed to their low thickness (≈10 nm for both) and low crystallinity (Table S7 and Fig. S16†).
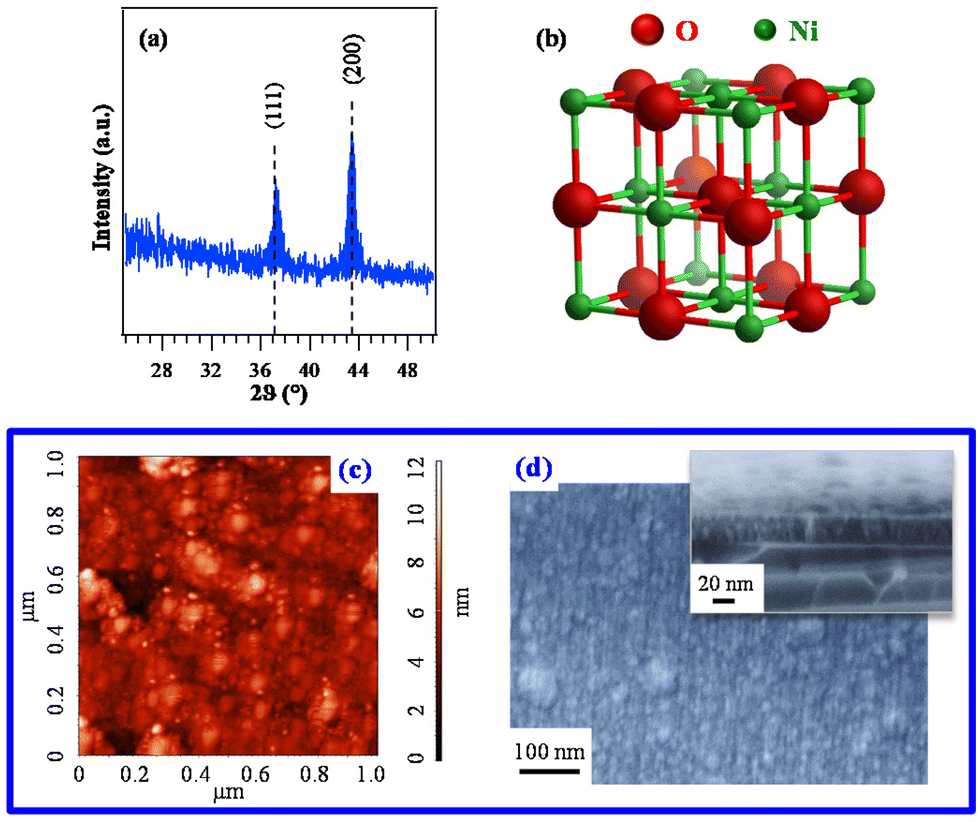 | ||
| Fig. 6 (a) XRD pattern of a specimen grown on Si(100) from 3. The observed peaks can be assigned to cubic NiO (reference peak positions70 are marked), whose structure is reported in (b). Corresponding AFM (c) and plane-view FE-SEM images (d). In the latter case, a representative cross-sectional image is shown as the inset (film thickness ≈ 37 nm). | ||
Since the deposit morphology and interface quality play an important role in various functional applications,12 AFM was utilized to investigate the film surface topography. The recorded images (Fig. 6c) evidenced a granular morphology, likely associated with a 3D growth mode, featuring a root-mean-square (RMS) roughness as low as 1.0 nm. This result, indicating the occurrence of very flat film surfaces, was in line with FE-SEM results that revealed the formation of compact systems free from cracks/pinholes and featuring a very good adhesion to the used substrate (Fig. 6d and S16†).
Overall, structural and morphological analyses indicated a much higher decomposition efficiency for 3 in comparison with fluorinated molecular sources, i.e.1 and 2. This conclusion was indeed unexpected based on the sole outcomes of thermal analyses, which indicated 3 as the least volatile family member. Such a result highlights that the sole volatility is not sufficient by itself to ensure an appreciable precursor decomposition efficiency on the growth surface – an issue apparently obvious, but not always foregone. The higher sublimation temperature of 3 (120 °C) with respect to 1 (75 °C) and 2 (80 °C) indicates that 3 is introduced in the CVD reactor chamber more vibrationally excited than 1 and 2, and thus potentially more reactive.50 Indeed, since deformation modes of the Ni coordination octahedron, which strongly involve metal–ligand distances, are found at ≈250 cm−1 (Tables S3–S5†), they are likely to be excited at 120 °C (kT = 273 cm−1). Additionally, the non-fluorinated complex 3 should be the one characterized by the weakest Ni–N bonds, as suggested by the Ni–N stretching frequency trend. A further key indication is the energetic cost of TMEDA loss computed for the three complexes (Table 2), which is the lowest for compound 3, providing a plausible explanation for the more efficient film growth obtained in the case of this compound.
The system chemical composition was preliminarily investigated by EDXS analyses. Regardless of the preparation conditions, the EDXS spectra (Fig. 7 and S17, S18†) were dominated by the lines of oxygen (O Kα at 0.52 keV) and nickel (Ni Lα and Kα at 0.85 and 7.47 keV), as well as the substrate Si signal (Si Kα at 1.74 keV). Adventitious carbon was also detected (C Kα at 0.28 keV). EDXS spectra recorded in different regions and in-plane compositional mapping pointed out an even O and Ni lateral distribution, indicating a homogeneous system composition. Quantitation yielded mean values of (42.0 ± 0.2) wt% O, (50.5 ± 0.2) wt% Ni, and (7.5 ± 0.2) wt% C. Additional X-ray photoelectron spectroscopy (XPS) analyses (see Fig. S19† and related observations) confirmed the formation of NiO films and revealed that, after Ar+ erosion, the C 1s signal was reduced to noise level, thus proving that contamination was mostly limited to the sample surface.
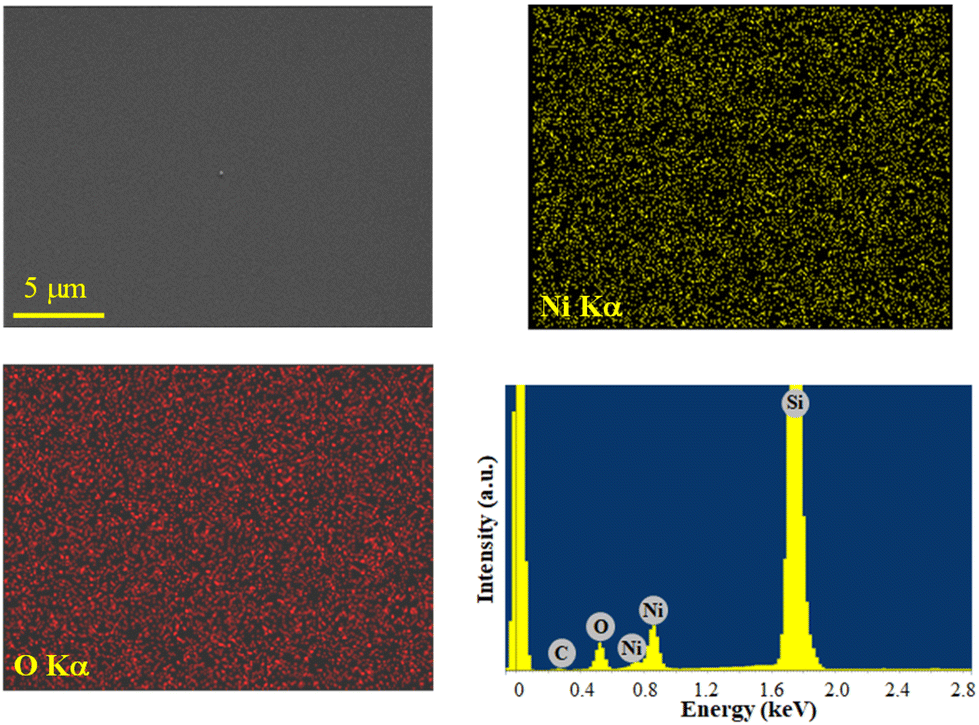 | ||
| Fig. 7 Ni and O EDXS elemental maps, recorded on the corresponding electron image, for a NiO specimen obtained from 3, and the representative spectrum. | ||
Based on the results presented so far, it appears that fluorinated derivatives in the present precursors’ family, in spite of their higher volatility, yield very thin films under the adopted CVD conditions. This result, in turn, indicates that 1 and 2 are highly stable in the vapor phase, and that the overall process bottleneck is their reaction at the growth surface. As a consequence, an important issue to be tackled is: how to increase the decomposition efficiency, and obtain a higher growth rate, from the target Ni precursors? As shown by our recent studies on Fe and Co tfa-TMEDA adducts,31,32 the introduction of water vapor in the reaction atmosphere plays an important role in the obtainment of thicker films, thanks to its promotional effect on precursors’ decomposition. Further efforts in this direction hold a significant promise towards the obtainment of thicker deposits.
Conclusions
A series of closely related nickel diketonate-diamine complexes were rationally developed and characterized in terms of structures, bonding and reactivity, in view of their eventual application as vapor phase precursors for NiO. The target compounds {[Ni(tfa)2TMEDA], (1); [Ni(fod)2TMEDA], (2); [Ni(thd)2TMEDA], (3)} were synthesized in high yields by a convenient route and subjected to comprehensive experimental–theoretical characterization. Particular attention was paid to the influence of functional groups in the diketonate ligand structure on the electronic and vibrational properties, as well as on the thermal behavior and fragmentation patterns of the compounds. The obtained heteroleptic complexes, united by a pseudo-octahedral coordination of Ni(II) centers, are all monomeric, water-free and air stable. The compounds’ thermal stability, volatility and fragmentation pathways, adjustable as a function of the ligand features, highlight their promising versatility as vapor phase precursors for NiO systems. A preliminary validation, performed under a dry O2 atmosphere at volatilization/growth temperatures lower than those previously employed, yielded pure and homogeneous NiO films with flat surfaces.Overall, the reported results highlight that the [NiL2TMEDA] family contributes to successfully expanding the library of CVD Ni precursors with improved properties. A strong basis to shed further light on the interplay between precursors’ chemistry and CVD process design will be set by additional work aimed at optimizing the CVD operating conditions and at testing the precursors’ feasibility for ALD end-uses. In this regard, special attention will be devoted to investigating the behavior of the developed compounds in the presence of different co-reactants (O2, H2O, and O3), also taking into account that precursors’ reactivity might appreciably vary under CVD vs. ALD conditions. The theoretical calculations will enable, in perspective, further important advancements, even in view of potential precursors’ applications in plasma-activated CVD/ALD processes.
Author contributions
M. B.: CVD of NiO films; D. B.: project administration, funding acquisition, and writing – original draft; E. F.: methodology and DFT studies; R. S. and M. R.: EI-MS analyses and interpretation; G. T.: methodologies, DFT studies, and supervision; G. P.: TGA analysis; C. I.: DFT calculations and interpretation; A. G.: XRD analysis and investigation; A. A. H. and A. P.: single crystal X-ray analysis; E. C. and S. D.: NMR, IR and DSC analyses; V. D. N.: TGA analysis, supervision, and writing – review and editing; S. B.: supervision, writing – review and editing; C. M.: conceptualization, funding acquisition, and FE-SEM/AFM analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from CNR (Progetti di Ricerca@CNR – avviso 2020 – ASSIST), Padova University (PDiSC#04BIRD2020-UNIPD EUREKA, DOR 2021–2023 and the project “ACHILLES”, prot. BIRD219831), INSTM Consortium (INSTM21PDGASPAROTTO-NANOMAT, INSTM21PDBARMAC-ATENA), AMGA Foundation (NYMPHEA project) and Insubria University (FAR2021). XPS measurements were performed with an ESCALAB™ QXi Spectrometer funded by “Sviluppo delle infrastrutture e programma biennale degli interventi del Consiglio Nazionale delle Ricerche (2019)”. The authors thank Dr L. Calore and Dr D. Canton for their valuable technical support.References
- S. D. Cosham, S. P. Richards, T. Manning, M. S. Hill, A. L. Johnson and K. C. Molloy, Eur. J. Inorg. Chem., 2017, 1868–1876 CrossRef CAS.
- A. S. Kondrateva, M. Mishin, A. Shakhmin, M. Baryshnikova and S. E. Alexandrov, Phys. Status Solidi C, 2015, 12, 912–917 CrossRef CAS.
- J. Tian, H. Jiang, X. Zhao, G. Shi, Y. Dai, X. Deng, H. Xie and W. Zhang, Sens. Actuators, B, 2022, 366, 131981 CrossRef CAS.
- H. Wang, G. Wu, X. P. Cai, Y. Zhao, Z. F. Shi, J. Wang, X. C. Xia, X. Dong, B. L. Zhang, Y. Ma and G. T. Du, Vacuum, 2012, 86, 2044–2047 CrossRef CAS.
- H. Wang, Y. Zhao, X. Li, C. Wu, X. Dong, Y. Ma, B. Zhang and G. Du, Vacuum, 2015, 119, 77–80 CrossRef CAS.
- M. Chandrakala, S. Raj Bharath, T. Maiyalagan and S. Arockiasamy, Mater. Chem. Phys., 2017, 201, 344–353 CrossRef CAS.
- T. M. Roffi, S. Nozaki and K. Uchida, J. Cryst. Growth, 2016, 451, 57–64 CrossRef CAS.
- M. Z. Sialvi, R. J. Mortimer, G. D. Wilcox, A. M. Teridi, T. S. Varley, K. G. U. Wijayantha and C. A. Kirk, ACS Appl. Mater. Interfaces, 2013, 5, 5675–5682 CrossRef CAS.
- R. L. Wilson, T. J. Macdonald, C.-T. Lin, S. Xu, A. Taylor, C. E. Knapp, S. Guldin, M. A. McLachlan, C. J. Carmalt and C. S. Blackman, RSC Adv., 2021, 11, 22199–22205 RSC.
- S. W. Han, I. H. Kim, D. H. Kim, K. J. Park, E. J. Park, M.-G. Jeong and Y. D. Kim, Appl. Surf. Sci., 2016, 385, 597–604 CrossRef CAS.
- N. Kitchamsetti, M. S. Ramteke, S. R. Rondiya, S. R. Mulani, M. S. Patil, R. W. Cross, N. Y. Dzade and R. S. Devan, J. Alloys Compd., 2021, 855, 157337 CrossRef CAS.
- D. Zywitzki, D. H. Taffa, L. Lamkowski, M. Winter, D. Rogalla, M. Wark and A. Devi, Inorg. Chem., 2020, 59, 10059–10070 CrossRef CAS.
- W.-J. An, E. Thimsen and P. Biswas, J. Phys. Chem. Lett., 2010, 1, 249–253 CrossRef CAS.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CrossRef CAS.
- N. Hussain, W. Yang, J. Dou, Y. Chen, Y. Qian and L. Xu, J. Mater. Chem. A, 2019, 7, 9656–9664 RSC.
- P. Salunkhe, M. A. A V and D. Kekuda, Mater. Res. Express, 2020, 7, 016427 CrossRef CAS.
- D. S. Kim and H. C. Lee, J. Appl. Phys., 2012, 112, 034504 CrossRef.
- D. Alders, F. C. Voogt, T. Hibma and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 7716–7719 CrossRef CAS.
- F. Hajakbari, J. Nanostruct. Chem., 2020, 10, 97–103 CrossRef CAS.
- R. S. Kate, S. C. Bulakhe and R. J. Deokate, Opt. Quantum Electron., 2019, 51, 319 CrossRef.
- S. Kerli and U. Alver, Crystallogr. Rep., 2014, 59, 1103–1106 CrossRef CAS.
- M. Utriainen, M. Kröger-Laukkanen, L.-S. Johansson and L. Niinistö, Appl. Surf. Sci., 2000, 157, 151–158 CrossRef CAS.
- M. Utriainen, M. Kröger-Laukkanen and L. Niinistö, Mater. Sci. Eng., B, 1998, 54, 98–103 CrossRef.
- E. Lindahl, M. Ottosson and J.-O. Carlsson, Chem. Vap. Deposition, 2009, 15, 186–191 CrossRef CAS.
- E. Lindahl, J. Lu, M. Ottosson and J. O. Carlsson, J. Cryst. Growth, 2009, 311, 4082–4088 CrossRef CAS.
- T. S. Yang, W. Cho, M. Kim, K.-S. An, T.-M. Chung, C. G. Kim and Y. Kim, J. Vac. Sci. Technol., A, 2005, 23, 1238–1243 CrossRef CAS.
- Y. Zhang, L. Du, X. Liu and Y. Ding, Nanoscale, 2019, 11, 3484–3488 RSC.
- M. Basato, E. Faggin, C. Tubaro and A. C. Veronese, Polyhedron, 2009, 28, 1229–1234 CrossRef CAS.
- S. Battiato, M. M. Giangregorio, M. R. Catalano, R. Lo Nigro, M. Losurdo and G. Malandrino, RSC Adv., 2016, 6, 30813–30823 RSC.
- G. Bandoli, D. Barreca, A. Gasparotto, C. Maccato, R. Seraglia, E. Tondello, A. Devi, R. A. Fischer and M. Winter, Inorg. Chem., 2009, 48, 82–89 CrossRef CAS.
- M. Klotzsche, D. Barreca, L. Bigiani, R. Seraglia, A. Gasparotto, L. Vanin, C. Jandl, A. Pöthig, M. Roverso, S. Bogialli, G. Tabacchi, E. Fois, E. Callone, S. Dirè and C. Maccato, Dalton Trans., 2021, 50, 10374–10385 RSC.
- D. Barreca, L. Bigiani, M. Klotzsche, A. Gasparotto, R. Seraglia, C. Jandl, A. Pöthig, E. Fois, L. Vanin, G. Tabacchi, M. Roverso, S. Bogialli, E. Callone, S. Dirè and C. Maccato, Mater. Chem. Phys., 2022, 277, 125534 CrossRef CAS.
- S. Mishra and S. Daniele, Chem. Rev., 2015, 115, 8379–8448 CrossRef CAS PubMed.
- G. Malandrino, L. M. S. Perdicaro, G. Condorelli, I. L. Fragalà, P. Rossi and P. Dapporto, Dalton Trans., 2006, 1101–1106 RSC.
- G. Fang, L. Xu, Y. Cao and A. Li, Coord. Chem. Rev., 2016, 322, 94–103 CrossRef CAS.
- V. Miikkulainen, M. Leskelä, M. Ritala and R. L. Puurunen, J. Appl. Phys., 2013, 113, 021301 CrossRef.
- C. Stienen, J. Grahl, C. Wölper, S. Schulz and G. Bendt, RSC Adv., 2022, 12, 22974–22983 RSC.
- F. Emmenegger, C. W. Schlaepfer, H. Stoeckli-Evans, M. Piccand and H. Piekarski, Inorg. Chem., 2001, 40, 3884–3888 CrossRef CAS PubMed.
- G. I. Zharkova, S. I. Dorovskikh, S. V. Sysoev, I. P. Asanov, A. V. Panin, N. B. Morozova and I. K. Igumenov, Surf. Coat. Technol., 2013, 230, 290–296 CrossRef CAS.
- Y. Zhang, L. Du, X. Liu and Y. Ding, Polyhedron, 2018, 156, 218–222 CrossRef CAS.
- K. C. Min, M. Kim, Y. H. You, S. S. Lee, Y. K. Lee, T. M. Chung, C. G. Kim, J. H. Hwang, K. S. An, N. S. Lee and Y. Kim, Surf. Coat. Technol., 2007, 201, 9252–9255 CrossRef CAS.
- S. I. Dorovskikh, E. A. Bykova, N. V. Kuratieva, L. N. Zelenina, Y. V. Shubin, N. B. Morozova and I. K. Igumenov, J. Organomet. Chem., 2012, 698, 22–27 CrossRef CAS.
- S. I. Dorovskikh, A. V. Alexeyev, N. V. Kuratieva, T. V. Basova, V. G. Kiselev, L. A. Sheludyakova, Y. V. Shubin, N. B. Morozova and I. K. Igumenov, J. Organomet. Chem., 2013, 741–742, 122–130 CrossRef CAS.
- D. Barreca, G. Carraro, E. Fois, A. Gasparotto, F. Gri, R. Seraglia, M. Wilken, A. Venzo, A. Devi, G. Tabacchi and C. Maccato, J. Phys. Chem. C, 2018, 122, 1367–1375 CrossRef CAS.
- C. Maccato, L. Bigiani, G. Carraro, A. Gasparotto, R. Seraglia, J. Kim, A. Devi, G. Tabacchi, E. Fois, G. Pace, V. Di Noto and D. Barreca, Chem. – Eur. J., 2017, 23, 17954–17963 CrossRef CAS PubMed.
- D. Barreca, G. Carraro, A. Devi, E. Fois, A. Gasparotto, R. Seraglia, C. Maccato, C. Sada, G. Tabacchi, E. Tondello, A. Venzo and M. Winter, Dalton Trans., 2012, 41, 149–155 RSC.
- D. Barreca, G. Carraro, A. Gasparotto, C. Maccato, R. Seraglia and G. Tabacchi, Inorg. Chim. Acta, 2012, 380, 161–166 CrossRef CAS.
- A. Gasparotto, D. Barreca, A. Devi, R. A. Fischer, E. Fois, A. Gamba, C. Maccato, R. Seraglia, G. Tabacchi and E. Tondello, ECS Trans., 2009, 25, 549–556 CrossRef CAS.
- G. Bandoli, D. Barreca, A. Gasparotto, R. Seraglia, E. Tondello, A. Devi, R. A. Fischer, M. Winter, E. Fois, A. Gamba and G. Tabacchi, Phys. Chem. Chem. Phys., 2009, 11, 5998–6007 RSC.
- E. Fois, G. Tabacchi, D. Barreca, A. Gasparotto and E. Tondello, Angew. Chem., Int. Ed., 2010, 49, 1944–1948 CrossRef CAS.
- D. Barreca, E. Fois, A. Gasparotto, R. Seraglia, E. Tondello and G. Tabacchi, Chem. – Eur. J., 2011, 17, 10864–10870 CrossRef CAS.
- G. Tabacchi, E. Fois, D. Barreca and A. Gasparotto, Phys. Status Solidi A, 2014, 211, 251–259 CrossRef CAS.
- D. Barreca, E. Fois, A. Gasparotto, C. Maccato, M. Oriani and G. Tabacchi, Molecules, 2021, 26, 1988 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- T. H. Dunning and P. J. Hay, Modern Theoretical Chemistry, Plenum Press, New York, 1976, ch. 1, vol. 2 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- R. O’ Donoghue, D. Peeters, D. Rogalla, H.-W. Becker, J. Rechmann, S. Henke, M. Winter and A. Devi, Dalton Trans., 2016, 45, 19012–19023 RSC.
- K. j. Puring, D. Zywitzki, D. H. Taffa, D. Rogalla, M. Winter, M. Wark and A. Devi, Inorg. Chem., 2018, 57, 5133–5144 CrossRef PubMed.
- In the originally reported crystal structure a wrong occupancy variable dependency was used for the disorder, resulting in a wrong reported ratio. When corrected, the ratio used here for discussion can be derived (see ESI, § S2.2†).
- M. Kruck, D. C. Sauer, M. Enders, H. Wadepohl and L. H. Gade, Dalton Trans., 2011, 40, 10406–10415 RSC.
- A. S. Al-Bogami, T. S. Saleh, A. E. M. Mekky and M. R. Shaaban, J. Mol. Struct., 2016, 1121, 167–179 CrossRef CAS.
- G. Carraro, C. Maccato, A. Gasparotto, D. Barreca, M. Walter, L. Mayrhofer, M. Moseler, A. Venzo, R. Seraglia and C. Marega, Phys. Chem. Chem. Phys., 2015, 17, 11174–11181 RSC.
- Y. Zhang, L. Du, X. Liu and Y. Ding, Appl. Surf. Sci., 2019, 481, 138–143 CrossRef CAS.
- Pattern no. 00-0047-1049, JCPDS, 2000.
Footnote |
| † Electronic supplementary information (ESI) available: Precursor single crystal XRD data, NMR, mass spectrometry and thermal characterization, and computational analysis; additional XRD, AFM, and FE-SEM results for NiO films. CCDC 2245880 (3) and 2245881 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01282d |
| This journal is © The Royal Society of Chemistry 2023 |