
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of germanium analogue of organic isonitrile with Cu(I) imide in side-on mode†
Shuai-Cong
Huo
a,
Yao
Li
a,
Peng-Fei
Ji
a,
De-Xiang
Zhang
a,
Ying
Yang
 *a and
Herbert W.
Roesky
*b
*a and
Herbert W.
Roesky
*b
aSchool of Chemistry and Chemical Engineering, Central South University, Lushannan Road 932, 410083 Changsha, China. E-mail: yangy@csu.edu.cn
bInstitut für Anorganische Chemie, Universität Göttingen, Tammannstrasse 4, 37077 Göttingen, Germany. E-mail: hroesky@gwdg.de
First published on 20th July 2023
Abstract
[NHC → GeN(Ar)Cu2NAr]2, the formal adduct of germanium analogue of organic isonitrile [GeNAr] with Cu(I) imide [(Cu2NAr)2] (Ar = 2,6-iPr2C6H3) was prepared from the N-heterocyclic carbene (NHC) stabilized acyclic germylene Ge[N(H)Ar]2 by reacting with two equivalents of nBuLi and CuCl(PPh3)3. As elucidated by X-ray crystal structural characterization, the two separated [GeNAr] moieties interacted with [(Cu2NAr)2] core in the side-on mode.
The synthesis of the heavier group 14 element analogues of organic isonitriles, in the form of MNR (M = Ge, Sn, Pb; R = organic group), is both structurally interesting and reactively attractive. Due to the large size of the M(II) elements as well as the weak π bonds, compared to the congeneric C, such heavier analogues mostly exist as oligomers (MNR)n (n = 2, 3, 4), rather than monomers like the organic isonitriles C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NR. For example, using the moderately sterically hindered diisopropyl phenyl ligand, Power and co-workers synthesized the trimeric cyclic (GeNR)3 (R = 2,6-iPr2C6H3), which contained a planar Ge3N3 six-membered ring.1 Other instances included cubanes (M4N4, M = Ge,2 Sn,2–7 and Pb5) or the double cubane (Sn7N8).8 However, germanium derivatives with lower aggregation, like dimers, which could involve multiple bonding of Ge(II) and N, are still rare. Limited available examples included the dimeric (GeNR)2, containing the Ge2N2 four-membered ring, supported by exceptionally bulky R ligands (R = 2,4,6-(CF3)3C6H2,9 2,4,6-tBu3C6H2,10 and 2,6-(2,4,6-Me3C6H2)2C6H3
NR. For example, using the moderately sterically hindered diisopropyl phenyl ligand, Power and co-workers synthesized the trimeric cyclic (GeNR)3 (R = 2,6-iPr2C6H3), which contained a planar Ge3N3 six-membered ring.1 Other instances included cubanes (M4N4, M = Ge,2 Sn,2–7 and Pb5) or the double cubane (Sn7N8).8 However, germanium derivatives with lower aggregation, like dimers, which could involve multiple bonding of Ge(II) and N, are still rare. Limited available examples included the dimeric (GeNR)2, containing the Ge2N2 four-membered ring, supported by exceptionally bulky R ligands (R = 2,4,6-(CF3)3C6H2,9 2,4,6-tBu3C6H2,10 and 2,6-(2,4,6-Me3C6H2)2C6H3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11). In relevant reactivity studies,10 the dimeric Ge(II) imido complexes (GeNR)2 were found to be reactive in oxidative addition or coordination to transition metals. So far, neither example of splitting the dimer (GeNR)2 into separated GeNR species has been reported, nor the interaction of GeNR with metals through both Ge and N sites. So far, the interaction mode in the side-on bonded isonitrile complex [Mo2(μ-η2-CNtBu)-(CO)4Cp2],12 is known.
11). In relevant reactivity studies,10 the dimeric Ge(II) imido complexes (GeNR)2 were found to be reactive in oxidative addition or coordination to transition metals. So far, neither example of splitting the dimer (GeNR)2 into separated GeNR species has been reported, nor the interaction of GeNR with metals through both Ge and N sites. So far, the interaction mode in the side-on bonded isonitrile complex [Mo2(μ-η2-CNtBu)-(CO)4Cp2],12 is known.
It is noteworthy that in the above reports of dimeric (GeNR)2,10,11 the key precursor Ge[N(H)R]2 was specially mentioned, which was found to spontaneously generate dimeric (GeNR)2 and release the primary amine H2NR, as shown in the reaction eqn (1).
 | (1) |
 | (2) |
This gives us a hint of whether a similar reaction would occur if the amine proton H in (1) is replaced by the monovalent metal M(I) (as shown by reaction eqn (2))? If so, would the in situ generated [GeNR] moiety preferentially coordinate with the in situ formal [M(I)2NR] to bring about the possibility of the two [GeNR] species being split? For this end, we planned to carry out the reaction exploration outlined in eqn (2) using Cu(I) of group 11 metals as the costar metal element, which has a good affinity for both germanium and nitrogen.13 To make this methodology more representative and comparable, we proposed to use the common commercially available primary amine H2NAr with large steric hindrance (typically Ar = 2,6-iPr2C6H3) rather than extra bulky ones9–11 to realize our synthetic intention. However, as known, it is more challenging to carry out the reaction using 2,6-diisopropyl aniline as the starting material due to two possible difficulties derived from the literature work. First, if the route of intermolecular deamination of primary amines is used, i.e., if the synthesis starts from Ge[N(SiMe3)2]2 with H2NAr (Ar = 2,6-iPr2C6H3), the more stable trimeric (GeNAr)3 rather than the expected Ge[N(H)Ar]2 is obtained regardless of the ratio of reactants. Second, if the route of intermolecular elimination of salt is used, it could result in very low yields of impure products, as suggested by the previous unsuccessful attempt in which GeCl2·dioxane was treated with even greatly hindered LiN(H)Ar# (Ar# = 2,6-(2,4,6-Me3C6H2)2C6H311). Thus, the two-electron-donor groups (D) was considered, which may offer additional steric protection and help the adduct D → Ge(NHAr)2 to be a much stronger Lewis base. Among such donors, N-heterocyclic carbene (NHC) has been applied in a number of previous studies on the synthesis and reactions of germylenes.14–19 Therefore, our work started with the synthesis of NHC → Ge[N(H)Ar]2.
The primary amine ArNH2 (Ar = 2,6-iPr2C6H3) was treated with the equivalent molar of nBuLi to generate the lithiated product [LiN(H)Ar] (Scheme 1). The latter with or without separation was further reacted with GeCl2·1,4-dioxane in the presence of NHC (:C[N(iPr)C(Me)]2) to give the expected product NHC → Ge[N(H)Ar]2 (1, Scheme 1) as yellow solids in relatively high yield (80%). Alternatively, 1 can also be obtained in moderated yield (74%) by the treatment of GeCl2·1,4-dioxane with equimolar [NHC → LiN(H)Ar]2 (2), which was formed by the addition of NHC to the solution of the intermediate [LiN(H)Ar].
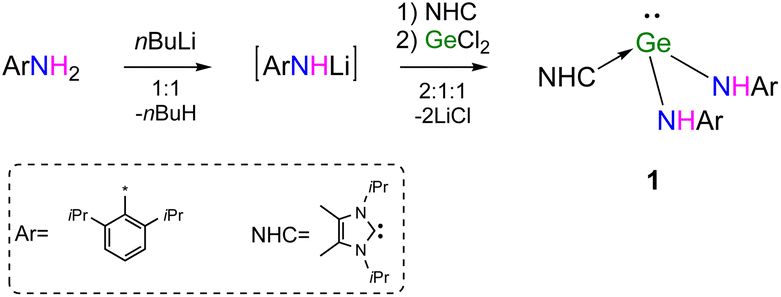 | ||
| Scheme 1 Synthesis of NHC → Ge[N(H)Ar]2 (1). | ||
The melting point of NHC → Ge[N(H)Ar]2 (1) is 135 °C, which is much lower than that of Ge[N(H)R]2 (R = 2,6-(2,4,6-Me3C6H2)2C6H3) (198 °C).11 In the FT-IR spectrum of 1, the strong adsorption at 3349 cm−1 was attributed to NH stretching. The 1H NMR spectrum of 1 exhibited resonances for amide protons observed at δ 5.98 ppm as a broad signal, while the septet at δ 3.51 ppm for the methine protons of isopropyl groups of Ar groups showed a correct integral ratio relative to that of NHC methine protons (2:1), consistent with the theoretically chemical formulation. The elemental analysis of 1 further confirmed the chemical compositions by comparing the measured elemental ratios with the theoretically calculated results.
The saturated solution of 1 in Et2O was stored at room temperature for one day to produce yellow crystals suitable for single-crystal X-ray diffraction measurement. Compound 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Its molecular structure is depicted in Fig. 1. Due to the steric hindrance among NHC and amine ligands, the molecular structure of 1 is not symmetric. The pyramidal geometry of the Ge(II) centre was completed by two amide nitrogen atoms and one carbene carbon atom, where Ge(II) was 1.0267(13) Å away from the C(35)–N(1)–N(2) plane. The sum of bond angles around Ge(II) is 288.19(25)°, indicating the presence of the lone pair of electrons on the vertex Ge(II) atom. In contrast, the carbene C(35) atom is located in an almost trigonal planar environment, as revealed by the sum of the angles at C(35) (360.00(47)°). The Ge(1)–C(35) bond length is 2.144(2) Å, which is longer than the Ge–C single bond in general (∼2.04 Å).20 (Fig. S1c†). The Ge–N bond lengths (1.950(2) and 1.951(2) Å) and N–Ge–N bond angle (97.04(9)°) of 1 are found to be larger than those of Ge[N(H)Ar#]2 (1.896(2) Å), (88.6(2)°).11
. Its molecular structure is depicted in Fig. 1. Due to the steric hindrance among NHC and amine ligands, the molecular structure of 1 is not symmetric. The pyramidal geometry of the Ge(II) centre was completed by two amide nitrogen atoms and one carbene carbon atom, where Ge(II) was 1.0267(13) Å away from the C(35)–N(1)–N(2) plane. The sum of bond angles around Ge(II) is 288.19(25)°, indicating the presence of the lone pair of electrons on the vertex Ge(II) atom. In contrast, the carbene C(35) atom is located in an almost trigonal planar environment, as revealed by the sum of the angles at C(35) (360.00(47)°). The Ge(1)–C(35) bond length is 2.144(2) Å, which is longer than the Ge–C single bond in general (∼2.04 Å).20 (Fig. S1c†). The Ge–N bond lengths (1.950(2) and 1.951(2) Å) and N–Ge–N bond angle (97.04(9)°) of 1 are found to be larger than those of Ge[N(H)Ar#]2 (1.896(2) Å), (88.6(2)°).11
 | ||
| Fig. 1 Molecular structure of NHC → Ge[N(H)Ar]2 (1). Ellipsoids are drawn at 30% probability. H atoms except for NH groups have been omitted for clarity. Selected bond lengths (Å) and bond angles (°): Ge(1)–C(35) 2.144(2), Ge(1)–N(1) 1.951(2), Ge(1)–N(2) 1.950(2); N(1)–Ge(1)–N(2) 97.04(9), N(1)–Ge(1)–C(35) 89.75(8), N(2)–Ge(1)–C(35) 101.40(8). | ||
Interestingly, the Ge(II) atom of the acyclic compound 1 possesses a highly similar environment to that in a cyclic germylene L′Ge ← NHC (L′ = HC[C(CH2)N(Ar)]C(Me)N(Ar), Ar = 2,6-iPr2C6H3; NHC = :C[N(Me)C(Me)]2), in terms of the bond lengths and bond angles of CGeN2 core with angle around Ge(II) (288.01°), Ge–C bond length (2.147(3) Å), Ge–N bond lengths (1.927(3) and 1.929(3) Å), and N–Ge–N bond angle (95.1(1)°).15
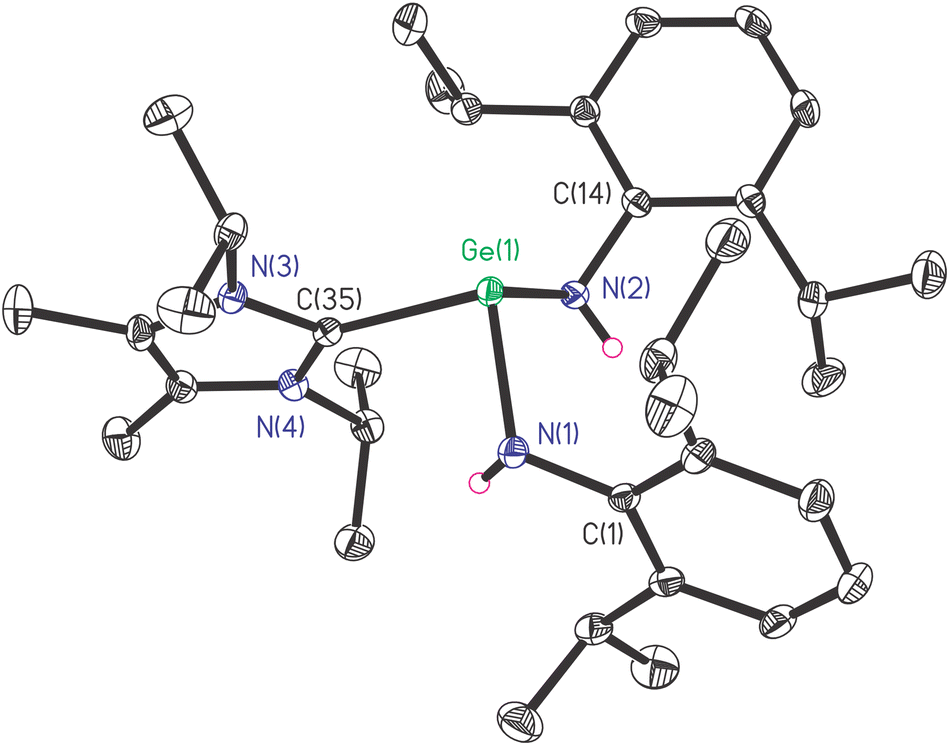
To gain a deeper understanding of the electronic nature of 1, the quantum chemical calculations were performed using PBE1PBE/genecp (def2-TZVP basis set for Ge and def2SVP basis set for C, N, H) with Gaussian 16 program suite,21 and analysed with Multiwfn.22 The optimized structure of 1′ is the global minimum and Cartesian coordinates were listed in Table S1 (ESI)† with a geometry similar to that determined experimentally. The frontier molecular orbitals, Ge(II)–C bond, and the interaction between carbene and germylene fragments were investigated. The HOMO and LUMO surfaces are depicted in Fig. 2.
 | ||
| Fig. 2 HOMO (a) and LUMO (b) of NHC → Ge[N(H)Ar]2 (1′) (H atoms are omitted for clarity). Optimized geometry: Ge(1)–C(35) 2.14101 Å, Ge(1)–N(1) 1.9537 Å, Ge(1)–N(2) 1.9467 Å; N(1)–Ge(1)–N(2) 97.2133°, N(1)–Ge(1)–C(35) 87.3829°, N(2)–Ge(1)–C(35) 100.3288°. | ||
The HOMO and LUMO energies are −4.73 and −0.30 eV, respectively, and the energy gap is 4.43 eV (426.99 kJ mol−1), which falls in the common range for germylenes.23
To investigate the electron transfer between the NHC and [ArN(H)]2Ge fragments, the extended charge decomposition analysis (ECDA) was performed, which suggested the 0.38 net electrons obtained by Ge[N(H)Ar]2 from NHC (Fig. S11†).
The lithiation of 1 was carried out in situ and the subsequent transmetallation was performed with CuCl(PPh3)3 to afford the separation of 3 as dark yellow solid in 40% yield (Scheme 2).
 | ||
| Scheme 2 Synthesis of [NHC → GeN(Ar)Cu2NAr]2 (3) and the possible intermediates (3A and 3B). | ||
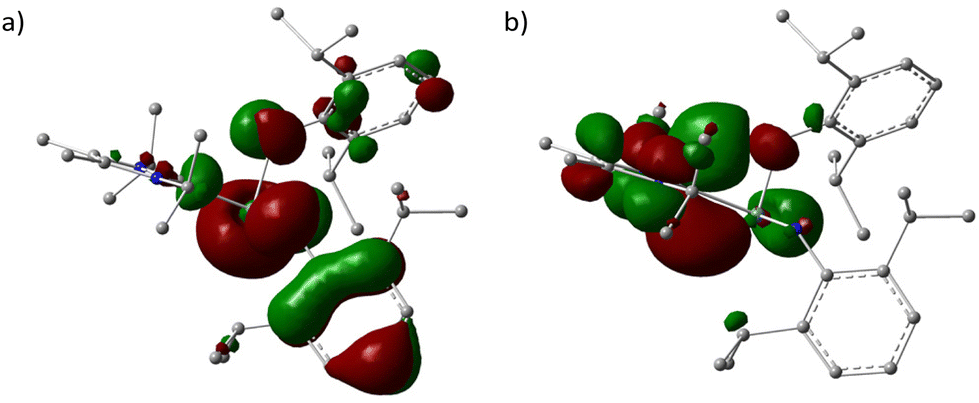
NMR characterizations and elemental analysis have shown that compound 3 may preclude the presence of PPh3 ligand and suggested a 2:1 composition of Ar and NHC moieties in it. As expected, X-ray single crystal diffraction proved this composition and further indicated the cleavage of Ge–N bonds and the concurrent formation Ge–Cu and N–Cu bonds (Fig. 3).
 | ||
| Fig. 3 The molecular structure of [NHC → GeN(Ar)Cu2NAr]2 (3). Ellipsoids are drawn at 30% probability. H atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (°): Ge(1)–C(1) 1.910(5), Ge(1)–N(3) 1.878(4), Ge(1)–Cu(1) 2.8909(8), Ge(1)–Cu(2) 2.6988(9), Cu(1)–N(3) 1.867(4), Cu(2)–N(3) 1.880(4), Cu(1)–N(4) 1.893(4), Cu(2)–N(4A) 1.927(4), Cu(1)–Cu(2) 2.6333(8), Cu(1)–Cu(2A) 2.7573(8); N(3)–Ge(1)–C(1) 175.68(19), Cu(1)–N(3)–Cu(2) 89.30(16), Cu(1)–N(4)–Cu(2A) 92.41(16); A1 − X, −Y, 1 − Z. | ||
It is therefore proposed as illustrated in Scheme 2 that, the reaction may proceed four steps, i.e. lithiation, transmetallation (3A), dimerization (3B), and rearrangement, to give [NHC → GeN(Ar)Cu2NAr]2 (3), as a unique adduct of (HNC → GeNAr) with (Cu2NAr)2 in a 2:1 ratio.
As shown in Fig. 3, compound [NHC → GeN(Ar)Cu2NAr]2 (3) crystallizes in the monoclinic P21/n space group with an inversion center, in which Ge(1) centre is located in a four-coordinated environments by bonding to C(1), N(3) and two Cu atoms. The Cu-linked N atoms adopted two different coordinate environments, in which the amino N(3) is four-coordinated in tetrahedral geometry, while the imino N(4) is three-coordinated in trigonal planar geometry; each of them bonded to two Cu atoms with shorter average Cu–N bond lengths (1.867(4), 1.880(4) Å) for the former and longer averages for the latter (1.893(4), 1.926(4) Å). The Cu4N4 core is highly planar, with nearly right angles of Cu–Cu–Cu (88.75(2)° and 91.25(2)°) and Cu–N–Cu (89.3(2)° and 92.4(2)°). This is significantly different from the previous related examples, where either ligands used were homoleptic,24,25 or the Cu4N4 plane was very distorted.26 The Cu–Cu separations (2.6334(9) Å and 2.7572(7) Å) in 3 are in the common range,24–26 while Ge–C (1.909(6) Å) and Ge–N (1.878(4) Å) bond lengths are greatly shortened compared to those in 1 (2.146(3) Å and 1.946(2)–1.950(2) Å, respectively), and can be seen to fall between the single and double Ge–C/N bonds.27,28 Interestingly, Ge(1) and N(3) were found to be right on the NHC backbone plane (0.002(9) Å and 0.078(15) Å deviation, respectively), and the C(1), Ge(1), and N(3) atoms were almost arranged in a straight line (C(1)–Ge(1)–N(3) 175.6(2)°). Compound 3 represents the first example separating the GeNR monomers by interaction with metals in the side-on bonded isonitrile mode.
Using the density functional method PBE1PBE/genecp (def2-TZVP basis set for Ge and Cu and def2SVP basis set for C, N, H), the optimized structure 3′ was obtained with constraints of heavy-atom positions. Calculation of the vibrational frequencies revealed no imaginary frequencies, indicating that this structure is at a minimum on the potential energy surface. The optimized Cartesian coordinates of 3′ were listed in Table S2.†Fig. 4 presented the HOMO and LUMO surfaces of 3′.
 | ||
| Fig. 4 HOMO (a) and LUMO (b) of [NHC → GeN(Ar)Cu2NAr]2 (3′) (H atoms are omitted for clarity). | ||
The HOMO/LUMO energies 3′ were −3.21 and −2.29 eV, respectively, leading to a HOMO–LUMO gap of 0.92 eV (89.14 kJ mol−1). The Wiberg bond order of the Ge(II)–N (0.92) and Ge(II)–C (0.98) bonds (Fig. S13†) in 3′ suggested the intermediate nature between single and double bonds, when compared with those in 1′ (0.56 and 0.62). The Ge(1)–N(3) bond is largely occupied (1.89e) (Fig. S14†), in which the Ge(II) centre uses sp0.78 hybrid orbital bonding to N (sp8.46) with high p character. The lone pair of electrons (1.33e) on Ge(1) is found somewhat reduced, in a natural bond orbital of high p character (sp29.91). The ECDA (notes in Fig. S15†) showed that each NHC gave 0.69 net electrons to the rest fragment of 3′, while each NHC → GeN(Ar) moiety rendered 0.35 net electrons, evidencing the donor role of NHC in carbene-germylene tandem interaction.
In conclusion, we have synthesized and characterized the N-heterocyclic carbene stabilized acyclic germylene Ge[N(H)Ar]2 (Ar = 2,6-iPr2C6H3) (1). The introduction of an extra auxiliary carbene donor proved to be effective in decreasing the bulky demand of the Ar group. The reaction of lithiated 1 with CuCl(PPh3)3 underwent rearrangement to form [NHC → GeN(Ar)Cu2NAr]2 (3), the formal adduct of germanium analogue of organic isonitrile GeNAr with Cu(I) imide Cu2NAr. Further exploration of the reactivity and properties of compound 3 can be expected to lead to certain discoveries. In addition, acyclic germylene 1 can be considered as an important and versatile precursor for the preparation of the title product and possibly more other M(I), M(II) or M(III) derivatives to bring out interesting compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21771194). We are grateful for resources from the High-Performance Computing Center of Central South University.References
- R. A. Bartlett and P. P. Power, J. Am. Chem. Soc., 1990, 112, 3660–3662 CrossRef CAS.
- W. J. Grigsby, T. Hascall, J. J. Ellison, M. M. Olmstead and P. P. Power, Inorg. Chem., 1996, 35, 3254–3261 CrossRef CAS PubMed.
- M. Veith, Angew. Chem., Int. Ed. Engl., 1987, 26, 1–14 CrossRef.
- R. E. Allan, M. A. Beswick, A. J. Edwards, M. A. Paver, M.-A. Rennie, P. R. Raithby and D. S. Wright, J. Chem. Soc., Dalton Trans., 1995, 1991–1994 RSC.
- H. Chen, R. A. Bartlett, H. V. R. Dias, M. M. Olmstead and P. P. Power, Inorg. Chem., 1991, 30, 3390–3394 CrossRef CAS.
- M. Veith and O. Recktenwald, Z. Naturforsch., B: Chem. Sci., 1983, 38, 1054–1061 CrossRef.
- M. Veith, M.-L. Sommer and D. Jäger, Chem. Ber., 1979, 112, 2581–2587 CrossRef CAS.
- F. Benevelli, E. L. Doyle, E. A. Harron, N. Feeder, E. A. Quadrelli, D. Sáez and D. S. Wright, Angew. Chem., Int. Ed., 2000, 39, 1501–1503 CrossRef CAS PubMed.
- J.-T. Ahlemann, H. W. Roesky, R. Murugavel, E. Parisini, M. Noltemeyer, H.-G. Schmidt, O. Müller, R. Herbst-Irmer, L. N. Markovskii and Y. G. Shermilovich, Chem. Ber., 1997, 130, 1113–1121 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and A. J. Thorne, J. Chem. Soc., Chem. Commun., 1990, 1587–1589 RSC.
- W. A. Merrill, R. J. Wright, C. S. Stanciu, M. M. Olmstead, J. C. Fettinger and P. P. Power, Inorg. Chem., 2010, 49, 7097–7105 CrossRef CAS PubMed.
- H. Adams, N. A. Bailey, C. Bannister, M. A. Faers, P. Fedorko, V. A. Osborn and M. J. Winter, J. Chem. Soc., Dalton Trans., 1987, 341–348 RSC.
- J. Hlina, H. Arp, M. Walewska, U. Flörke, K. Zangger, C. Marschner and J. Baumgartner, Organometallics, 2014, 33, 7069–7077 CrossRef CAS PubMed.
- J. A. Cabeza, P. Garcia-Alvarez and C. J. Laglera-Gandara, Eur. J. Inorg. Chem., 2020, 784–795 CrossRef CAS.
- S. Yao, Y. Xiong and M. Driess, Chem. Commun., 2009, 6466–6468 RSC.
- C. Jones, A. Sidiropoulos, N. Holzmann, G. Frenking and A. Stasch, Chem. Commun., 2012, 48, 9855–9857 RSC.
- N. Katir, D. Matioszek, S. Ladeira, J. Escudié and A. Castel, Angew. Chem., Int. Ed., 2011, 50, 5352–5355 CrossRef CAS PubMed.
- A. Jana, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2013, 52, 12179–12182 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, G. Tan, S. Inoue and M. Driess, J. Am. Chem. Soc., 2013, 135, 5004–5007 CrossRef CAS PubMed.
- P. Jutzi, S. Keitemeyer, B. Neumann and H. G. Stammler, Organometallics, 1999, 18, 4778–4784 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- L. Wang, Y. S. Lim, Y. Li, R. Ganguly and R. Kinjo, Molecules, 2016, 21, 990 CrossRef PubMed.
- P. Reiß and D. Fenske, Z. Anorg. Allg. Chem., 2000, 626, 1317–1331 CrossRef.
- M. Veith and K. L. Woll, Chem. Ber., 1993, 126, 2383–2388 CrossRef CAS.
- M. K. Davies, P. R. Raithby, M.-A. Rennie, A. Steiner and D. S. Wright, J. Chem. Soc., Dalton Trans., 1995, 2707–2709 RSC.
- S. Kundu, P. P. Samuel, A. Luebben, D. M. Andrada, G. Frenking, B. Dittrich and H. W. Roesky, Dalton Trans., 2017, 46, 7947–7952 RSC.
- M. J. Evans, F. M. Burke, P. M. Chapple and J. R. Fulton, Inorg. Chem., 2021, 60, 8293–8303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, calculation details and outputs, characterisation data, NMR spectra, Cartesian coordinates, crystal data and refinement. CCDC 2201109-2201112. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02094k |
| This journal is © The Royal Society of Chemistry 2023 |