
Profiling MAP kinase cysteines for targeted covalent inhibitor design†
Ruibin
Liu
 ,
Neha
Verma‡
,
Jack A.
Henderson
,
Shaoqi
Zhan
and
Jana
Shen
*
,
Neha
Verma‡
,
Jack A.
Henderson
,
Shaoqi
Zhan
and
Jana
Shen
*
University of Maryland School of Pharmacy, Baltimore, MD, USA. E-mail: jana.shen@rx.umaryland.edu
First published on 3rd November 2021
Abstract
Mitogen-activated protein kinases (MAPK) are important therapeutic targets, and yet no inhibitors have advanced to the market. Here we applied the GPU-accelerated continuous constant pH molecular dynamics (CpHMD) to calculate the pKa's and profile the cysteine reactivities of all 14 MAPKs for assisting the targeted covalent inhibitor design. The simulations not only recapitulated but also rationalized the reactive cysteines in the front pocket of JNK1/2/3 and the extended front pocket of p38α. Interestingly, the DFG − 1 cysteine in the DFG-in conformation of ERK1/ERK2 was found somewhat reactive or unreactive; however, simulations of MKK7 showed that switching to the DFG-out conformation makes the DFG − 1 cysteine reactive, suggesting the advantage of type II covalent inhibitors. Additionally, the simulations prospectively predicted several druggable cysteine and lysine sites, including the αH head cysteine in JNK1/3 and DFG + 6 cysteine in JNK2, corroborating the chemical proteomic screening data. Given the low cost and the ability to offer physics-based rationales, we envision CpHMD simulations to complement the chemo-proteomic platform for systematic profiling cysteine reactivities for targeted covalent drug discovery.
Introduction
Mitogen-activated protein kinases (MAPKs) are a family of kinases downstream of the important MAPK signal transduction pathway which contains three tiers. Stimulus signals such as growth factors, mitogens, cellular stresses, and inflammatory cytokines are first received by MAPKK kinases (MAPKKKs), e.g., BRAF and CRAF, which activate MAPK kinases (MAPKKs), e.g., MEK1/2, through phosphorylation. The activated MAPKKs transfer the signals further to activate the MAP kinases (MAPKs), which elicit biological responses for cell growth, survival, differentiation, development, inflammation, and apoptosis.1,2 Thus, targeting the MAPK pathway is an important therapeutic strategy for many diseases. In recent years, the U.S. Food and Drug Administration (FDA) approved several inhibitors targeting the first two tiers of the MAPK pathway (e.g., BRAF inhibitors vemurafenib and dabrafenib and MEK inhibitors trametinib and binimetinib3); however, no inhibitor of the downstream MAPKs has gained the regulatory approval as of July 2021. Development of inhibitors against the MAPKs is important, particularly because of the innate and acquired resistance to RAF or MEK inhibitors.4,5The MAPKs contain 14 genes, of which 10 are the classical and 4 are the atypical MAPKs (Fig. 1a). The well-characterized classical MAPKs have a consensus TXY motif in the activation loop for dual phosphorylation.2 They include three subfamilies, the extracellular signal-regulated kinases ERK1/2/5, c-Jun N-terminal kinases JNK1/2/3, and p38 enzymes p38α/β/γ/δ (Fig. 1a enlarge). The less studied atypical MAPKs have no consensus motif for phosphorylation and they include ERK3/4/7 and nemo-like kinase (NLK).2
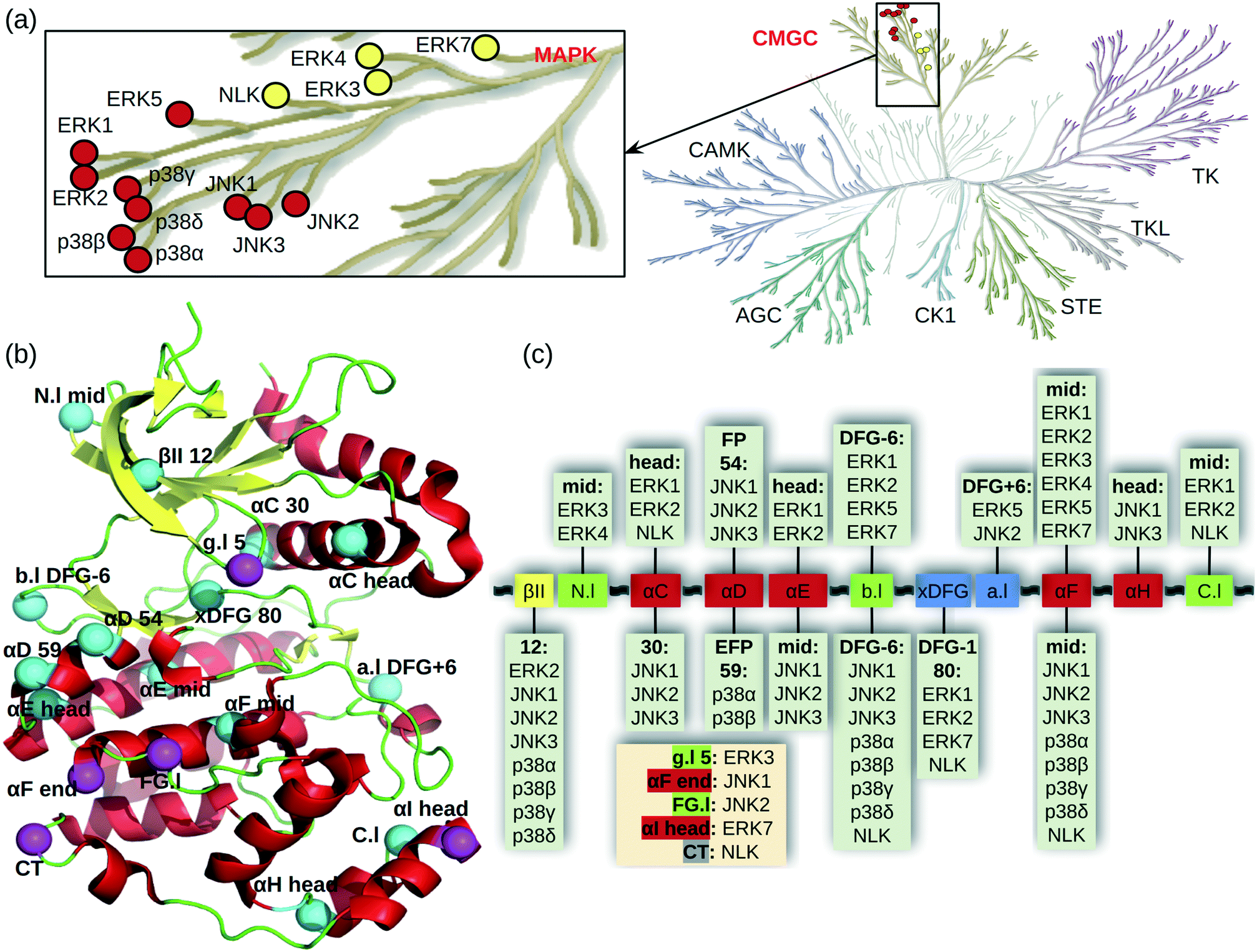 | ||
| Fig. 1 Cysteine locations in the MAPKs. (a) MAPK family in the human kinome (kinome tree generated with KinMap11). A zoomed-in view shows the classical (red) and atypical (yellow) MAPKs. (b) Locations of cysteines shown in a typical kinase structure. Cysteines are shown as cyan (conserved) and magenta (unique) spheres. The numbers refer to the positions in the 85 kinase ligand binding site residues (aligned by KLIFS based on all available kinase crystal structures12). For residues not in the ligand binding site, their positions relative to the nearest secondary structure or the DFG motif are given. Secondary structure annotations are as follows, Gly-rich or P loop (g.l), β-II strand (βII), N-lobe loop between β-II and β-III strands (N.l), αC/D/E/F/G/H/I helices (αC/D/E/F/G/H/I), back loop between β-VII and β-VIII strands (b.l), X-Asp-Phe-Glu motif (xDFG), activation loop (a.l), loop between αF and αG helices (FG.l), C-lobe loop between αH and αI helices (C.l), and C terminal (CT). (c) MAPK cysteines mapped onto the kinase ligand binding site structural motifs. Helix, β strand, and loop are colored red, yellow, and green, respectively. The xDFG motif and activation loop are colored blue. The unique cysteine positions are listed in a beige box. | ||
ERK1/2 signaling regulates cell proliferation, survival, growth, metabolism, migration, and differentiation in response to extracellular stimuli.2 As such, they are attractive anticancer drug targets, with several inhibitors entering the clinical trials in recent years.3,6 JNKs play important roles in neurodegenerative diseases, inflammation, and cancer progression, and the phase II clinical trial of a selective JNK inhibitor AS602801 is ongoing.1,7 p38 kinases have received more attention than ERKs and JNKs, with over 20 candidate drugs in clinical trials.8 Since SARS-CoV-2 virus often induces severe inflammation, inhibition of p38 has been hypothesized to attenuate COVID-19 infection, as upregulation of p38 MAPK pathway activates pro-inflammatory cytokines.9 The physiological role of atypical MAPKs is poorly understood, but some studies showed that they are also involved in cell proliferation, differentiation and the production of cytokines.10
While most MAPK inhibitors are reversible noncovalent binders, targeted covalent kinase inhibitors (TCKIs) have emerged in recent years. TCKIs carry an electrophilic warhead to enable chemical reaction with a nucleophilic residue once a noncovalent enzyme/inhibitor complex is formed.13 When designed properly, a TCKI has the potential to improve potency, pharmacodynamics, and target selectivity over the reversible analog;14–16 in recent years, over a dozen TCKIs have been approved for the treatment of various cancer conditions.
Cysteine is the most nucleophilic amino acid and the most frequently targeted site in a TCKI design.14–17 Kinetic experiments showed that the apparent or observed rate constant of a thiol nucleophilic addition is inversely related to the thiol pKa.18–21 This can be explained, as a decrease in pKa leads to an increase in the availability of the highly nucleophilic thiolate form.14,22,23 A cysteine in the fully solvent exposed condition (e.g., in the model pentapeptide AACAA) has a pKa around 8.6,24 and thus mainly protonated at physiological pH 7.4. Therefore, a pKa downshift relative to the model value promotes the nucleophilicity of a cysteine, and as such may be used as a proxy for estimating the reactivity of cysteine. Note, the reactivity of a thiol (defined as the apparent rate constant) is not to be confused with the intrinsic nucleophilicity of a thiolate which may, dependent on the particular molecule, increase with the thiol pKa.18,21 A recent experiment demonstrated that the electrophilic fragments are more reactive towards the cysteine in cathepsin B which has a lower pKa than that in MurA.25
MAPKs have 2–8 cysteines, mostly near the ligand binding site and shared among several members of the MAPK family (Fig. 1b). Fig. 1c shows the locations of the MAPK cysteines relative to the kinase secondary structures mapped onto the aligned ligand binding site sequence taken from KLIFS.12 Nearly all secondary structures have one cysteine conserved to a varying degree. The cysteine in the middle of the αF helix is conserved among all MAPKs. The second most common cysteine location is DFG-6 in the back loop (b.l) that connects the β-VII and β-VIII strands near the ATP binding pocket; this Cys exists in all MAPKs except for ERK3/7 (Fig. 1c). By contrast, ERK3 has a unique cysteine on the Gly-rich loop (g.l, also known as the P loop), and JNK2 has a unique cysteine in the loop connecting the αF and αG helices. Some of the MAPK cysteine locations have already been exploited in TCKI design, e.g., the front pocket (FP) N-terminal capping (Ncap) cysteine of the αD helix in JNKs, the extended FP cysteine in p38α, and the DFG − 1 cysteine in ERK1/2.
The knowledge of highly nucleophilic cysteine locations is valuable in TCKI design, because pairing a highly reactive cysteine with a weakly reactive electrophilic warhead may lower the risk of off-target toxicities and increase selectivity. In traditional noncovalent drug discovery, binding site difference is a main driver for inhibitor selectivity; in targeted covalent drug discovery, the variation in cysteine reactivity offers an additional parameter for selective design. Thus, the knowledge of relative cysteine reactivity is also important. For example, directing at a cysteine that is conserved among a protein family may only be worthwhile if the binding site and/or the cysteine reactivity varies.
Molecular dynamics (MD) is a type of computer simulation that offers atomically detailed view of conformational dynamics of proteins using the physics-based energy function (force field). The continuous constant pH MD (CpHMD) allows sampling of microscopic conformational states while simultaneously titrating all ionizable groups.26 Due to the direct coupling between conformational dynamics and proton titration, it has emerged as one of the most accurate protein pKa predictors.27 Recently, the generalized Born (GB) implicit solvent based CpHMD method in the independent pH mode or the pH replica-exchange protocol (for accelerated convergence)28,29 have been validated for cysteine pKa predictions.30,31 For 24 proteins with NMR-determined cysteine pKa's that significantly deviate from the solution (model) value, the root-mean-square error (RMSE) of the CpHMD predicted pKa's is 1.2, which is more than two units smaller than the RMSE's from the traditional pKa calculations based on the Poisson–Boltzmann (PB) or empirical PROPKA method.31 The accuracy (percentage of true positives and negatives) of predicting thiolates at physiological pH is about 81% with replica-exchange CpHMD, compared to the accuracy below 50% with the PB or empirical methods.31 Importantly, the CpHMD simulations were able to recapitulate the mutation induced changes of the cysteine pKa's of several proteins.31 Most recently, CpHMD was applied to systematically examine how the local environment affects the reactivity of the front-pocket N-terminal cap cysteine for all human kinases.32
To assist the TCKI discovery efforts against MAPKs, here we employed the GPU-accelerated continuous constant pH molecular dynamics (CpHMD) titration simulations33,34 to systematically profile and rationalize the reactivities of cysteines in all 14 MAPKs using their pKa as a proxy. We identified several reactive cysteines in JNK1/2/3, p38α/β, ERK1/2/5, and the atypical kinases ERK3 and NLK. With the exception of the DFG − 1 cysteine in ERK1/2, all previously targeted cysteine locations either by TCKIs or probes/fragments in chemo-proteomic screening35,36 are recapitulated. We discuss the physical basis for the increased cysteine reactivities and the conformational factor that modulates the reactivity of the DFG − 1 cysteine. Additionally, we report prospectively predicted locations that may be targeted by TCKIs.
Results and discussion
Reactivity assessment of cysteines in La antigen
To further demonstrate the CpHMD method for discriminating reactive from nonreactive cysteines, we tested it on the La antigen protein which is used in the NMR-based thio reactivity assay called ALARM NMR.37 In contrast to glutathione (a tripeptide γGlu-Cys-Gly), which is used in the fluorescence-based thio reactivity assay, La antigen contains two cysteines Cys232 and Cys245. While Cys232 was modified by reactive compounds, Cys245 was considered exceptionally reactive on the basis of NMR shifts.37 Using a similar definition as in our previous work,30 we consider a cysteine hyper reactive if >50% of the population is deprotonated at physiological pH 7.4, which typically corresponds to a pKa < 7.4. We consider a cysteine reactive if the deprotonated fraction is 10–50% (typical pKa 7.4–8.4), and somewhat reactive if the deprotonated fraction is 1–10% (typical pKa 8.4–9.4).CpHMD titration of La antigen yielded a slightly upshifted pKa of 8.8 ± 0.1 for Cys232 and a highly downshifted pKa of 3.7 ± 0.1 for Cys245. Thus, according to our definition, Cys232 is somewhat reactive and Cys245 is hyper reactive, in agreement with the NMR-based assessment of their reactivities.37 The 5-unit difference in the pKa's of the two cysteines can be explained by the local conformational and electrostatic environment. Cys232 is located on the unstructured N-terminal loop and pointing toward solvent without significant interactions with other residues (Fig. 2a). Therefore, its pKa is similar to the model value of 8.6. In contrast, Cys245 is the Ncap residue of a helix (Fig. 2a). It is widely known that the helix dipole stabilizes a negative charge on the N terminus,38,39 and it was hypothesized that Ncap cysteines are in the highly reactive thiolate form prone to chemical modification.22 The latter was used to explain why Cys is the least frequent Ncap residue.22 In addition to the helix dipole stabilization, the deprotonated Cys245 in La antigen can form a salt bridge with Arg266 on the opposite loop (Fig. 2b). The pH-dependent deprotonation of Cys245 is well correlated with the pH-dependent formation of the salt-bridge between Cys245 and Arg266 (Fig. 2c and d), suggesting that the latter is a major contributor to the pKa downshift. These data further demonstrate that CpHMD distinguishes and provides rationale.
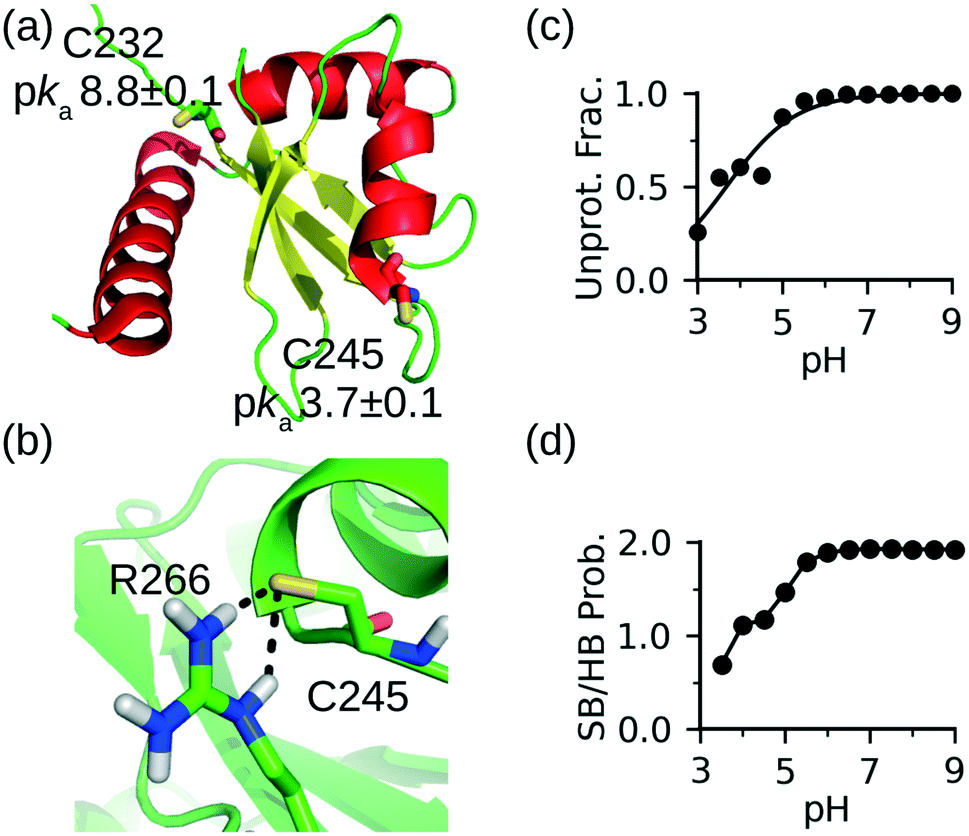 | ||
| Fig. 2 CpHMD simulations recapitulated the relative reactivities of the two cysteines in La antigen. (a) Solution NMR structure of La antigen (PDB: 1owx).40 The two cysteine locations and calculated pKa's are shown. (b) A zoomed-in view of the local environment of Cys245 taken from the simulation replica at pH 7. (c) Deprotonated fraction of Cys245 at different pH values. (d) Probability of the salt-bridge formation between Cys245 and Arg266. Salt bridge is defined by a distance cutoff 4.0 Å between the sulfur of cysteine and a nitrogen of arginine. | ||
Reactivities of the previously targeted MAPK cysteines
We next performed replica-exchange CpHMD titration to calculate the pKa's and identify reactive cysteines in the 14 MAPKs (Tables 1 and S1†). Our simulations predicted that the FP Ncap cysteine of the αD helix (position 54 in KLIFS alignment12): Cys116 in JNK1/2 or Cys154 in JNK3 is reactive or hyper-reactive, with the highly downshifted pKa's of 7.5, 8.0, or 6.3, respectively. Consistent with the predictions, the FP Ncap cysteine of all three JNKs has been covalently modified.41,42 This is not surprising, as the FP Ncap cysteine is the most frequently targeted site of covalent modification in kinases (see ref. 15 and unpublished work by Liu, Zhan, Shen, et al.).| Kinase name | PDB | Targeted location and reactivity | Predicted new location and reactivity |
|---|---|---|---|
| a Cysteines that have been targeted by TCKIs are in bold and indicated by references. Except for the DFG − 1 cysteine in ERK1/2, all these cysteines are predicted as reactive by CpHMD. Reactive cysteines that have been identified by the chemical proteomic screening studies are indicated by references. Residue locations in a kinase are given in parentheses: FP (front pocket), EFP (extended front pocket), and C terminal (CT). Other names are explained in Fig. 1. ERK4/7 and NLK were simulated with the homology models (HMs). For JNK2, simulations starting from a different X-ray structure (PDB: 3e7o) gave very similar results except for C177 which is mutated to Ser. | |||
| Classical MAPKs | |||
| JNK1 (MAPK8) | 2xrw | C116 (FP)41 reactive | C245 (αH head)36 reactive |
| JNK2 (MAPK9) | 3npc | C116 (FP)41 reactive | C177 (a.l DFG + 6)35,36 hyper, K153 (HRD + 2) somewhat |
| JNK3 (MAPK10) | 6emh | C154 (FP)42 hyper | C283 (αH head)36 reactive |
| p38α (MAPK14) | 5tbe | C119 (EFP)43 hyper | |
| p38β (MAPK11) | 3gc8 | C119 (EFP) somewhat | |
| p38γ (MAPK12) | 1cm8 | ||
| p38δ (MAPK13) | 5ekn | ||
| ERK1 (MAPK3) | 6ges | C183 (DFG − 1)44 somewhat | C144 (αE head) somewhat, C271 (C.l) reactive |
| ERK2 (MAPK1) | 6g54 | C166 (DFG − 1)44 unreactive | C254 (C.l) hyper |
| ERK5 (MAPK7) | 4ic8 | C208 (a.l DFG + 6) hyper, C359 (CT) reactive | |
| Atypical MAPKs | |||
| ERK3 (MAPK6) | 2i6l | C28 (g.l 5) reactive, C42 (N.l) somewhat | |
| ERK4 (MAPK4) | HM | K151 (HRD + 2) somewhat | |
| ERK7 (MAPK15) | HM | K29 (βII) somewhat, K42 (catalytic) reactive, C249 (αI head) hyper | |
| NLK (NLK) | HM | K167 (catalytic) reactive, C179 (αC head) somewhat, C369 (C.l) hyper | |
For the p38 kinases, CpHMD simulations predicted that Cys119 located in the middle of the αD helix of p38α (position 59 in KLIFS alignment,12) is hyper reactive, with a downshifted pKa of 6.9 (Table 1). Consistent with this prediction, Cys119, which is also known as the extended front pocket (EFP) cysteine,15 has been covalently modified in p38α,43 although its location is further away from the ATP binding pocket as compared to the FP cysteine. Interestingly, the EFP cysteine also exists in p38β, but CpHMD simulations predicted it as unreactive, and no TCKI directed at this site has been developed so far.
As to the ERKs, CpHMD simulation predicted Cys183 located at the DFG − 1 position in ERK1 as somewhat reactive, with the calculated pKa of 9.1; however, the simulation predicted the analogous Cys166 in ERK2 as unreactive, with the calculated pKa of 10.0. Note, both cysteines have been recently targeted by the same TCKI.44 We will come back to the discussion of the DFG − 1 cysteines.
Identification of new reactive cysteine and lysine locations
In addition to retrospectively predicting the previously targeted cysteines, we also prospectively predicted reactive cysteines at various other locations that can be potential target sites for MAPK TCKI design. Some of these reactive cysteines are in the proximity of the ATP binding site. Consistent with the chemical proteomic studies,35,36 the JNK2 Cys177 located at the DFG + 6 position on the activation loop is predicted as hyper-reactive with the pKa shifted below 7.0. Interestingly, the CpHMD titration also predicted the DFG + 6 Cys208 of ERK5 as hyper-reactive with the pKa shifted below 6.0. Cys144 on the αE helix of ERK1 is somewhat reactive with a calculated pKa of 8.8. Cys28 on the P-loop (g.l 5) which is unique to the atypical ERK3 is also reactive with a downshifted pKa of 7.8. Note, the P-loop and DFG + 6 cysteines are popular sites of covalent modification in non-MAP kinases; a 2018 survey found at least 35 and 7 TCKIs directed at the P-loop and DFG + 6 cysteines, respectively.15 Aside from the ATP binding site, CpHMD simulations also identified several reactive cysteines in the C-terminal domain, including the αH head cysteines: JNK1 Cys245 and JNK3 Cys283, and the cysteine on the C-lobe loop between αH and αI helices (C.l): ERK1 Cys271, ERK2 Cys254, and NLK Cys369. Cys245 in JNK1 and Cys283 in JNK3 have been covalently labeled in a recent chemo-proteomic study.36 Besides cysteines, the simulations also predicted several reactive or somewhat reactive lysines, including the catalytic lysine in ERK7 (Lys42) and NLK (Lys167), HRD + 2 lysine in JNK2 (Lys153) and ERK4 (Lys151), and βII lysine in ERK7 (Lys29). Below we will examine the physical determinants that decrease the pKa values of the reactive cysteines we identified.Front-pocket cysteines in JNK1/2/3
The FP Ncap cysteine conserved among JNKs (Fig. 1c) is the most frequently targeted site of covalent modification.15 Analysis of the CpHMD trajectories found that a common contributor to thiolate stabilization is the hydrogen bond (h-bond) formation between the thiolate and the side chains of Gln at the i + 1 position (Gln117 in JNK1/2 or Gln155 in JNK3) and Ser at the i + 39 position (Ser155 in JNK1/2 and Ser193 in JNK3), where i indicates the FP cysteine position (Fig. 3a and S1†). Additionally, Asn114 in JNK1, Tyr185 in JNK2, or Ser72 in JNK3 can also donate a h-bond, stabilizing the thiolate form of the FP cysteine.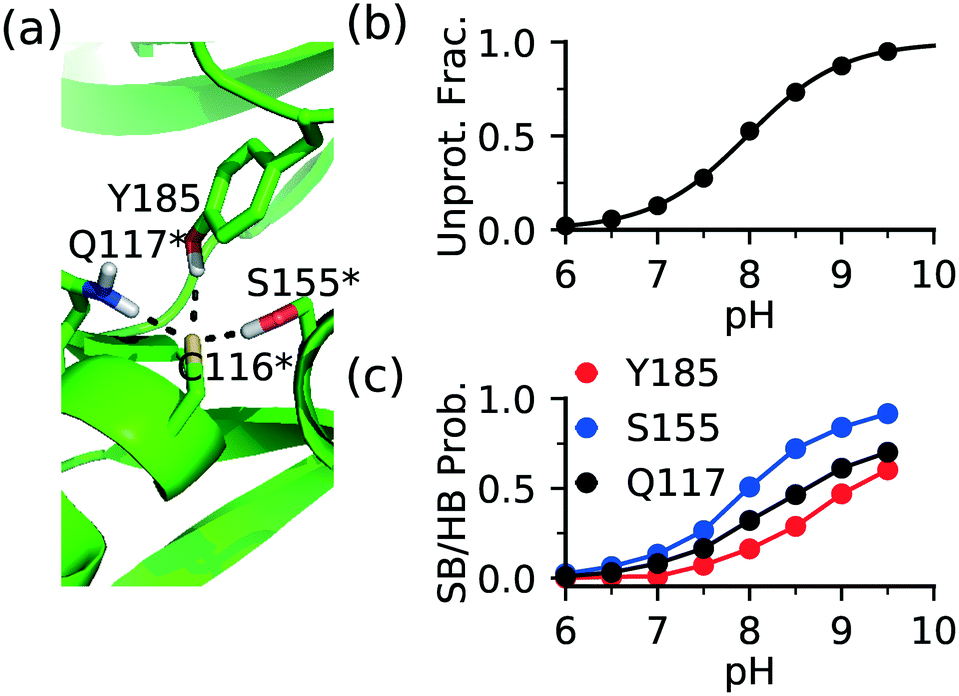 | ||
| Fig. 3 The FP thiolate in JNKs is stabilized by local h-bonds. (a) A zoomed-in view of the local environment of the FP Cys116 in JNK2. The Gln117-Cys116, Ser155-Cys116, and Tyr185-Cys116 h-bonds are shown. A snapshot from the simulation replica at pH 9.0 is used. Note, Cys116, Gln117, and Ser155 indicated by an asterisk are conserved among JNK1/2/3. (b) Deprotonated fraction of Cys116 at different pH values. The curve represents the best fit to the Henderson–Hasselbalch equation. (c) The probability of the h-bond formation between Cys116 thiolate and Gln117, Ser155, or Tyr185 is shown at different pH values. A h-bond is defined using a donor–acceptor heavy-atom distance cutoff of 3.5 Å and the donor-hydrogen-acceptor angle cutoff of 150°. | ||
To examine the effect of h-bond formation on cysteine deprotonation, we calculated the deprotonated fraction of JNK2 Cys116 and the individual h-bond occupancies as a function of pH (Fig. 3b and c). The Cys116 thiolate fraction increases with the increasing occupancies of the h-bonds with Gln117, Ser155, and Tyr185. For JNK1 and JNK3, similar correlations can be seen from the analysis (Fig. S1†). It is worthwhile noticing that the total h-bond occupancy at pH 7.5 is the highest in JNK3, which corroborates the lowest pKa of the FP cysteine (Cys154) in JNK3 (6.3) as compared to JNK1/2 (7.5/8.0).
Extended front-pocket cysteines in p38α/β
An extended front pocket (EFP) cysteine15 is located on the C-terminal end of the αD helix in p38α/β at position 59 in the KLIFS alignment.12 The EFP Cys119 in p38α has been covalently modified by a TCKI,43 and no covalent inhibitor targeting this position has been reported for p38β. Consistently, the CpHMD simulations predicted Cys119 as hyper-reactive in p38α and somewhat reactive (lower limit of our reactivity scale) in p38β, with the respective pKa's of 6.9 and 9.4 (Table S1†).To understand the three-unit difference between the pKa's of the EFP cysteine Cys119 in p38α and P38β, we examined the local environment of Cys119 in the X-ray structures. Interestingly, unlike in p38β and other kinases, the αD helix (residues 112–120) in p38α is largely unwound. For example, in the PDB entry 5tbe45 residues 113 to 116 form a hydrogen bonded turn, and in the PDB entry46 these residues form a 310 helix with Cys119 located on a loop (Fig. 4a). During the simulation based on the PDB entry 5tbe, residues 113–116 sampled both 310 helix and hydrogen-bonded turn. Due to the different secondary structure environment in p38β and p38α, the thiol group of Cys119 points to different directions. In p38β, it extends to solvent, whereas in p38α it points towards a loop located near αF, allowing it to form h-bond or salt-bridge interactions with Arg220 and Thr221 (Lys220 and Ala221 in p38β) upon deprotonation (Fig. 4a). Trajectory analysis showed that the probability of the salt-bridge formation between Arg220 and the Cys119 thiolate increases from pH 7 to pH 8, which correlates with the increase in the deprotonation of Cys119 in this pH range (Fig. 4b and c). Thr221 is found to donate hydrogen bonds via its sidechain hydroxyl and backbone amide groups to Cys119 with an overall probability above 0.5 in the entire pH range (Fig. 4c), which further stabilizes the thiolate form.
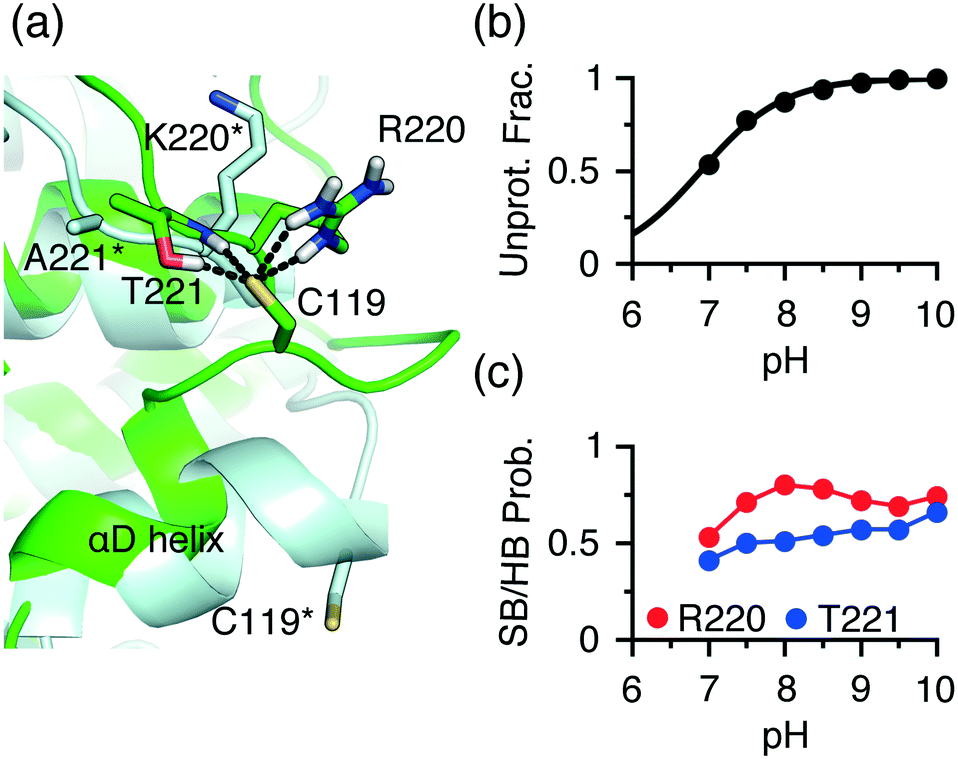 | ||
| Fig. 4 Different local environments of the EFP cysteines in p38α and p38β result in the different reactivities. (a) A zoomed-in view of the local environment of the EFP Cys119 in p38α (green, taken from the simulation replica at pH 10 started from the X-ray structure PDB 5tbe45) and p38β (light blue, taken from the X-ray structure PDB 3gc8 (ref. 47)). In p38α, Cys119 forms salt-bridge and h-bond interactions with Arg220 and Thr221. The analogous residues, Lys220 and Ala221 in p38β are labeled with an asterisk. (b) The deprotonated fraction of Cys119 in p38α is shown at different pH values. The curve is the best fit to the Henderson–Hasselbalch equation. (c) The probabilities of the Thr221-Cys119 (side chain and backbone) h-bond (blue) and the Arg220-Cys119 (red) salt-bridge interactions at different pH values. A salt bridge is considered formed if the minimum distance between the Cys119 sulfur and the guanidinium nitrogen atoms of Arg220 is below 4 Å. A h-bond is considered form if the minimum distance between the Cys119 sulfur and the side chain hydroxyl or the backbone amide hydrogen of Thr221 is below 2.4 Å. | ||
A question arises as to why Cys119 in p38β has a pKa shifted above the model value of 8.5, despite being largely exposed to solvent and not in the vicinity of any negatively charged residue. We suggest that the pKa upshift is due to the position of Cys119, i.e., C terminal cap of the αD helix in p38β. At the C terminus of a helix, a negative thiolate is destabilized by the helix dipole, as the latter can be approximated as placing a negative 0.5–0.7 unit charge near the C terminus.38 This is in contrast to the FP cysteine, the N-terminal cap of the αD helix in JNKs, which experiences the helix dipole interaction that stabilizes the thiolate state. Note, the Cys119 thiolate in p38α does not experience repulsion with the helix dipole, as the αD helix is unwound.
DFG − 1 cysteines in ERK1/2, ERK7, and NLK
The DFG − 1 cysteine (position 80 in KLIFS alignment12) is the second most targeted site in kinases for covalent modification15 and it is present in ERK1 (Cys183), ERK2 (Cys166), as well as the atypical MAPKs ERK7 (Cys154) and NLK (Cys281). The DFG − 1 cysteine in ERK1/2 has been targeted by covalent inhibitors.44,48 However, the CpHMD titration predicted the DFG − 1 cysteine in ERK1 as somewhat reactive and that in ERK2 as unreactive, with the estimated pKa's of 9.1 and 10.0, respectively. The DFG − 1 cysteines in the atypical MAPKs ERK7 and NLK were also predicted as unreactive, with pKa's above 9.5. Note, the structures of ERK7 and NLK were obtained through homology modeling and will be excluded in the following discussion.Trajectory analysis showed that Cys183 of ERK1 deprotonates as pH increases above 8.5 and the thiolate form is stabilized by the hydrogen bonding with the sidechain of Asn171 (on the C-loop) and the salt-bridge interactions with the HRD Arg165 as well as the catalytic lysine Lys71 (Fig. 5a and b). By contrast, the DFG − 1 Cys166 in ERK2 does not form a h-bond with the analogous Asn154, which may explain the increased pKa as compared to that in ERK1. It is puzzling that the DFG − 1 cysteines in ERK1/2 have been covalently modified44 but our simulations predicted low reactivities. One possibility is that the nearby DFG Asp acts as a general base to assist the thiol-Michael addition reaction. Such a base-assisted mechanism has been suggested for the FP Ncap cysteine in EGFR whereby the Ncap + 3 Asp may deprotonate the cysteine in the first stage of the reaction.49
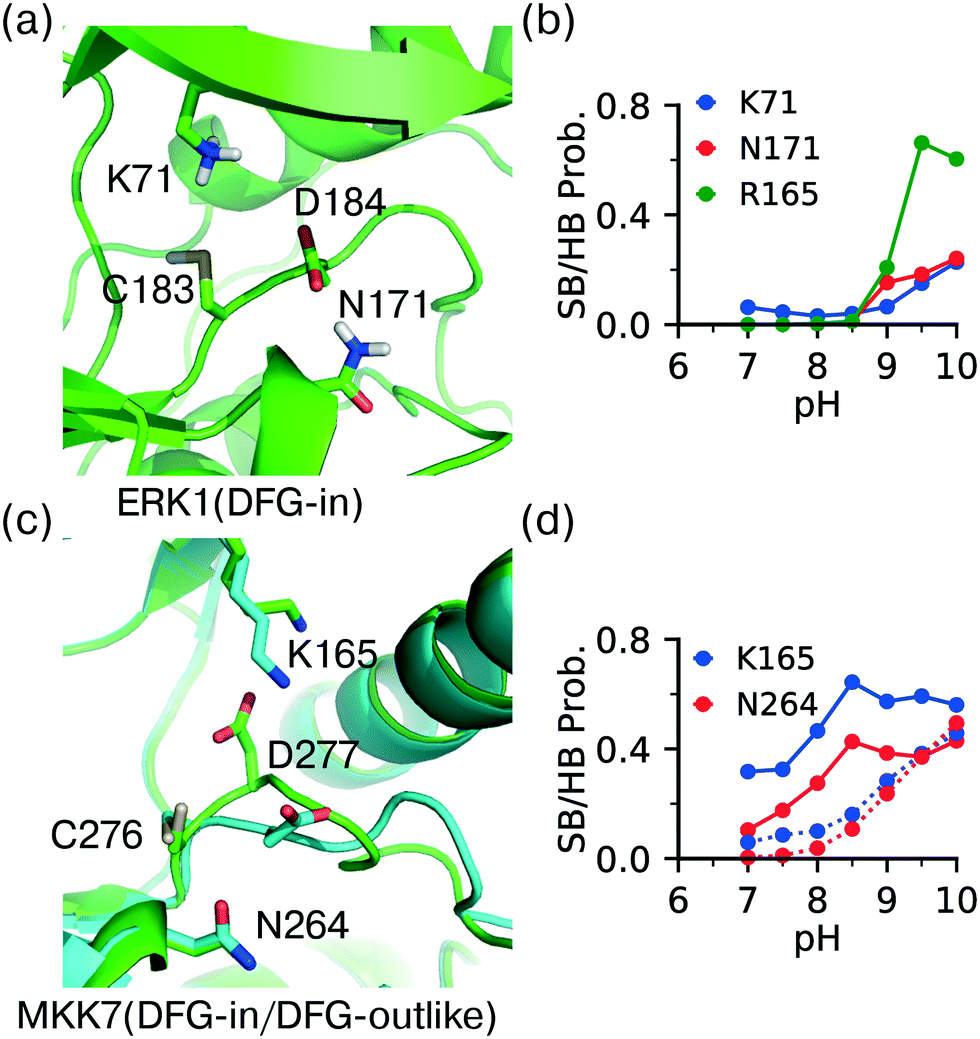 | ||
| Fig. 5 DFG − 1 Cys has different local environment depending on the DFG conformation. (a) Zoomed-in view of the protonated DFG − 1 cysteine (Cys183) in ERK1 taken from the simulation replica at pH 7.5 (started from the X-ray structure in the DICI conformation; PDB: 6ges).44 (b) The probabilities of the salt-bridge and h-bond interactions of Cys183 in ERK1 at different pH values. (c) An overlay between the MKK7 crystal structures in the DFG-in (PDB: 6qft, cyan)50 and DFG-outlike (PDB: 6qfr, green)50 conformations. The important residues are labeled. (d) The probabilities of the salt-bridge and h-bond interactions of the DFG − 1 cysteine (Cys276) in MKK7 at different pH values. Solid lines are for the DFG-in conformation (PDB: 6qfr) and dotted lines are for the DFG-out conformation (PDB: 6qft). | ||
Conformational dependent reactivity of the DFG − 1 cysteine
Analysis of crystal structures suggests that unlike other locations, reactivity of the DFG − 1 cysteine may be modulated by the conformation of the DFG motif. Located at the bottom of the catalytic pocket, the DFG − 1 cysteine is nearly entirely buried in ERK1/2. Thus, desolvation promotes the neutral thiol state and contributes to a significant pKa upshift relative to the model cysteine pKa of 8.6.24 Additionally, the charged thiolate state may be disfavored through the potential electrostatic repulsion from the neighboring sidechain of the DFG Asp. On the contrary, the thiolate state may be stabilized by the electrostatic attraction with the catalytic lysine and hydrogen bonding with the nearby Asn on the C-loop. The latter favorable interactions are dependent on the DFG conformation.To test the hypothesis that the cysteine reactivity is modulated by the DFG conformation, we examined MKK7, for which crystal structures with both DFG-in and DFG-outlike conformations are available. Note, there is no ERK1/2 crystal structure with the alternative DFG conformations in the PDB. For the DFG-in structure (PDB: 6qft,50Fig. 5c) the CpHMD predicted pKa for the DFG − 1 cysteine (C276) is 9.1, consistent with the results for ERK1/2. However, for the DFG-outlike structure (PDB: 6qfr,50Fig. 5c) the predicted pKa is 8.2, suggesting that the DFG − 1 cysteine is reactive. Note, the structure in PDB 6qfr is classified as DFG-outlike by the KLIFS database based on manual structure superposition,12 but as DFG-out based on the pseudo torsion angle defined by the Cα atoms of DFG − 2, DFG − 1 (Cys), DFG Asp, and DFG Phe residues.51
Analysis of the trajectories based on the DFG-in structure of MKK7 (PDB: 6qft,50Fig. 5c) showed that the salt-bridge interaction with the catalytic lysine (Lys165) and the hydrogen bonding with the C-loop asparagine near HRD (Asn264) are the major driving forces for the deprotonation of Cys276 (Fig. 5d, dashed lines), consistent with the simulations of ERK1 based on the DFG-in structure (Fig. 5b). Importantly, the probabilities of the Lys165-Cys276 salt-bridge and Asn264-Cys276 h-bond formation are significantly increased at pH 7–8.5 in the trajectories based on the DFG-outlike structure of MKK7 (PDB: 6qfr,50Fig. 5d, solid lines). This suggests that the change in the DFG Asp conformation modulates the interactions of the DFG − 1 cysteine and therefore its pKa value. These data are consistent with our previous simulation51 and experimental work52 which suggested that the pH-dependent binding kinetics of imatinib is due to the pH-dependent conformational dynamics of the DFG motif.
Concluding discussion
Irreversible inhibition is an important and actively pursued strategy against MAPKs, which have no FDA approved inhibitors. To help the medicinal chemistry efforts, we examined and rationalized the reactivities of the cysteine locations in all 14 MAPKs. MAPKs possess 2–8 cysteines, and most of them are near the binding site and shared by several MAPK family members. Three of the cysteine locations (FP Ncap, EFP, and DFG − 1) have been targeted by covalent inhibitors.41,43 Consistently, our simulations predicted that the FP Ncap cysteine (Cys116) conserved in JNK1/2/3 is reactive or hyper-reactive. The EFP cysteine (Cys119) common to p38α/β was predicted as hyper-reactive in p38α and somewhat reactive in p38β. The former is consistent with a reported irreversible inhibitor.43 Analysis of the MAPK trajectories demonstrated that the increased cysteine reactivity, i.e., pKa downshift, is mainly due to the availability of nearby hydrogen bond acceptors or positive charges, corroborating our previous findings regarding the physical determinants of cysteine pKa's,30,31Based on the simulations of ERK1/2 as well as MKK7, we suggest that the reactivity of the DFG − 1 cysteine is modulated by the DFG conformation. In the DFG-in conformation, where the DFG Asp is pointing into the ATP binding site, the DFG − 1 cysteines in ERK1/2 as well as MKK7 were found somewhat reactive or unreactive, whereas in the DFG-outlike conformation, where the DFG Asp is pointing outward, the DFG − 1 cysteine in MKK7 was found reactive. The increased thiol reactivity can be attributed to the enhanced tendency of the DFG − 1 thiolate to form salt-bridge with the catalytic lysine and hydrogen bonding with the HRD + 6 Asn on the C-loop. These data suggest that a DFG − 1 cysteine may be covalently modified by type II inhibitors (that target DFG out conformations) with weaker electrophilic warheads.
Together with our recent study of the FP Ncap cysteine reactivities,32 the current data suggests that the presence of a proximal basic residue (e.g. Lys or Arg) makes a Cys more reactive; however, the net effect of a nearby acid group (e.g., Asp) on the Cys pKa is unclear and may be sensitive to the local conformation. For example, the DFG-outlike conformation brings the HRD + 6 Asn closer to the DFG − 1 Cys, allowing the formation of a hydrogen bond that stabilizes the thiolate state. It is also possible that in the DFG-in conformation, the DFG Asp is positioned such that it can serve as a general base to abstract the proton from the neighboring cysteine, reminiscent of the mechanism proposed49 for the FP Ncap cysteine in EGFR, whereby the Ncap + 3 Asp may serve as a proton acceptor in a base-assisted thiol-Michael addition. This might explain why the crystal structures of the covalently labeled ERK1/2 display the DFG-in conformation.44,48 Another type of base-catalyzed reaction may occur in which a basic amine group on the inhibitor abstracts the proton from the DFG − 1 thiol.53,54 Finally, a direct proton transfer from the cysteine thiol to the electrophilic warhead may occur, as demonstrated by a recent combined quantum-mechanics/molecular mechanics (QM/MM) study for the covalent modification of BTK by ibrutinib.55 To thoroughly understand the reactivities of the DFG − 1 cysteines, all-atom MD simulations and QM/MM calculations will be conducted in the future.
The current work also prospectively predicted locations of reactive cysteines that may be covalently targeted. In particular, the DFG + 6 cysteine which only exist in JNK2 and ERK5 is predicted as hyper-reactive in both. The former was covalently labeled in the chemo-proteomic profiling studies.35,36 Our predicted reactivity status of the αH head cysteine in JNK1 (Cys245) and JNK3 (Cys283) is also in agreement with the chemo-proteomic data.36 Thus, except for the DFG − 1 cysteine in ERK1/ERK2, all previously targeted MAPK cysteines either by TCKIs or probes/fragments in proteomic screening are retrospectively predicted by the current CpHMD simulations as reactive or hyper-reactive. Additionally, the simulations suggest that the P-loop cysteine (Cys28) unique to the atypical ERK3 is reactive. Several lysine locations are also predicted to be reactive or somewhat reactive, including the catalytic lysine in ERK7 (Lys42) and NLK (Lys167), HRD + 2 lysine in JNK2 (Lys153) and ERK4 (Lys151), and βII lysine in ERK7 (Lys29). Identification and rationalization of reactive and hyper-reactive cysteine sites may assist medicinal chemists in the design of weakly electrophilic warheads with increased selectivity and reduced off-target effects. CpHMD simulation requires target structural information; however, it demands significantly less resources than chemo-proteomic screening and provides physics-based rationales not available from experiment. Thus, CpHMD offers a complementary tool that can expedite the effort to discover covalently druggable sites on a proteome-wide level.
Methods and protocols
Structure preparation
The starting structures were taken from the protein data bank (PDB). The following X-ray structures were used for the MAPKs: JNK1 (PDB 2xrw);56 JNK2 (PDB: 3e7o and 3npc);57 JNK3 (PDB: 6emh);41 p38α (PDB 5tbe);45 p38β (PDB 3gc8);47 p38γ (PDB 1 cm8);58 p38δ (PDB 5ekn);59 ERK1 (PDB: 6ges);44 ERK2 (PDB: 3w55);60 ERK5 (PDB: 4ic8);61 ERK3 (PDB: 2i6l).62 For MKK7, two crystal structures (PDB: 6qfr and 6qft)50 were used. As X-ray crystal structures are unavailable for the atypical kinases ERK4, ERK7, and NLK, homology models were built using SWISS-MODEL,63 with the X-ray structures of ERK3 (PDB code: 2i6l, 73.8% sequence identity with ERK4), ERK2 (PDB code: 3oz6, 51.6% sequence identity with ERK7), and ERK5 (PDB code: 5o7i, 46.5% sequence identity with NLK) as respective templates. The open source tool pdbfixer from the OpenMM package64 was used to remove water, inhibitor, and to add missing residues, atoms, acetylated N terminus, amidated C terminus, and disulfide bonds (if present), as well as hydrogen atoms. Lys and Cys had one dummy hydrogen, while His had two dummy hydrogens. tLeap from the Amber18 package65 was then used to generate input files. The structures were first subject to energy minimization using 500 steps of steepest descent and 500 steps of conjugate gradient method with a harmonic force constant of 50 kcal mol−1 Å2 applied to the heavy atoms. Four stages of equilibration were performed at 300 K using 2000 steps for each stage. A harmonic force constant applied to the heavy atoms was gradually decreased from 5.0, 2.0, 1.0, to 0 kcal mol−1 Å2.pH replica-exchange CpHMD simulations
Simulations were performed using the GPU-accelerated pmemd program in AMBER 18,65 and a CpHMD patch was applied to enable the CpHMD titration functionality.33,34 The proteins were represented by the ff14SB force field66 and water was represented by the GB-Neck2 implicit-solvent model67 (igb = 8, mbondi3 intrinsic Born radii). The ionic strength was set to 0.15 M. Following the previous studies,30 the scaling parameter (Sx) of sulfur was set to that of oxygen (1.061039) and the intrinsic Born radius of sulfur was set to 2.0 Å.Unless otherwise noted, one set of pH replica-exchanged CpHMD simulations was conducted for each structure, following our previous protocol.30,31 In brief, trajectories were initiated with the same conformation but different pH conditions in the pH range 7.0–11.0 with an interval of 0.5 pH unit. pH exchanges between adjacent replicas were attempted at every 500 or 1000 steps (1 or 2 ps) according to the Metropolis criterion. Each replica was run for 20–80 ns until convergence. The converged portion (the last 20 or 30 ns per replica) was used for analysis. His, Cys, and Lys sidechains were allowed to titrate. Since simulation pH was above 7, Asp and Glu were fixed in the charged state.
Model parameters and pKa calculations
The model pKa values for His, Cys, and Lys are 6.5, 8.5, and 10.4, respectively. The same model titration parameters were used as derived in our previous studies.30,31,33 At different pH conditions, the deprotonated fraction of cysteine was calculated according to our definition of protonated (λ < 0.2) and deprotonated (λ > 0.8) states.30,31,33 The pKa was calculated by fitted the deprotonated fractions at different pH to the generalized Henderson–Hasselbalch equation.Conflicts of interest
The findings in this work might be of interest to ComputChem LLC, for which J. S. is a co-founder and scientific advisor.Acknowledgements
Financial support from the National Institutes of Health (R01GM098818 and R01CA256557) is acknowledged.Notes and references
- D. K. Morrison, Cold Spring Harbor Perspect. Biol., 2012, 4, a011254 CrossRef.
- H. Lavoie, J. Gagnon and M. Therrien, Nat. Rev. Mol. Cell Biol., 2020, 21, 607–632 CrossRef CAS PubMed.
- I. Smalley and K. S. Smalley, Cancer Discovery, 2018, 8, 140–142 CrossRef CAS PubMed.
- C. J. Caunt, M. J. Sale, P. D. Smith and S. J. Cook, Nat. Rev. Cancer, 2015, 15, 577–592 CrossRef CAS PubMed.
- A. M. Kidger, J. Sipthorp and S. J. Cook, Pharmacol. Ther., 2018, 187, 45–60 CrossRef CAS.
- H. Chin, D. Lai and G. Falchook, Journal of Immunotherapy and Precision Oncology, 2019, 2, 10 CrossRef.
- Q. Wu, W. Wu, V. Jacevic, T. C. C. Franca, X. Wang and K. Kuca, J. Enzyme Inhib. Med. Chem., 2020, 35, 574–583 CrossRef CAS PubMed.
- V. Haller, P. Nahidino, M. Forster and S. A. Laufer, Expert Opin. Ther. Pat., 2020, 30, 453–466 CrossRef CAS.
- J. M. Grimes and K. V. Grimes, J. Mol. Cell. Cardiol., 2020, 144, 63–65 CrossRef CAS PubMed.
- P. Coulombe and S. Meloche, Biochim. Biophys. Acta, 2007, 1773, 1376–1387 CrossRef CAS.
- S. Eid, S. Turk, A. Volkamer, F. Rippmann and S. Fulle, BMC Bioinf., 2017, 18, 16 CrossRef PubMed.
- O. P. J. van Linden, A. J. Kooistra, R. Leurs, I. J. P. de Esch and C. de Graaf, J. Med. Chem., 2014, 57, 249–277 CrossRef CAS PubMed.
- J. Pettinger, K. Jones and M. D. Cheeseman, Am. Ethnol., 2017, 56, 15200–15209 CAS.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- Z. Zhao and P. E. Bourne, Drug Discovery Today, 2018, 23, 727–735 CrossRef CAS PubMed.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- A. Chaikuad, P. Koch, S. A. Laufer and S. Knapp, Angew. Chem., Int. Ed., 2018, 57, 4372–4385 CrossRef CAS PubMed.
- G. Bulaj, T. Kortemme and D. P. Goldenberg, Biochemistry, 1998, 37, 8965–8972 CrossRef CAS.
- A. V. Peskin and C. C. Winterbourn, Free Radical Biol. Med., 2001, 30, 572–579 CrossRef CAS PubMed.
- S. C. E. Tosatto, V. Bosello, F. Fogolari, P. Mauri, A. Roveri, S. Toppo, L. Flohé, F. Ursini and M. Maiorino, Antioxid. Redox Signaling, 2008, 10, 1515–1526 CrossRef CAS PubMed.
- G. Ferrer-Sueta, B. Manta, H. Botti, R. Radi, M. Trujillo and A. Denicola, Chem. Res. Toxicol., 2011, 24, 434–450 Search PubMed.
- T. A. Anderson and R. T. Sauer, Biophys. Chem., 2002, 100, 341–350 CrossRef.
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–377 CrossRef CAS PubMed.
- R. L. Thurlkill, G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2006, 15, 1214–1218 CrossRef CAS PubMed.
- P. Ábrányi-Balogh, L. Petri, T. Imre, P. Szijj, A. Scarpino, M. Hrast, A. Mitrović, U. P. Fonovič, K. Németh, H. Barreteau, D. I. Roper, K. Horváti, G. G. Ferenczy, J. Kos, J. Ilaš, S. Gobec and G. M. Keserű, Eur. J. Med. Chem., 2018, 160, 94–107 CrossRef PubMed.
- W. Chen, B. H. Morrow, C. Shi and J. K. Shen, Mol. Simul., 2014, 40, 830–838 CrossRef CAS PubMed.
- E. Alexov, E. L. Mehler, N. Baker, A. Baptista, Y. Huang, F. Milletti, J. E. Nielsen, D. Farrell, T. Carstensen, M. H. M. Olsson, J. K. Shen, J. Warwicker, S. Williams and J. M. Word, Proteins, 2011, 79, 3260–3275 CrossRef CAS PubMed.
- J. A. Wallace and J. K. Shen, J. Chem. Theory Comput., 2011, 7, 2617–2629 CrossRef CAS.
- J. A. Henderson, N. Verma, R. C. Harris, R. Liu and J. Shen, J. Chem. Phys., 2020, 153, 115101 CrossRef CAS.
- R. Liu, Z. Yue, C.-C. Tsai and J. Shen, J. Am. Chem. Soc., 2019, 141, 6553–6560 CrossRef CAS.
- R. C. Harris, R. Liu and J. Shen, J. Chem. Theory Comput., 2020, 16, 3689–3698 CrossRef CAS PubMed.
- R. Liu, S. Zhan, Y. Che and J. Shen, J. Med. Chem., 2021 DOI:10.1021/acs.jmedchem.1c01186.
- Y. Huang, R. C. Harris and J. Shen, J. Chem. Inf. Model., 2018, 58, 1372–1383 CrossRef CAS PubMed.
- R. C. Harris and J. Shen, J. Chem. Inf. Model., 2019, 59, 4821–4832 CrossRef CAS PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- K. Senkane, E. V. Vinogradova, R. M. Suciu, V. M. Crowley, B. W. Zaro, J. M. Bradshaw, K. A. Brameld and B. F. Cravatt, Am. Ethnol., 2019, 11507–11511 Search PubMed.
- J. R. Huth, R. Mendoza, E. T. Olejniczak, R. W. Johnson, D. A. Cothron, Y. Liu, C. G. Lerner, J. Chen and P. J. Hajduk, J. Am. Chem. Soc., 2005, 127, 217–224 CrossRef CAS.
- W. G. J. Hol, Adv. Biophys., 1985, 19, 133–165 CrossRef CAS PubMed.
- L. Serrano and A. R. Fersht, Nature, 1989, 342, 296–299 CrossRef CAS PubMed.
- A. Jacks, J. Babon, G. Kelly, I. Manolaridis, P. D. Cary, S. Curry and M. R. Conte, Structure, 2003, 11, 833–843 CrossRef CAS.
- T. Zhang, F. Inesta-Vaquera, M. Niepel, J. Zhang, S. B. Ficarro, T. Machleidt, T. Xie, J. A. Marto, N. Kim, T. Sim, J. D. Laughlin, H. Park, P. V. LoGrasso, M. Patricelli, T. K. Nomanbhoy, P. K. Sorger, D. R. Alessi and N. S. Gray, Chem. Biol., 2012, 19, 140–154 CrossRef CAS.
- F. Muth, A. El-Gokha, F. Ansideri, M. Eitel, E. Do, A. Sievers-Engler, A. Lange, F. M. Boeckler, M. Lam and S. A. Laufer, J. Med. Chem., 2017, 60, 594–607 CrossRef CAS PubMed.
- J. Li, T. S. Kaoud, J. LeVieux, B. Gilbreath, S. Moharana, K. N. Dalby and S. M. Kerwin, ChemBioChem, 2013, 14, 66–71 CrossRef CAS PubMed.
- S. Rao, D. Gurbani, G. Du, R. A. Everley, C. M. Browne, A. Chaikuad, L. Tan, M. Schröder, S. Gondi, S. B. Ficarro, T. Sim, N. D. Kim, M. J. Berberich, S. Knapp, J. A. Marto, K. D. Westover, P. K. Sorger and N. S. Gray, Cell Chem. Biol., 2019, 26, 818–829.e9 CrossRef CAS PubMed.
- H. K. Wentsch, N. M. Walter, M. Bührmann, S. Mayer-Wrangowski, D. Rauh, G. J. R. Zaman, N. Willemsen-Seegers, R. C. Buijsman, M. Henning, D. Dauch, L. Zender and S. Laufer, Angew. Chem., Int. Ed., 2017, 56, 5363–5367 CrossRef CAS PubMed.
- S. C. Koeberle, J. Romir, S. Fischer, A. Koeberle, V. Schattel, W. Albrecht, C. Grütter, O. Werz, D. Rauh, T. Stehle and S. A. Laufer, Nat. Chem. Biol., 2012, 8, 141–143 CrossRef CAS PubMed.
- S. B. Patel, P. M. Cameron, S. J. O'Keefe, B. Frantz-Wattley, J. Thompson, E. A. O'Neill, T. Tennis, L. Liu, J. W. Becker and G. Scapin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 777–785 CrossRef CAS PubMed.
- R. A. Ward, N. Colclough, M. Challinor, J. E. Debreczeni, K. Eckersley, G. Fairley, L. Feron, V. Flemington, M. A. Graham, R. Greenwood, P. Hopcroft, T. D. Howard, M. James, C. D. Jones, C. R. Jones, J. Renshaw, K. Roberts, L. Snow, M. Tonge and K. Yeung, J. Med. Chem., 2015, 58, 4790–4801 CrossRef CAS.
- L. Capoferri, A. Lodola, S. Rivara and M. Mor, J. Chem. Inf. Model., 2015, 55, 589–599 CrossRef CAS PubMed.
- P. Wolle, J. Engel, S. Smith, L. Goebel, E. Hennes, J. Lategahn and D. Rauh, J. Med. Chem., 2019, 62, 5541–5546 CrossRef CAS.
- C.-C. Tsai, Z. Yue and J. Shen, J. Am. Chem. Soc., 2019, 141, 15092–15101 CrossRef CAS PubMed.
- Y. Shan, M. A. Seeliger, M. P. Eastwood, F. Frank, H. Xu, M. O. Jensen, R. O. Dror, J. Kuriyan and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 139–144 CrossRef CAS PubMed.
- A. Wissner, E. Overbeek, M. F. Reich, M. B. Floyd, B. D. Johnson, N. Mamuya, E. C. Rosfjord, C. Discafani, R. Davis, X. Shi, S. K. Rabindran, B. C. Gruber, F. Ye, W. A. Hallett, R. Nilakantan, R. Shen, Y.-F. Wang, L. M. Greenberger and H.-R. Tsou, J. Med. Chem., 2003, 46, 49–63 CrossRef CAS PubMed.
- E. R. Wood, L. M. Shewchuk, B. Ellis, P. Brignola, R. L. Brashear, T. R. Caferro, S. H. Dickerson, H. D. Dickson, K. H. Donaldson, M. Gaul, R. J. Griffin, A. M. Hassell, B. Keith, R. Mullin, K. G. Petrov, M. J. Reno, D. W. Rusnak, S. M. Tadepalli, J. C. Ulrich, C. D. Wagner, D. E. Vanderwall, A. G. Waterson, J. D. Williams, W. L. White and D. E. Uehling, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2773–2778 CrossRef CAS PubMed.
- A. T. Voice, G. Tresadern, R. M. Twidale, H. van Vlijmen and A. J. Mulholland, Chem. Sci., 2021, 12, 5511–5516 RSC.
- S. D. Chamberlain, A. M. Redman, J. W. Wilson, F. Deanda, J. B. Shotwell, R. Gerding, H. Lei, B. Yang, K. L. Stevens, A. M. Hassell, L. M. Shewchuk, M. A. Leesnitzer, J. L. Smith, P. Sabbatini, C. Atkins, A. Groy, J. L. Rowand, R. Kumar, R. A. Mook, G. Moorthy and S. Patnaik, Bioorg. Med. Chem. Lett., 2009, 19, 360–364 CrossRef CAS PubMed.
- A. Kuglstatter, M. Ghate, S. Tsing, A. G. Villaseñor, D. Shaw, J. W. Barnett and M. F. Browner, Bioorg. Med. Chem. Lett., 2010, 20, 5217–5220 CrossRef CAS PubMed.
- S. Bellon, M. J. Fitzgibbon, T. Fox, H. M. Hsiao and K. P. Wilson, Structure, 1999, 7, 1057–1065 CrossRef CAS PubMed.
- Z. Yurtsever, D. A. Patel, D. L. Kober, A. Su, C. A. Miller, A. G. Romero, M. J. Holtzman and T. J. Brett, Biochim. Biophys. Acta, 2016, 1860, 2335–2344 CrossRef CAS.
- M. Ohori, T. Kinoshita, S. Yoshimura, M. Warizaya, H. Nakajima and H. Miyake, Biochem. Biophys. Res. Commun., 2007, 353, 633–637 CrossRef CAS PubMed.
- G. Glatz, G. Gógl, A. Alexa and A. Reményi, J. Biol. Chem., 2013, 288, 8596–8609 CrossRef CAS PubMed.
- M. Schröder, P. Filippakopoulos, M. P. Schwalm, C. A. Ferrer, D. H. Drewry, S. Knapp and A. Chaikuad, Int. J. Mol. Sci., 2020, 21, 7953 CrossRef PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks and V. S. Pande, PLoS Comput. Biol., 2017, 13, e1005659 CrossRef.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. Cheatham, III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, Technical Report, AMBER 2018, University of California, 2018 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- H. Nguyen, D. R. Roe and C. Simmerling, J. Chem. Theory Comput., 2013, 9, 2020–2034 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI tables contain the calculated pKa's of the reactive cysteines and lysines in MAPKs as well as the DFG − 1 cysteines in ERK1/2 and MKK7. ESI figures contain the titration plots and analysis of pH-dependent h-bonding and electrostatic interactions. See DOI: 10.1039/d1md00277e |
| ‡ Current affiliation: Enzymaster GmbH, Düsseldorf, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |