
An overview of the development of EED inhibitors to disable the PRC2 function
Kai-Lu
Liu
,
Kongkai
Zhu
 * and
Hua
Zhang
*
* and
Hua
Zhang
*
School of Biological Science and Technology, University of Jinan, Jinan 250022, China. E-mail: hkhhh.k@163.com; bio_zhangh@ujn.edu.cn
First published on 21st October 2021
Abstract
Polycomb repressive complex 2 (PRC2) catalyzes the methylation of histone H3 lysine 27 (H3K27) and the enrichment of its catalytic product H3K27me3 is responsible for the silencing of tumor suppressor genes and the blocking of transcripts related to immunity and cell terminal differentiation. Aberrations of PRC2 components, such as mutation and overexpression, have been observed in various cancers, which makes PRC2 a potential therapeutic target for cancer. Up to now, targeting the enhancer of zeste homolog 2 (EZH2), the catalytic subunit of PRC2, represents the main strategy in the development of PRC2 inhibitors. Although significant progress has been made, new problems also emerge, e.g. the drug resistance caused by secondary mutations. In recent years, more and more efforts have shifted to another new strategy – targeting embryonic ectoderm development (EED) to disrupt its major interactions with other components, which are necessary to the PRC2 function, and some promising results have been obtained. This review summarizes the recent development of EED inhibitors as possible chemotherapy for cancer treatment, which could help accelerate future related research work.
 Kailu Liu | Kailu Liu received her B. S. degree in Pharmacy from Shandong University of Traditional Chinese Medicine in 2019. Currently, she is pursuing her M.S. degree at the School of Biological Science and Technology, University of Jinan. Her research interests mainly focus on cell biology and protein engineering. |
 Kongkai Zhu | Kongkai Zhu received his Ph.D. from Shanghai Institute of Materia Medica, Chinese Academy of Sciences. He joined the School of Biological Science and Technology, University of Jinan, Jinan, China, in August 2016. Currently, he is working on the discovery of lead compounds targeting cancer disease. |
 Hua Zhang | Hua Zhang obtained his Ph.D. in Medicinal Chemistry from Shanghai Institute of Materia Medica (SIMM, 2001–2006) and did his postdoctoral research at The University of Queensland (2006–2010). He continued his academic career at SIMM for another four years and then moved to the University of Jinan in 2015. He is now a full-time professor of Natural Products and Medicinal Chemistry at the School of Biological Science and Technology. In addition to carrying on his efforts in natural product biodiscovery, his group has also been involved in rational design and synthesis of enzyme (e.g. PRMT and HDAC families) inhibitors for cancer treatment in recent years. |
Introduction
Epigenetics is closely related to the occurrence and development of cancer. Histone modification, DNA modification, chromatin remodeling, non-coding RNA and so on are all important epigenetic machinery.1,2 Polycomb repressive complex 2 (PRC2), one of the two major polycomb complexes with transcriptional inhibition in mammals, is the only one that can catalyze the methylation of histone H3 lysine 27 (H3K27) among more than 60 histone methyltransferases (HMTs) in humans, and three different methylation products, H3K27me1, H3K27me2 and H3K27me3, could be generated.3–7 This catalytic process is similar to those of other HMTs. It has been demonstrated that the enrichment of H3K27me3 is related to the silencing of tumor suppressor genes and the blocking of transcripts related to immunity and cell terminal differentiation, leading to tumorigenesis.7–9The minimal core of PRC2 consists of four protein components, namely, one catalytic subunit enhancer of zeste homolog 2 (EZH2), two regulatory subunits of embryonic ectoderm development (EED) and a suppressor of zeste 12 (SUZ12), and histone binding protein RbAp46/48 (also known as RBBP7/4). In addition, EZH1 (a homolog of EZH2) is also capable of interacting with EED, SUZ12 and RbAp46/48 to form a noncanonical PRC2 complex but with lower methyltransferase activity than PRC2-EZH2.10 Since 2012, small-molecule disruptors have been reported one after another to disable the PRC2 function, while most of them have been designed to target EZH2. Although these reported molecules have shown promising efficacy, drug resistance caused by secondary mutations appears inevitable.11–13 Therefore, exploration of new strategies to regulate the activity of PRC2 remains a challenge and pressing task.
In fact, the basal PRC2 activity requires the combination of EZH2, EED and SUZ12, and depletion of any one of them will completely abrogate the PRC2 function.14–17 Furthermore, the binding of H3K27me3 to EED can enhance the PRC2 activity, which is important for the methylation of unmodified H3K27.18–22 Therefore, developing inhibitors that target EED could be an attractive strategy to modulate the function of PRC2, which has received increasing interest from researchers since 2014. In this review, we summarize the prominent EED inhibitors and their activities.
The aberration of PRC2 in cancer
Aberrations of PRC2 components, especially EZH2, have been intimately implicated in the oncogenesis and progression of various neoplasms. In diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL), some EZH2 mutations have been unveiled in the catalytic su(var), enhancer-of-zeste and trithorax (SET) domain, including Y641F, Y641S, Y641N, Y641H, A677G, A682G, A687V and A692V, among which Y641 mutations are the most prevalent.8,23–28 The catalytic substrate preferences of the different EZH2 mutations are diverse. Y641 mutations greatly favor the H3K27me2 substrate, while the A687V mutation mainly enhances the methylation of H3K27me1.29 Although the A677G mutation increases the H3K27me3 level similarly to Y641 mutations, the catalytic efficiencies of A677G toward all three substrates are almost equal.30 Apart from EZH2 mutations, EZH2 overexpression, which was initially observed in prostate and breast cancers,31–33 is found in many other solid tumors as well, including ovarian carcinoma,34–36 esophageal squamous cell carcinoma (ESCC),37 cutaneous melanoma,38 renal cell carcinoma (RCC),39 endometrial cancer,33 gastric cancer40 and lung carcinoma.41 Because the increased expression of EZH2 is highly correlated with tumor grade and poor prognosis, it can be used to assess tumor progression and serve as a new biomarker to predict the prognosis of some tumors.39 Furthermore, high EZH2 expression is related not only to the high proliferation rate of tumor cells but also to the transcriptional inhibition and migration. For example, in ovarian carcinoma, the EZH2 overexpression might be involved in the transforming growth factor-b1 (TGF-b1) associated pathway to promote cell migration via upregulating the TGF-b1 expression.35 Tumor suppressor gene E-cadherin was repressed by high EZH2 expression in advanced prostate cancer31 and the transcriptional inactivation of p53 was also associated with the upregulated expression of the EZH2 protein in ESCC.37Several representative EZH2 disruptors
Developing EZH2 inhibitors to block the function of PRC2 is currently the main strategy of researchers. At present, there are several reviews on EZH2 inhibitors7,28,42–46 and some representative molecules are shown in Fig. 1.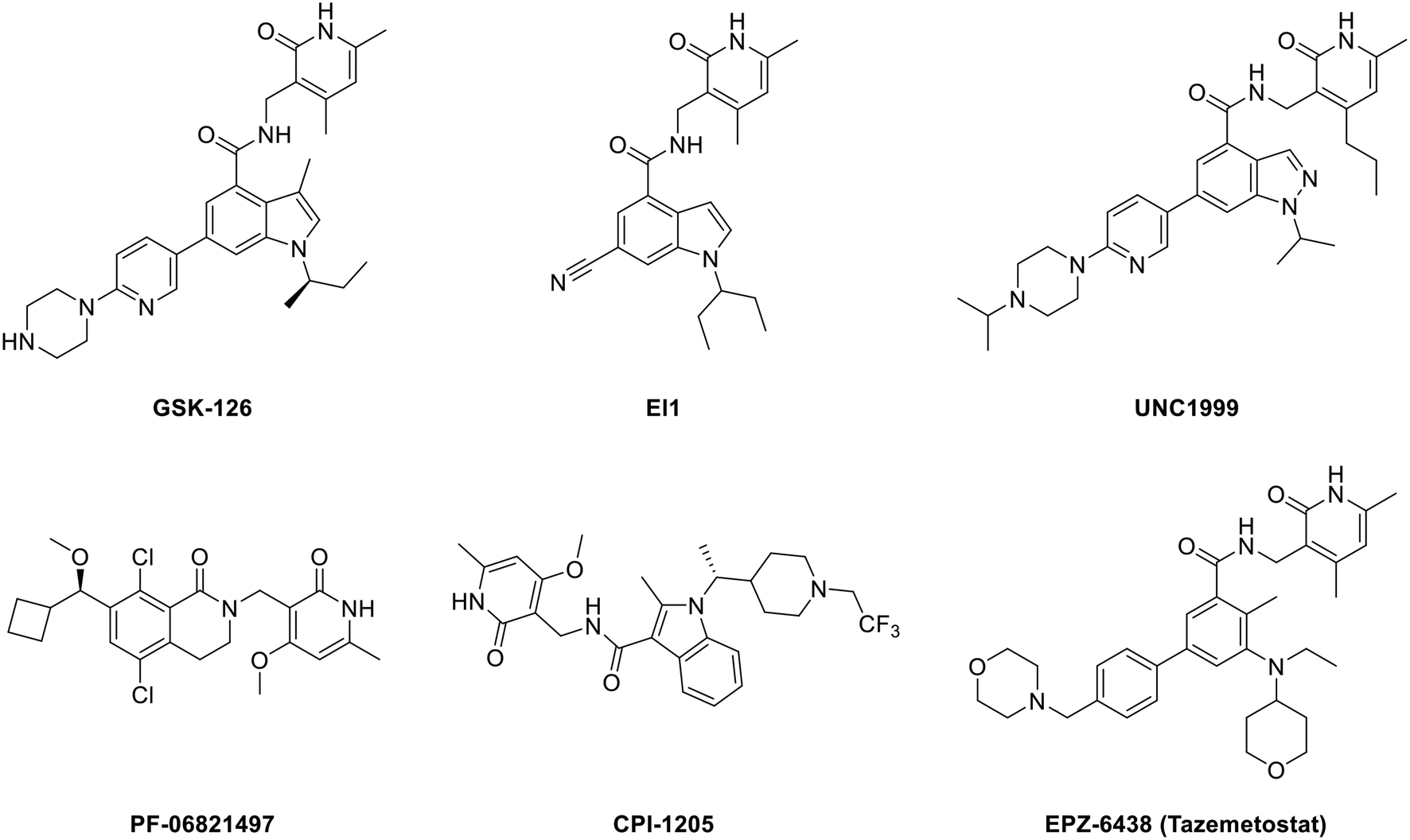 | ||
| Fig. 1 Structures of representative EZH2 inhibitors. CPI-1205 and PF-06821497 are in the clinical trial stage, while EPZ-6438 (tazemetostat) has been approved by the U.S. FDA. | ||
All the structures in Fig. 1 contain a pyridone unit which is essential to exert activity. GSK126 and EI1 are selective and potent SAM-competitive EZH2 inhibitors, exhibiting 150-fold and 90-fold selectivity for EZH2 over EZH1, respectively,25,47 while UNC1999, the first dual EZH2/1 inhibitor, is approximately 10-fold less potent for EZH1 than for EZH2.48 CPI-1205 (ref. 49) and PF-06821497 (ref. 50) are already in the clinical trial stage. While PF-06821497 is in the recruiting status for the phase 1 study (NCT03460977), the phase 2 clinical study for evaluating CPI-1205 in advanced solid tumors has been stopped as a result of no patients (NCT03525795), and the study of CPI-1205 on progressive B-cell lymphoma has been accomplished in the phase 1 clinical trial (NCT02395601). EPZ-6438 (tazemetostat), just approved by the U.S. FDA in January 2020, is the first EZH2 inhibitor for the treatment of advanced epithelioid sarcoma.51–53 Though these EZH2 modulators show promising efficacy, there are still some disadvantages. For example, GSK126, as a drug that entered clinical trials but was eventually terminated, suffered from low membrane permeability with an oral bioavailability of only 0.2% in wild type (WT) mice,54 which is another new problem to be solved. The disadvantage of CPI-1205, though exhibiting a better oral bioavailability, is attributed to its high clearance rate.49 What's more, cells sensitive to these inhibitors can acquire drug resistance via secondary mutations.55–57 For instance, Karpas422, a DLBCL cell line with EZH2 Y641N mutation, became also resistant to EI1 owing to the new EZH2 mutations of Y111L and Y661D caused by prolonged dosing.47,56
The pockets of the EED protein
With a pocket at each end, the EED protein contains seven copies of the WD40 repeat folding into a canonical β-propeller domain (Fig. 2).3,18,58 H3K27me3 is recognized and binds to the smaller top pocket with its trimethylated ammonium group inserting deeply into the pocket and being stabilized by van der Waals (vdW) and cation–π interactions. Besides, hydrophobic interactions are also observed between the aliphatic chain of trimethyllysine and the aromatic amino acid of W364. The binding of H3K27me3 makes the EZH2 subdomain stimulation-responsive motif (SRM) ordered, and through the SRM and another EZH2 element SET activation loop (SAL), the binding signal can be transmitted to the SET catalytic domain of EZH2, resulting in a 10–20 fold increase of PRC2 activity, which addresses the methylation of unmodified H3K27 of neighboring nucleosomes.18–22 The bottom pocket recognizes and interacts with the N-terminal α helix of EZH2 (residues 39–68, named as the EED-binding domain (EBD)).58,59 When the C-terminus of the EBD is mainly in vdW contact with EED, the major connection made by the N-terminus of the EBD with EED is hydrogen bonding. Further through EED, the SAL of EZH2 is brought proximally to the back of the SET domain, endowing PRC2 with basal enzymatic activity.60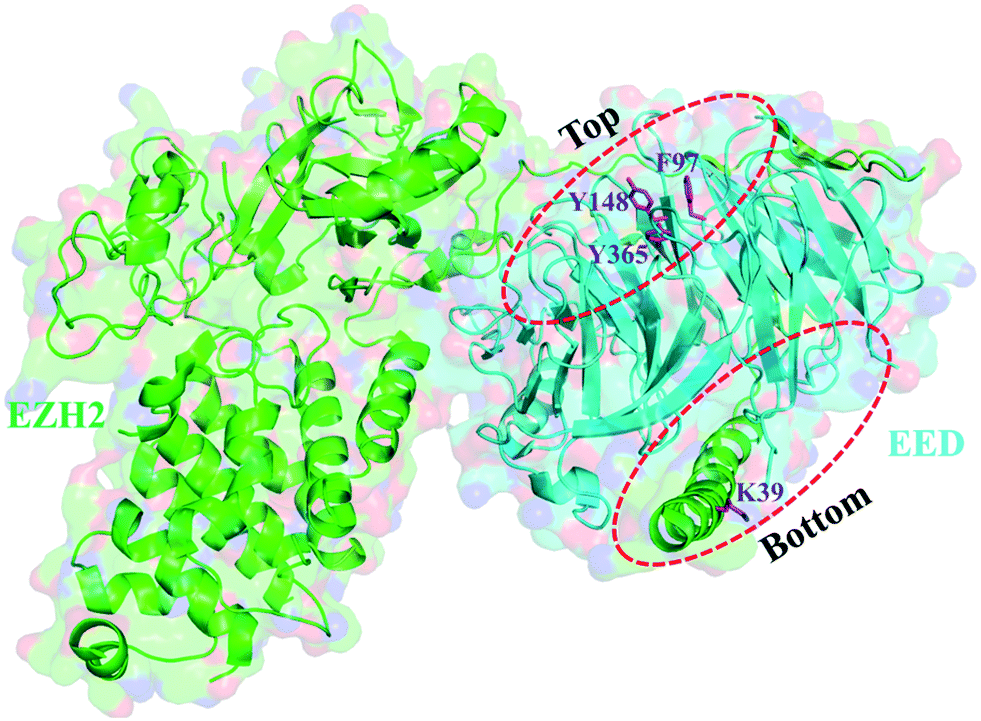 | ||
| Fig. 2 Two pockets in EED. The smaller top pocket is referred to as the “aromatic cage”, also known as the “Me3 pocket” with three sides formed from three aromatic residues F97, Y148 and Y365. The bottom pocket is mostly lined with hydrophobic residues that recognize and interact with the N-terminal α helix of EZH2. | ||
The efforts to develop EED inhibitors
Inhibitors targeting the top pocket of the EED protein (Table 1)
| Compound | Structure | Selectivity | Binding to EED | Associated tumor type | In vivo | Publication year | Ref. |
|---|---|---|---|---|---|---|---|
| EED226 |
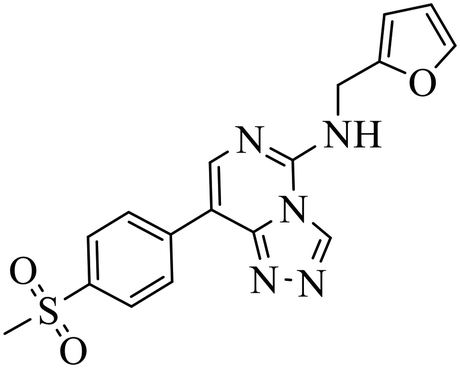
|
(1) Selective inhibition for PRC2 over 20 other methyltransferases with IC50 = 23.40 nM, H3K27me0 peptide as substrate | (1) ΔTm > 8 °C | DLBCL containing EZH2 mutations and WT EZH2: SU-DHL4, SU-DHL6, DB, Karpas422, WSU-DLCL2; GA10, OCI-LY19, Toledo | Karpas422 xenograft model | 2017 | 61 |
| (2) 25-Fold selective for EZH2 over EZH1 | (2) KD = 82.00 nM | ||||||
| A-395 |
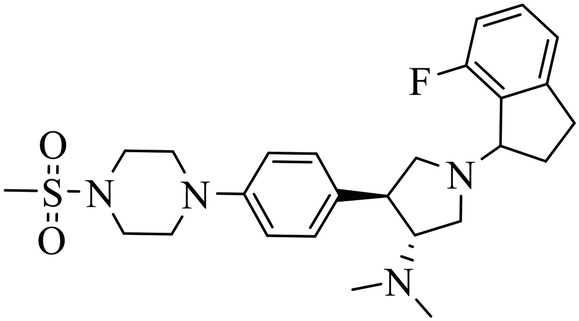
|
Selective inhibition for PRC2 over 32 other methyltransferases with IC50 = 18.00 nM | (1) ΔTm = 13−18 °C | (1) DLBCL containing EZH2 mutations and WT EZH2: SU-DHL4, DB, RL, UDHL6, Pfeiffer; SU-DHL8, Toledo, U2940 | Pfeiffer xenograft model | 2017 | 65 |
| (2) KD = 1.50 nM. | (2) Myeloma cell lines: RPMI 8226, MM.1s, L-363, AMO-1, NCI-H929, JJN-3, OPM-2, U266B1, Karpas 620, KMS-11 | ||||||
| (3) Breast cancer: SK-BR-3 | |||||||
| (4) Malignant rhabdoid tumor with SNF5 mutations: A204, G401 | |||||||
| BR-001 |
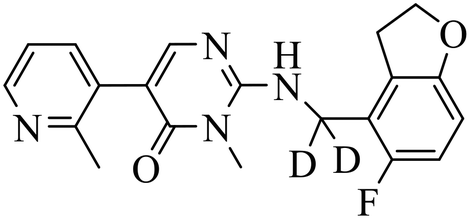
|
(1) selective inhibition for PRC2 over 371 wild-type kinases | ΔTm = 6−10 °C | DLBCL with EZH2 mutations: Karpas422, Pfeiffer, WSU-DLBCL | Karpas422 and Pfeiffer xenograft model | 2019 | 66 |
| (2) IC50 = 4.50 nM in displacement of H3K27me3 from EED | |||||||
| EEDi-5285 |
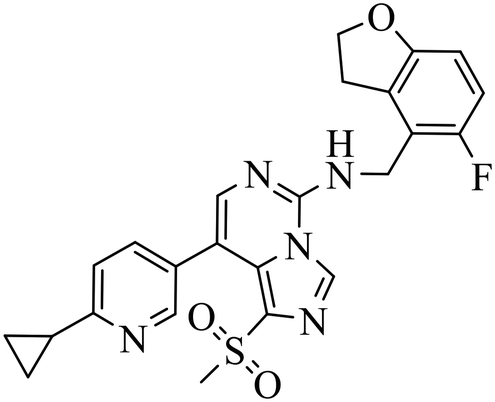
|
— | — | DLBCL with EZH2 mutations: Karpas422, Pfeiffer | Karpas422 xenograft model | 2020 | 67 |
| EEDi-1056 |
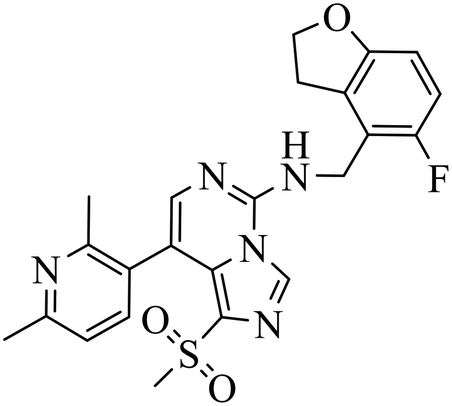
|
— | — | DLBCL with EZH2 mutations: Karpas422, Pfeiffer | Karpas422 xenograft model | 2020 | 67 |
| MAK683 |
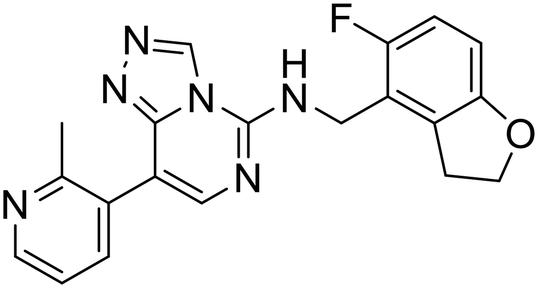
|
— | — | — | — | — | 68 |
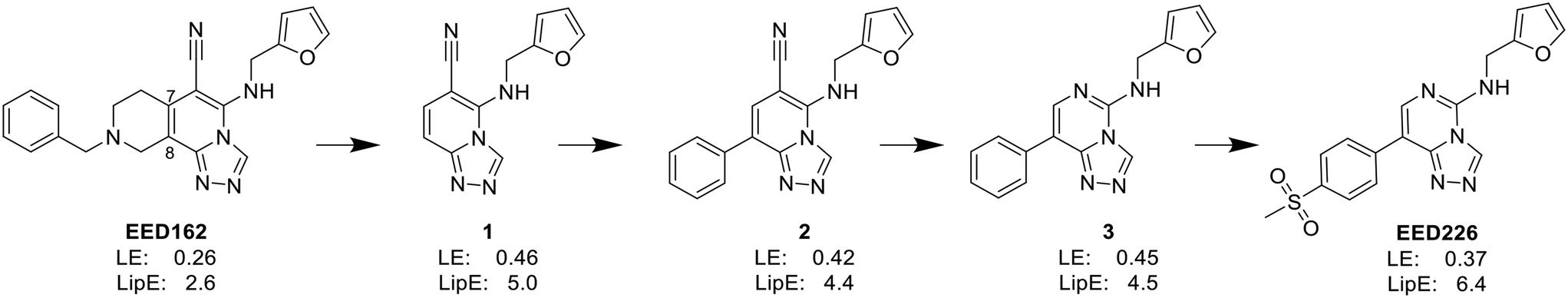 | ||
| Fig. 3 The optimization process of EED162 to EED226. Firstly, the benzylhydropyridine ring on C-7 and C-8 of EED162 was removed to afford compound 1 with improved ligand efficiency (LE) and lipophilic efficiency (LipE). Secondly, under the assumption that the substitution of an aryl group at the C-8 position can obtain edge-to-face interactions, compound 2 with phenyl substitution at C-8 was synthesized. Thirdly, the ensuing scaffold hopping – replacing the cyano group with nitrogen – was performed, and compound 3 was then obtained. Finally, in the optimization of pharmacokinetic potency, EED226 with methylsulfonyl substitution was selected because of its low clearance, moderate half-life and high oral bioavailability. | ||
In the enzyme-level assay, EED226 exhibited nanomolar inhibition potency against PRC2, compared with the half-maximal inhibitory concentration (IC50) of >100 μM toward other 20 protein methyltransferases. Moreover, it specifically inhibited the methylation of H3K27 in G401 cells. The crystal structure of EED226 in complex with EED and EZH2 (Protein Data Bank (PDB) ID: 5GSA) showed that it inserted into a much larger and deeper cage, which was newly formed from the conformational changes of residues F97, Y148, W364 and Y365 after the compound competitively bound to the H3K27me3 site in EED, and its furan group was in the innermost part of the pocket while its methylsulfonylphenyl group pointed to the solvent. When tested in a series of DLBLC cells, EED226 inhibited the proliferation more effectively in EZH2-mutant cells than in cells with WT EZH2, and the sensitivity of all tested cells to EED226 was at the same level as that to EI1. In Karpas422 cells, EED226 and EI1 implemented similar alteration of gene expression, and more importantly, their co-administration showed synergistic effects in terms of growth blockade and H3K27me3 reduction, with synergistic scores of 1.67 and 2.17, respectively, which was partly attributed to their simultaneous binding to PRC2. However, EED226 was still efficacious in EI1-resistant WSU-DLCL2 cells. With promising pharmacokinetic characteristics, EED226 achieved 100% tumor growth inhibition (TGI) at 40 mg kg−1 after 32 days of oral gavage in a mouse subcutaneous xenograft model of Karpas422. These findings provide insights into the unique mechanism of action (MOA) of EED226 and the potential combination therapy of EED and EZH2 inhibitors.
Novartis Institutes also reported another four novel EED binders (EED210, EED396, EED666, and EED709) along with EED162 (Fig. 4).63 Both the basal and H3K27me3-stimulated PRC2 catalytic activities were restrained by them via direct and H3K27me3-competitive binding to EED.
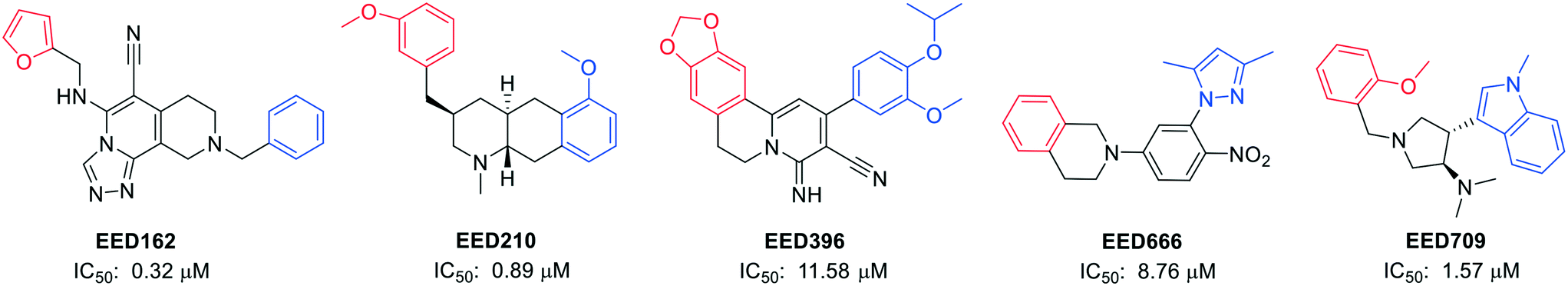 | ||
| Fig. 4 Structures of EED162, EED210, EED396, EED666 and EED709, together with their IC50 values in an EED-H3K27me3 AlphaScreen competition binding assay. The red parts in the structures interact with the R367 side chain by penetrating into the deep pocket, while the central rings are located among F97, Y148 and Y365, and the blue parts are at the edge of the pocket and still interact with EED.63 | ||
In another report, Lingel et al. from the same company deconstructed EED210 to compound 4. Through step-by-step optimization, five aminoimidazole compounds (11–15, Fig. 5), competitive with H3K27me3 for binding to the H3K27me3 pocket, were then obtained.64 Based on the co-crystal structure of EED in complex with compound 15, further optimization efforts can be focused on the isopropyl group owing to its exposure to the solvent. Although no intracellular and/or in vivo studies of the aforementioned hits have been carried out, they provide structural insights for future development of new EED inhibitors and could also serve as tool compounds.
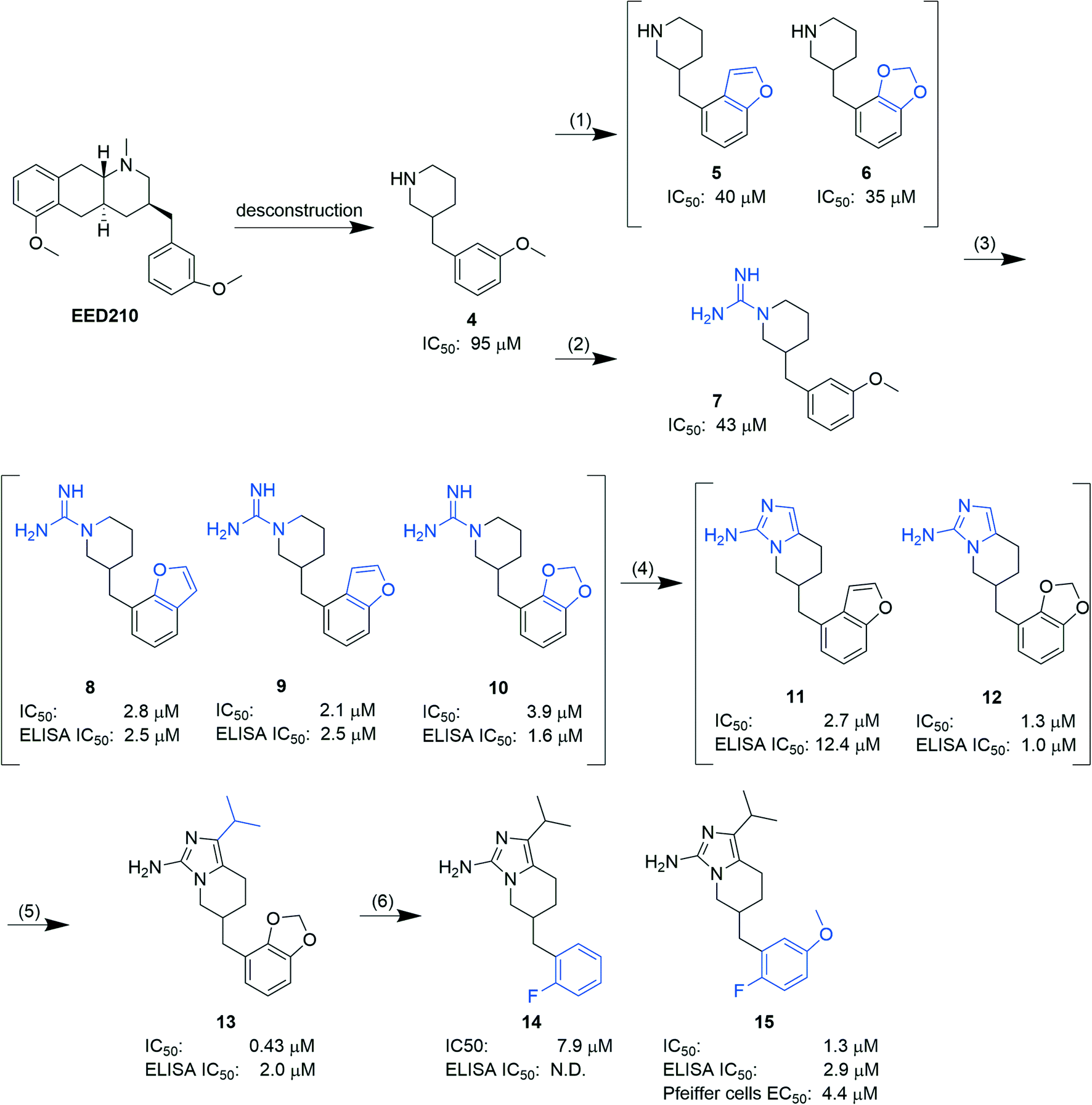 | ||
| Fig. 5 Structures of five aminoimidazole compounds (11–15) and the optimization process. (1) Replacing the methoxy group with an oxygen-containing ring; (2) optimizing the amine group in the piperidine ring; (3) combining (1) and (2); (4) substituting the guanidinium group with 2-aminoimidazolyl to improve permeability; (5) introducing an aliphatic group; (6) mitigating the potential liabilities of the phenyl ring. IC50 values represent the capability to inhibit the PRC2 activity; ELISA IC50 values represent the capability to inhibit the H3K27 methylation in G401 cells. N.D.: not determined. | ||
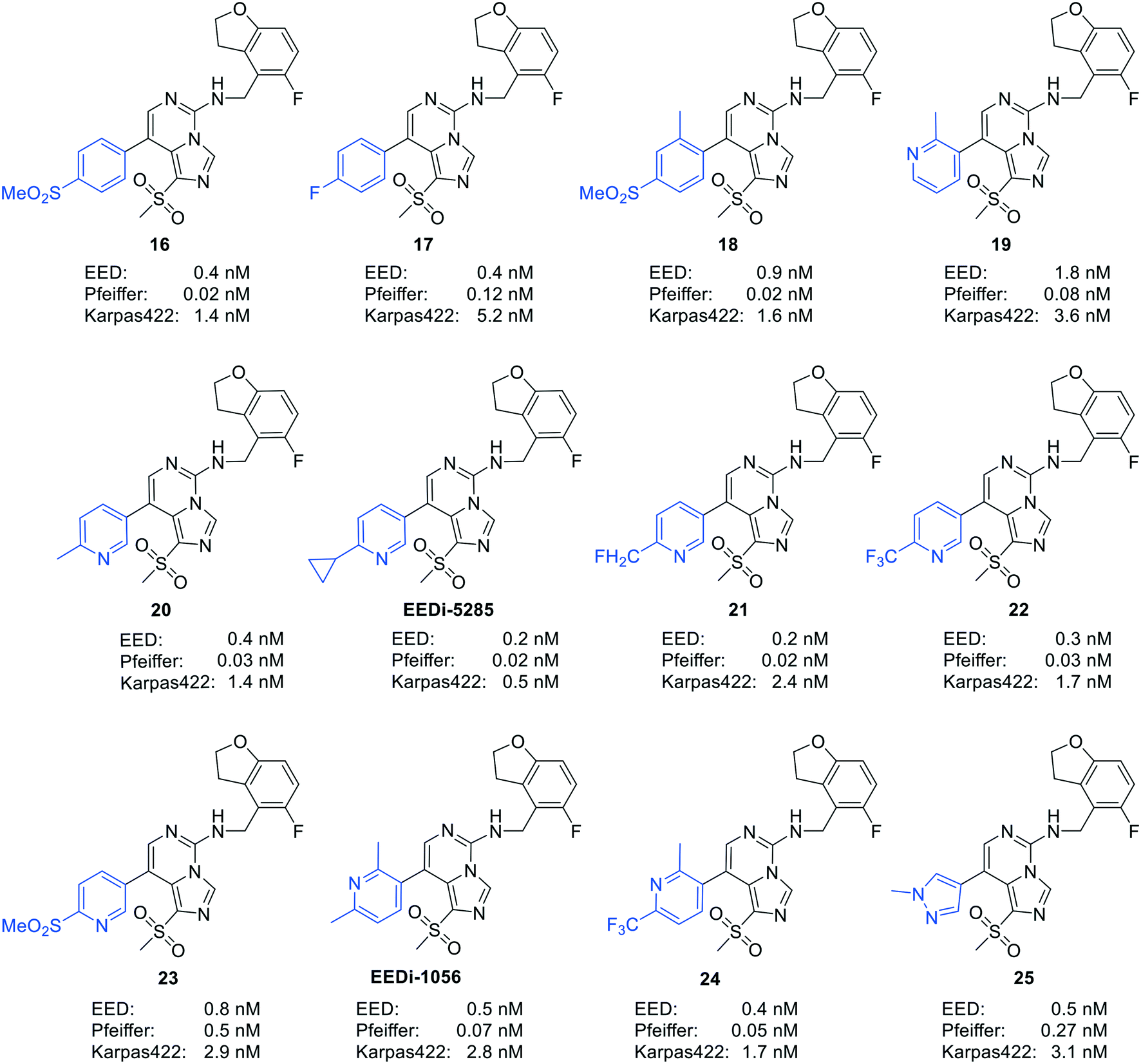 | ||
| Fig. 6 Structures of a number of highly efficacious EED inhibitors. | ||
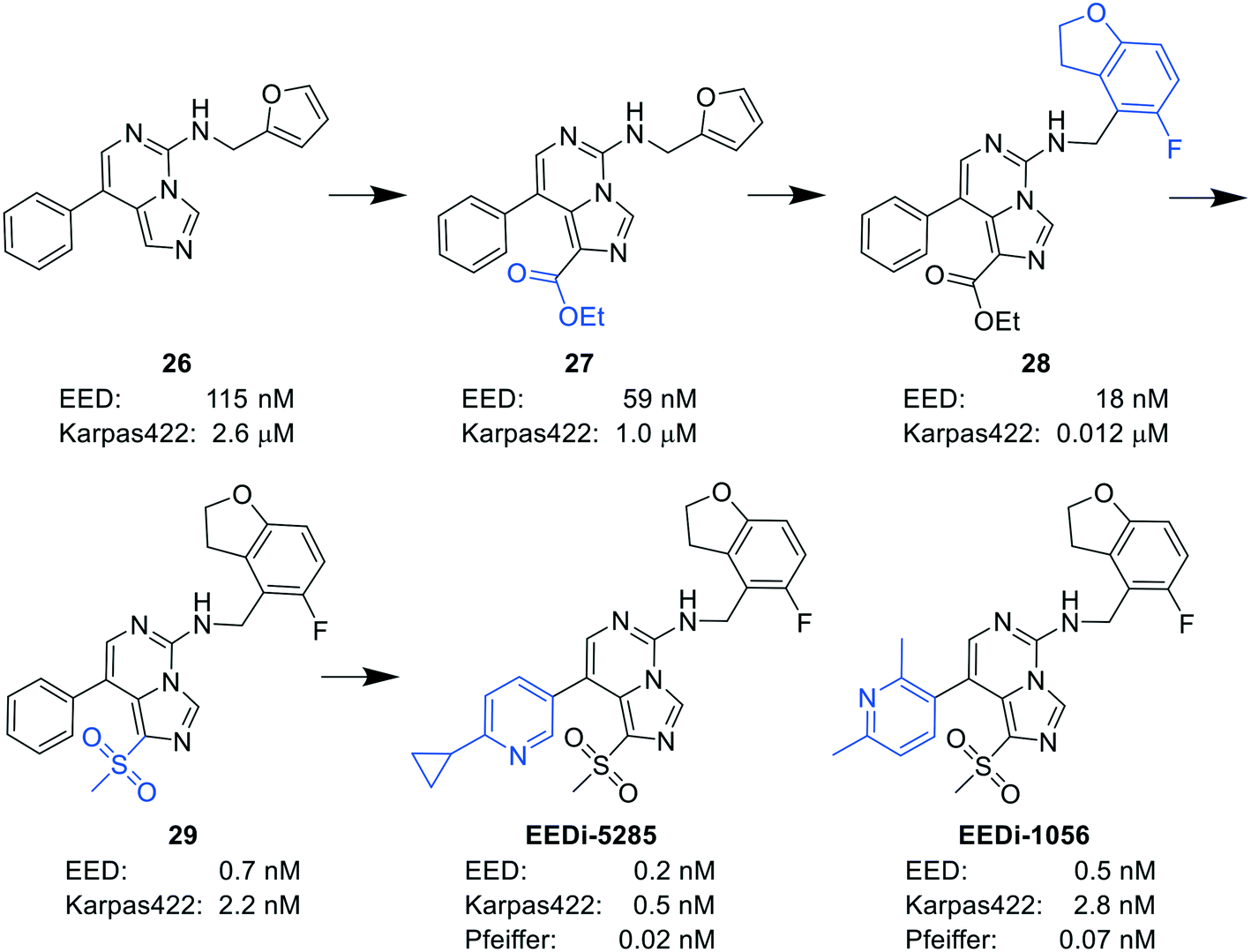 | ||
| Fig. 7 The optimization process of compound 26 to EEDi-5285 and EEDi-1056. First, by introducing an ethoxycarbonyl group into 26, compound 27 with enhanced π–π stacking interactions with EED was obtained. To improve cation–π interactions, the replacement of the furan moiety with another aromatic ring was then performed, and compound 28 exhibiting high affinity for EED (IC50 = 18 nM) and good inhibition against Karpas422 cells (IC50 = 0.012 μM) was thus acquired. Next, compound 29 was generated by displacing the ethoxycarbonyl unit in 28 with a methylsulfonyl group. Ultimately, considering the improvement of the solubility, the replacement of the phenyl group in 29 with 2-cyclopropyl-5-pyridinyl and 2,6-dimethyl-3-pyridinyl was conducted to yield EEDi-5285 and EEDi-1056, respectively. | ||
By comparing the co-crystal structures of EED with EEDi-5285 and EEDi-1056 (PDB ID: 6W7F and 6W7G, respectively) to that with EED226 (PDB ID: 5GSA), a series of similar interactions between the inhibitor and protein of the three complexes were disclosed, namely, cation–π interactions with residue R367, π–π interactions with Y148 and Y365 residues, and two hydrogen bonds with N194 and K211 residues. While in the former two co-crystal structures, the cation–π interactions were stronger than those in the latter one, the Y365 residue exhibited more extensive interactions owing to its position, which may account for their higher activities. Indeed, EEDi-5285 showed 88-fold and 364-fold more potency than EED226 in binding to EED (0.2 nM vs. 17.6 nM) and restraining the growth of Karpas422 cells (0.5 nM vs. 182 nM), respectively. Furthermore, when tested in the same in vivo model as EED226, EEDi-5285 and EEDi-1056 orally administered at 50 mg kg−1 displayed excellent efficacy, including 100% TGI sustained for another 72 days after the end of treatment (day 28) with minimal change of body weight.
Inhibitors targeting the bottom pocket of the EED protein (Table 2)
| Compound | Sequence or structure | Selectivity and binding | Associated tumor type | Publication year | Ref. |
|---|---|---|---|---|---|
| SAH-EZH2 | FSSNRXKILXRTEILNQEWKQRRIQPV | (1) Selective inhibition for PRC2 | (1) DLBCL with EZH2 mutations: Karpas422, Pfeiffer | 2014 | 69 |
| (2) Binding to EED with KD = 0.32 nM | (2) Breast cancer: MDA-MB-231 | ||||
| (3) Prostate cancer: DU145 | |||||
| (4) Leukemia: MLL-AF9 | |||||
| Astemizole |
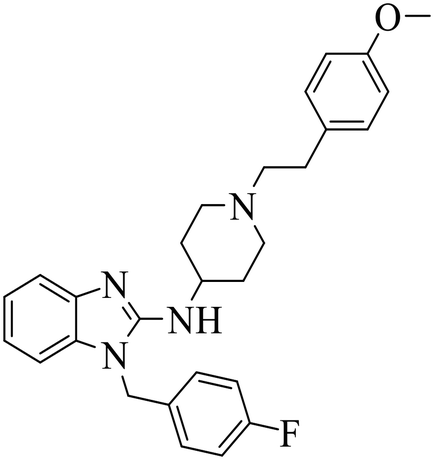
|
(1) Selective inhibition for PRC2 | DLBCL containing EZH2 mutations and WT EZH2: SU-DHL4, SU-DHL6, DB, Pfeiffer; Toledo | 2014 | 72 |
| (2) Binding to EED with ΔTm >4 °C | |||||
| Wedelolactone |
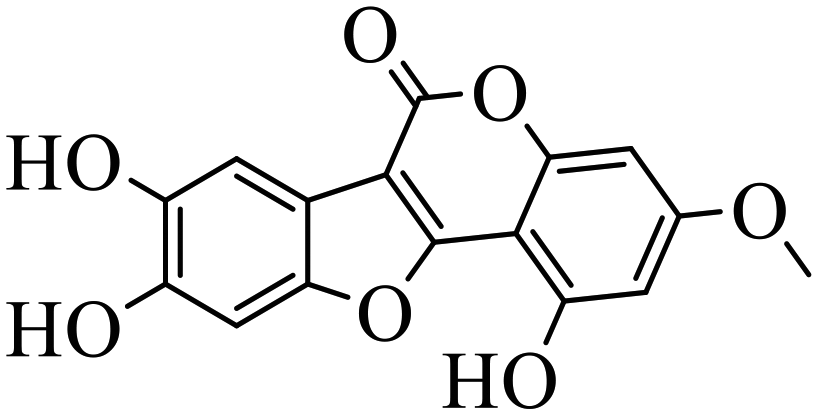
|
Binding to EED with KD = 2.82 μM | (1) Hepatocellular lines HepG2 | 2015 | 77 |
| (2) Monocytic leukemia cells THP1 | |||||
| (3) Myeloid leukemia cells K562 | |||||
| DC-PRC2in-01 |
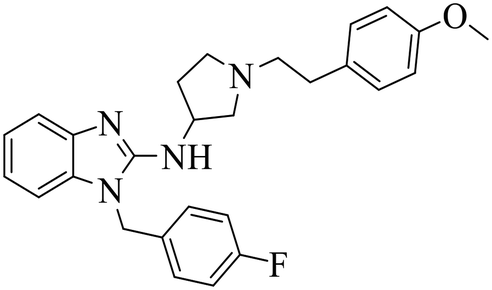
|
(1) Selective inhibition for PRC2 | DLBCL with EZH2 mutations: SU-DHL4, DB, Pfeiffer, Karpas422 | 2021 | 73 |
| (2) Binding to EED with KD = 4.56 μM |
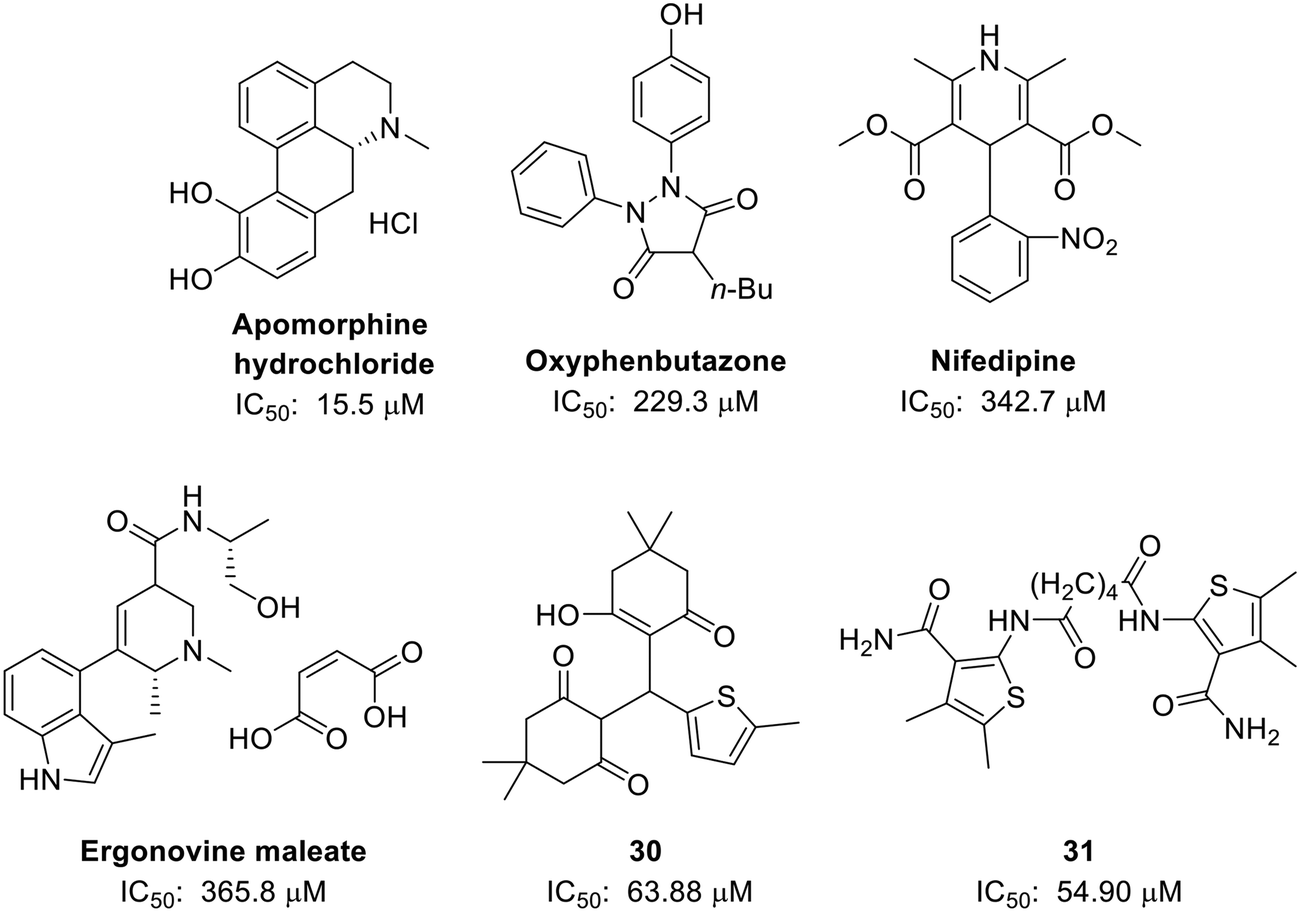 | ||
| Fig. 8 Structures of six initially identified EED-EZH2 inhibitors. In the FP experiment, apomorphine hydrochloride exhibited the best activity with an IC50 value of 15.50 μM. Both compounds 31 (IC50 = 54.90 μM) and 30 (IC50 = 63.88 μM) showed better activities than astemizole (IC50 = 93.80 μM). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 838 compounds and an FP experiment in 2019.82
838 compounds and an FP experiment in 2019.82
Several EZH2 and EED PROTACs
Bivalent chemical protein degraders, also known as proteolysis targeting chimeras (PROTACs), are molecules consisting of three parts, namely, an E3 ligase ligand, a protein of interest (POI) ligand and a linker concatenating the two ligands. The degradation of a POI is achieved via recruiting the E3 ubiquitin ligase to its proximity by a PROTAC. Recently, the development of bivalent degraders targeting EED or EZH2 has emerged as another new strategy for the inhibition of PRC2 activity. In early 2020, UNC6852 and PROTACs 1 and 2 were reported as active PROTAC molecules targeting EED by the University of North Carolina and AstraZeneca, respectively.83,84 In the same year, Ma et al. from Icahn School of Medicine at Mount Sinai described the discovery of MS1943 as the first-in-class EZH2 selective degrader.85 Then in 2021, a series of EZH2-targeted PROTACs were designed and synthesized by Liu et al. from Sichuan University, and among them, PROTAC E7 was assessed as the most effective PROTAC molecule.86 Their structures are shown in Fig. 9. In various cancer cells tested, although all the above-mentioned PROTACs can induce the degradation of H3K27me3, EZH2 and SUZ12, MS1943 failed to affect the EED level while the others could. Furthermore, while these PROTACs display cytotoxicity towards DLBCL cell lines with EZH2-activating mutations, such as DB, Pfeiffer, and Karpas422 cells, MS1943 has superior anti-proliferative efficacy against EZH2-dependent triple-negative breast cancer (TNBC) cells. Also, in mice bearing MDA-MB-468 tumor xenografts, complete suppression of tumor growth can be achieved with 150 mg kg−1 intraperitoneal (i.p.) injection of MS1943 once daily. Collectively, pharmacological degradation of EZH2 and EED is a new strategy to block the PRC2 function. EED- or EZH2-targeted PROTACs may become one of the key research directions in the development of chemotherapy targeting PRC2.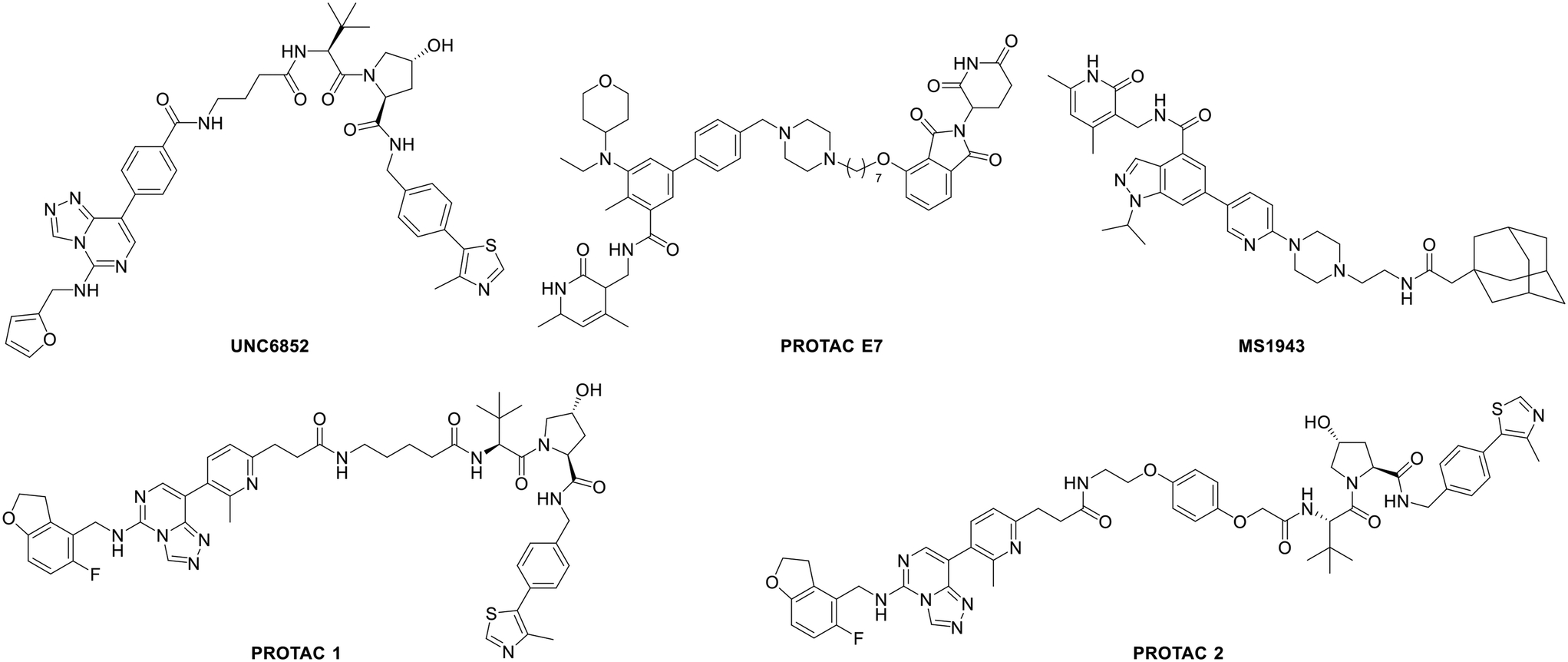 | ||
| Fig. 9 Structures of several EZH2 and EED PROTACs. | ||
Discussion
With the rapid development of epigenetic therapy in recent years, the PRC2 complex has become one of the significant targets and its inhibitors have thus been widely concerned in the drug discovery field. Among all the inhibitors, those directly targeting EZH2 have been mostly studied and some of them exhibit impressive profiles in preclinical and clinical practices. Nevertheless, it has to be noted that the secondary mutations caused by their long-term administration and the subsequent drug resistance have become new challenges to be solved. Also, due to their similar MOA – competing for methyl donor SAM – their combined treatment may not be a good solution. Therefore, it is necessary to explore other new strategies to regulate the PRC2 activity.Considering that EED is required for the basic activity of PRC2 and the binding of H3K27me3 with the EED top pocket can further improve the PRC2 activity, looking for inhibitors targeting EED could be a new strategy to disable the PRC2 activity in addition to targeting EZH2. SAH-EZH2, as a landmark discovery, proved the feasibility of this approach for the first time. It is gratifying that EED inhibitors themselves not only show good activity in repressing the growth of related cancers but also possess a synergistic effect with EZH2 inhibitors in impairing the viability of cancer cells sensitive to EZH2 inhibitors, suggesting potential combined therapeutic value between them. In addition, EED inhibitors may have advantages in the treatment of EZH1-dependent or both EZH1 and EZH2-dependent tumors, because both EZH1 and EZH2 in the PRC2 complex can be inhibited by some EED inhibitors, such as EED226. More importantly, the drug resistance caused by competitive EZH2 inhibitors may be overcome by EED inhibitors.
Nevertheless, since EZH2 has many non-PRC2 activities in cancers,87–90 the detailed mechanism of the synergistic effect of inhibitors targeting EZH2 and EED remains to be studied. Specifically, although the combined administration of SAH-EZH2 and GSK126 can enhance the anti-proliferative activity towards MLL-AF9 and Karpas422 cells compared with SAH-EZH2 alone, and that of astemizole and EPZ005687 towards Toledo and DB cells compared with astemizole alone, it is unclear whether this is due to additional inhibition of non-PRC2 related pathways by EZH2 inhibitors, or more complete blocking of PRC2, or both, which is worthy of further study. Another example is that the synergistic effect of EED226 and EI1 on Karpas422 cells can be in part attributed to a more complete blocking of PRC2, which was supported by the simultaneous binding of the two inhibitors to PRC2, as well as their combined effect on the protein level of H3K27me3 by CalcuSyn analysis.
As the old saying goes, every coin has two sides. EED inhibitors could also have their own issues. With more and more efforts being put into the development of new EED inhibitors, our understanding toward these molecules will also improve, and additional advantages as well as disadvantages of them will surely be disclosed in future work. For example, problems such as bioavailability, long-term toxicity and potential drug resistance are all significant issues to be considered and solved. We believe that there are always more solutions than difficulties, and the present efforts on EED inhibitors could be rewarding in the future.
In summary, there is no doubt that although this strategy of developing EED inhibitors is still in its infancy and needs a long way to go, it is bound to be a new hot spot of investigations for PRC2 disruptors and to be the complementary direction of development of EZH2 inhibitors, especially after the approval of the world's first EZH2 inhibitor. This review briefly introduces the research progress on EED inhibitors that disable the PRC2 function, hoping to provide some references for pharmaceutical workers who intend to understand this development trend and carry out relevant research studies.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the Innovation Team Project of Jinan Science & Technology Bureau [No. 2018GXRC003], the Young Taishan Scholars Program (tsqn20161037) and the National Natural Science Foundation of China (81803438) for financial support.References
- A. P. Feinberg, M. A. Koldobskiy and A. Gondor, Epigenetic modulators, modifiers and mediators in cancer aetiology and progression, Nat. Rev. Genet., 2016, 17, 284–299 CrossRef CAS PubMed.
- K. Skvortsova, N. Iovino and O. Bogdanovic, Functions and mechanisms of epigenetic inheritance in animals, Nat. Rev. Mol. Cell Biol., 2018, 19, 774–790 CrossRef CAS PubMed.
- P. Chammas, I. Mocavini and L. Di Croce, Engaging chromatin: PRC2 structure meets function, Br. J. Cancer, 2020, 122, 315–328 CrossRef CAS PubMed.
- E. Lavarone, C. M. Barbieri and D. Pasini, Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity, Nat. Commun., 2019, 10, 1679 CrossRef PubMed.
- J. W. Hojfeldt, A. Laugesen, B. M. Willumsen, H. Damhofer, L. Hedehus and A. Tvardovskiy, et al., Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2, Nat. Struct. Mol. Biol., 2018, 25, 225–232 CrossRef PubMed.
- E. Conway, E. Healy and A. P. Bracken, PRC2 mediated H3K27 methylations in cellular identity and cancer, Curr. Opin. Cell Biol., 2015, 37, 42–48 CrossRef CAS PubMed.
- R. Fioravanti, G. Stazi, C. Zwergel, S. Valente and A. Mai, Six years (2012-2018) of researches on catalytic EZH2 inhibitors: The boom of the 2-pyridone compounds, Chem. Rec., 2018, 18, 1818–1832 CrossRef CAS PubMed.
- K. H. Kim and C. W. Roberts, Targeting EZH2 in cancer, Nat. Med., 2016, 22, 128–134 CrossRef CAS PubMed.
- C. Ru, L. Wang, H. Wang, L. Xia, H. Erdjument-Bromage and P. Tempst, et al., Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing, Science, 2002, 298, 1039–1043 CrossRef PubMed.
- X. Shen, Y. Liu, Y. J. Hsu, Y. Fujiwara, J. Kim and X. Mao, et al., EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency, Mol. Cell, 2008, 32, 491–502 CrossRef CAS PubMed.
- M. Bisserier and N. Wajapeyee, Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas, Blood, 2018, 131, 2125–2137 CrossRef CAS PubMed.
- T. Baker, S. Nerle, J. Pritchard, B. Zhao, V. M. Rivera and A. Garner, et al., Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors, Oncotarget., 2015, 6, 32646–32655 CrossRef PubMed.
- V. Gibaja, F. Shen, J. Harari, J. Korn, D. Ruddy and V. Saenz-Vash, et al., Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors, Oncogene, 2016, 35, 558–566 CrossRef CAS PubMed.
- S. Chen, L. Jiao, M. Shubbar, X. Yang and X. Liu, Unique Structural platforms of Suz12 dictate distinct classes of PRC2 for chromatin binding, Mol. Cell, 2018, 69, 840–852 CrossRef CAS PubMed.
- D. Pasini, A. P. Bracken, M. R. Jensen, E. Lazzerini Denchi and K. Helin, Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity, EMBO J., 2004, 23, 4061–4071 CrossRef CAS PubMed.
- N. D. Montgomery, D. Yee, A. Chen, S. Kalantry, S. J. Chamberlain and A. P. Otte, et al., The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation, Curr. Biol., 2005, 15, 942–947 CrossRef CAS.
- R. Cao and Y. Zhang, SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex, Mol. Cell, 2004, 15, 57–67 CrossRef CAS.
- C. Xu, C. Bian, W. Yang, M. Galka, H. Ouyang and C. Chen, et al., Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2), Proc. Natl. Acad. Sci. U. S. A., 2011, 107, 19266–19271 CrossRef.
- N. Justin, Y. Zhang, C. Tarricone, S. R. Martin, S. Chen and E. Underwood, et al., Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2, Nat. Commun., 2016, 7, 11316–11326 CrossRef CAS PubMed.
- L. Jiao and X. Liu, Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2, Science, 2015, 350, aac4383 CrossRef PubMed.
- R. Margueron, N. Justin, K. Ohno, M. L. Sharpe, J. Son and W. J. Drury 3rd., Role of the polycomb protein EED in the propagation of repressive histone marks, Nature, 2009, 461, 762–767 CrossRef CAS PubMed.
- K. H. Hansen, A. P. Bracken, D. Pasini, N. Dietrich, S. S. Gehani and A. Monrad, et al., A model for transmission of the H3K27me3 epigenetic mark, Nat. Cell Biol., 2008, 10, 1291–1300 CrossRef CAS PubMed.
- R. D. Morin, N. A. Johnson, T. M. Severson, A. J. Mungall, J. An and R. Goya, et al., Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin, Nat. Genet., 2010, 42, 181–185 CrossRef CAS PubMed.
- C. Bödör, C. O'Riain, D. Wrench, J. Matthews, S. Iyengar and H. Tayyib, et al., EZH2 Y641 mutations in follicular lymphoma, Leukemia, 2011, 25, 726–729 CrossRef PubMed.
- M. T. McCabe, H. M. Ott, G. Ganji, S. Korenchuk, C. Thompson and G. S. Van Aller, et al., EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations, Nature, 2012, 492, 108–112 CrossRef CAS PubMed.
- C. Bödör, V. Grossmann, N. Popov, J. Okosun, C. O'Riain and K. Tan, et al., EZH2 mutations are frequent and represent an early event in follicular lymphoma, Blood, 2013, 122, 3165–3168 CrossRef PubMed.
- B. Li and W. J. Chng, EZH2 abnormalities in lymphoid malignancies: underlying mechanisms and therapeutic implications, J. Hematol. Oncol., 2019, 12, 118–131 CrossRef.
- M. L. Eich, M. Athar, J. E. Ferguson and S. Varambally, EZH2-Targeted therapies in cancer: hype or a reality, Cancer Res., 2020, 80, 5449–5458 CrossRef PubMed.
- C. R. Majer, L. Jin, M. P. Scott, S. K. Knutson, K. W. Kuntz and H. Keilhack, et al., A687V EZH2 is a gain-of-function mutation found in lymphoma patients, FEBS Lett., 2012, 586, 3448–3451 CrossRef CAS PubMed.
- M. T. Mccabe, A. P. Graves, G. Ganji, E. Diaz, W. S. Halsey and Y. Jiang, et al., Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27), Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2989–2994 CrossRef CAS.
- S. Varambally, S. M. Dhanasekaran, M. Zhou, T. R. Barrette, C. Kumar-Sinha and M. G. Sanda, et al., The polycomb group protein EZH2 is involved in progression of prostate cancer, Nature, 2002, 419, 624–629 CrossRef CAS PubMed.
- C. G. Kleer, Q. Cao, S. Varambally, R. Shen, I. Ota and S. A. Tomlins, et al., EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11606–11611 CrossRef CAS PubMed.
- I. M. Bachmann, O. J. Halvorsen, K. Collett, I. M. Stefansson, O. Straume and S. A. Haukaas, et al., EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast, J. Clin. Oncol., 2006, 24, 268–273 CrossRef CAS PubMed.
- C. Lu, H. D. Han, L. S. Mangala, R. Ali-Fehmi, C. S. Newton and L. Ozbun, et al., Regulation of tumor angiogenesis by EZH2, Cancer Cell, 2010, 18, 185–197 CrossRef CAS PubMed.
- Z. Rao, M. Cai, G. Yang, L. He, S. Mai and W. Hua, et al., EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-β 1 and is a predictor of outcome in ovarian carcinoma patients, Carcinogenesis, 2010, 31, 1576–1583 CrossRef CAS PubMed.
- H. Li, Q. Cai, A. K. Godwin and R. Zhang, Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells, Mol. Cancer Res., 2010, 12, 1610–1618 CrossRef PubMed.
- A. Yamada, S. Fujii, H. Daiko, M. Nishimura, T. Chiba and A. Ochiai, Aberrant expression of EZH2 is associated with a poor outcome and P53 alteration in squamous cell carcinoma of the esophagus, Int. J. Oncol., 2011, 38, 345–353 CAS.
- J. B. Mchugh, D. R. Fullen, L. Ma, C. G. Kleer and L. D. Su, Expression of polycomb group protein EZH2 in nevi and melanoma, J. Cutaneous Pathol., 2007, 34, 597–600 CrossRef PubMed.
- H. W. Lee and M. Choe, Expression of EZH2 in renal cell carcinoma as a novel prognostic marker, Pathol. Int., 2012, 62, 735–741 CrossRef CAS PubMed.
- J. Xu, Z. Wang, W. Lu, H. Jiang, J. Lu and J. Qiu, et al., EZH2 promotes gastric cancer cells proliferation by repressing p21 expression, Pathol., Res. Pract, 2019, 215, 152374 CrossRef CAS PubMed.
- C. Behrens, L. M. Solis, H. Lin, P. Yuan, X. Tang and H. Kadara, et al., EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma, Clin. Cancer Res., 2013, 19, 6556–6565 CrossRef CAS PubMed.
- F. Shahabipour, M. Caraglia, M. Majeed, G. Derosa, P. Maffioli and A. Sahebkar, Naturally occurring anti-cancer agents targeting EZH2, Cancer Lett., 2017, 400, 325–335 CrossRef CAS PubMed.
- G. Stazi, C. Zwergel, A. Mai and S. Valente, EZH2 inhibitors: a patent review (2014-2016), Expert Opin. Ther. Pat., 2017, 27, 797–813 CrossRef CAS PubMed.
- N. Gulati, W. Beguelin and L. Giulino-Roth, Enhancer of zeste homolog 2 (EZH2) inhibitors, Leuk. Lymphoma, 2018, 59, 1574–1585 CrossRef CAS.
- J. K. Lue and J. E. Amengual, Emerging EZH2 inhibitors and their application in lymphoma, Curr. Hematol. Malig. Rep., 2018, 13, 369–382 CrossRef.
- R. Duan, W. F. Du and W. J. Guo, EZH2: a novel target for cancer treatment, J. Hematol. Oncol., 2020, 13, 104–115 CrossRef.
- W. Qi, H. M. Chan, L. Teng, L. Li, S. N. Chuai and R. P. Zhang, et al., Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21360–21365 CrossRef CAS.
- K. D. Konze, A. Ma, F. Li, D. Barsyte-Lovejoy, T. Parton and C. J. Macnevin, et al., An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1, ACS Chem. Biol., 2013, 8, 1324–1334 CrossRef CAS.
- R. G. Vaswani, V. S. Gehling, L. A. Dakin, A. S. Cook, C. G. Nasveschuk and M. Duplessis, et al., Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-Cell lymphomas, J. Med. Chem., 2016, 59, 9928–9941 CrossRef CAS PubMed.
- P. P. Kung, P. Bingham, A. Brooun, M. Collins, Y. L. Deng and D. Dinh, et al., Optimization of orally bioavailable Enhancer of Zeste Homolog 2 (EZH2) inhibitors using ligand and property-based design strategies: identification of development candidate (R)-5,8-dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497), J. Med. Chem., 2018, 61, 650–665 CrossRef CAS PubMed.
- S. K. Knutson, S. Kawano, Y. Minoshima, N. M. Warholic, K. C. Huang and Y. H. Xiao, et al., Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma, Mol. Cancer Ther., 2014, 13, 842–854 CrossRef CAS PubMed.
- S. M. Hoy, Tazemetostat: First Approval, Drugs, 2020, 80, 513–521 CrossRef PubMed.
- F. Morschhauser, H. Tilly, A. Chaidos, P. Mckay, T. Phillips and S. Assouline, et al., Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial, Lancet Oncol., 2020, 21, 1433–1442 CrossRef CAS PubMed.
- P. Zhang, M. C. Gooijer, L. C. M. Buil, J. H. Beijnen, G. Li and O. Tellingen, ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-Inhibitors, Int. J. Cancer, 2015, 137, 2007–2018 CrossRef CAS PubMed.
- T. Baker, S. Nerle, J. Pritchard, B. Y. Zhao, V. M. Rivera and A. Garner, et al., Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors, Oncotarget, 2015, 6, 32646–32655 CrossRef.
- V. Gibaja, F. Shen, J. Harari, J. Korn, D. Ruddy and V. Saenz-Vash, et al., Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors, Oncogene, 2016, 35, 558–566 CrossRef CAS PubMed.
- M. Bisserier and N. Wajapeyee, Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas, Blood, 2018, 131, 2125–2137 CrossRef CAS PubMed.
- V. Kasinath, M. Faini, S. Poepsel, D. Reif, X. A. Feng and G. Stjepanovic, et al., Structures of human PRC2 with its cofactors AEBP2 and JARID2, Science, 2018, 359, 940–944 CrossRef CAS PubMed.
- Z. F. Han, X. M. Xing, M. Hu, Y. Zhang, P. Y. Liu and J. J. Chai, Structural basis of EZH2 recognition by EED, Structure, 2007, 15, 1306–1315 CrossRef CAS PubMed.
- A. Brooun, K. S. Gajiwala, Y. L. Deng, W. Liu, B. Bolanos and P. Bingham, et al., Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance, Nat. Commun., 2016, 7, 11384 CrossRef CAS PubMed.
- W. Qi, K. H. Zhao, J. Gu, Y. Huang, Y. Z. Wang and H. L. Zhang, et al., An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED, Nat. Chem. Biol., 2017, 13, 381–388 CrossRef CAS PubMed.
- Y. Huang, J. Zhang, Z. T. Yu, H. L. Zhang, Y. Z. Wang and A. Lingel, et al., Discovery of first-in-class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy, J. Med. Chem., 2017, 60, 2215–2226 CrossRef CAS PubMed.
- L. Li, H. L. Zhang, M. Zhang, M. X. Zhao, L. J. Feng and X. Luo, et al., Discovery and molecular basis of a diverse set of polycomb repressive complex 2 inhibitors recognition by EED, PLoS One, 2017, 12, e0169855 CrossRef.
- A. Lingel, M. Sendzik, Y. Huang, M. D. Shultz, J. Cantwell and M. P. Dillon, et al., Structure-guided design of EED binders allosterically inhibiting the epigenetic polycomb repressive complex 2 (PRC2) methyltransferase, J. Med. Chem., 2017, 60, 415–427 CrossRef CAS PubMed.
- Y. P. He, S. Selvaraju, M. L. Curtin, C. G. Jakob, H. Z. Zhu and K. M. Comess, et al., The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex, Nat. Chem. Biol., 2017, 13, 389–395 CrossRef CAS PubMed.
- H. P. Dong, S. J. Liu, X. J. Zhang, S. Chen, L. J. Kang and Y. N. Chen, et al., An Allosteric PRC2 inhibitor targeting EED suppresses tumor progression by modulating the immune response, Cancer Res., 2019, 79, 5587–5596 CrossRef CAS PubMed.
- R. K. Rej, C. W. Wang, J. F. Lu, M. Wang, E. Petrunak and K. P. Zawacki, et al., EEDi-5285: An exceptionally potent, efficacious, and orally active small-molecule inhibitor of embryonic ectoderm development, J. Med. Chem., 2020, 63, 7252–7267 CrossRef CAS PubMed.
- D. D. Huang, S. Z. Tian, Y. F. Qi and J. Z. H. Zhang, Binding Modes of Small-Molecule Inhibitors to the EED Pocket of PRC2, ChemPhysChem, 2020, 21, 263–271 CrossRef CAS PubMed.
- W. Kim, G. H. Bird, T. Neff, G. J. Guo, M. A. Kerenyi and L. D. Walensky, et al., Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer, Nat. Chem. Biol., 2013, 9, 643–650 CrossRef CAS PubMed.
- J. F. Lyu, E. J. Yang, S. A. Head, N. N. Ai, B. Y. Zhang and C. J. Wu, et al., Astemizole Inhibits mTOR Signaling and Angiogenesis by Blocking Cholesterol Trafficking, Int. J. Biol. Sci., 2018, 14, 1175–1185 CrossRef CAS PubMed.
- J. J. Tian, L. Vandermosten, S. Peigneur, L. Moreels, J. Rozenski and J. Tytgat, et al., Astemizole analogues with reduced hERG inhibition as potent antimalarial compounds, Bioorg. Med. Chem., 2017, 25, 6332–6344 CrossRef CAS PubMed.
- X. Kong, L. Chen, L. Jiao, X. Jiang, F. Lian and J. Lu, et al., Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2, J. Med. Chem., 2014, 57, 9512–9521 CrossRef CAS PubMed.
- D. H. Du, D. D. Xu, L. C. Zhu, G. L. Stazi, C. Zwergel and Y. L. Liu, et al., Structure-Guided Development of Small-Molecule PRC2 Inhibitors Targeting EZH2-EED Interaction, J. Med. Chem., 2021, 64, 8194–8207 CrossRef CAS PubMed.
- J. García-Quiroz and J. Camacho, Astemizole: an old anti-histamine as a new promising anti-cancer drug, Anti-Cancer Agents Med. Chem., 2011, 11, 30714 CrossRef PubMed.
- R. E. García-Ferreiro, D. Kerschensteiner, F. Major, F. Monje, W. Stühmer and L. A. Pardo, Mechanism of block of hEag1 K+ channels by imipramine and astemizole, J. Gen. Physiol., 2004, 124, 30117 CrossRef PubMed.
- S. Matsumoto and Y. Yamazoe, Involvement of multiple human cytochromes P450 in the liver microsomal metabolism of astemizole and a comparison with terfenadine, Br. J. Clin. Pharmacol., 2001, 51, 133–142 CAS.
- H. M. Chen, S. J. Gao, J. D. Li, D. Liu, C. J. Sheng and C. Yao, et al., Wedelolactone disrupts the interaction of EZH2-EED complex and inhibits PRC2-dependent cancer, Oncotarget, 2015, 6, 13049–13059 CrossRef.
- A. Svrlanska, A. Ruhland, M. Marschall, N. Reuter and T. Stamminger, Wedelolactone inhibits human cytomegalovirus replication by targeting distinct steps of the viral replication cycle, Antiviral Res., 2020, 174, 104677 CrossRef CAS PubMed.
- H. Pan, Y. Lin, J. Dou, Z. Fu, Y. Yao and S. Ye, et al., Wedelolactone facilitates Ser/Thr phosphorylation of NLRP3 dependent on PKA signalling to block inflammasome activation and pyroptosis, Cell Proliferation, 2020, 53, e12868 CAS.
- S. Sharma, S. Trivedi, T. Pandey, S. Ranjan and R. Pandey, Wedelolactone Mitigates Parkinsonism Via Alleviating Oxidative Stress and Mitochondrial Dysfunction Through NRF2/SKN-1, Mol. Neurobiol., 2021, 58, 65–77 CrossRef PubMed.
- M. R. Zhu, D. H. Du, J. C. Hu, L. C. Li, J. Q. Liu and H. Ding, et al., Development of a high-throughput fluorescence polarization assay for the discovery of EZH2-EED interaction inhibitors, Acta Pharmacol. Sin., 2018, 39, 302–310 CrossRef CAS PubMed.
- K. K. Zhu, D. H. Du, R. Yang, H. R. Tao and H. Zhang, Identification and assessments of novel and potent small-molecule inhibitors of EED-EZH2 interaction of polycomb repressive complex 2 by computational methods and biological evaluations, Chem. Pharm. Bull., 2020, 68, 58–63 CrossRef PubMed.
- F. Potjewyd, A. W. Turner, J. Beri, J. M. Rectenwald, J. L. Norris-Drouin and S. H. Cholensky, et al., Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader, Cell Chem. Biol., 2020, 27, 47–56.e15 CrossRef CAS PubMed.
- J. H. Hsu, T. Rasmusson, J. Robinson, F. Pachl, J. Read and S. Kawatkar, et al., EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex, Cell Chem. Biol., 2020, 16(27), 41–46.e17 CrossRef PubMed.
- A. Ma, E. Stratikopoulos, K. S. Park, J. Wei, T. C. Martin and X. Yang, et al., Discovery of a first-in-class EZH2 selective degrader, Nat. Chem. Biol., 2020, 16, 214–222 CrossRef CAS PubMed.
- Z. Liu, X. Hu, Q. Wang, X. Wu, Q. Zhang and W. Wei, et al., Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2, J. Med. Chem., 2021, 64, 2829–2848 CrossRef CAS PubMed.
- K. Xu, Z. J. Wu, A. C. Groner, H. H. He, C. Cai and R. T. Lis, et al., EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent, Science, 2012, 14(338), 1465–1469 CrossRef PubMed.
- A. Davies, S. Nouruzi, D. Ganguli, T. Namekawa, D. Thaper and S. Linder, et al., An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer, Nat. Cell Biol., 2021, 23, 1023–1034 CrossRef CAS.
- Y. Zhao, Z. Hu, J. Li and T. Hu, EZH2 Exacerbates Breast Cancer by Methylating and Activating STAT3 Directly, J. Cancer, 2021, 12, 5220–5230 CrossRef CAS PubMed.
- J. Wang and G. G. Wang, No Easy Way Out for EZH2: Its Pleiotropic, Noncanonical Effects on Gene Regulation and Cellular Function, Int. J. Mol. Sci., 2020, 21, 9501 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |