
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyanine-based near infra-red organic photoredox catalysis†
Anne Roly
Obah Kosso
,
Nicolas
Sellet
,
Alexandre
Baralle
,
Morgan
Cormier
 * and
Jean-Philippe
Goddard
*
* and
Jean-Philippe
Goddard
*
Laboratoire d’Innovation Moléculaire et Applications (LIMA), UMR 7042, Université de Haute-Alsace (UHA), Université de Strasbourg, CNRS, 68100 Mulhouse, France. E-mail: morgan.cormier@uha.fr; jean-philippe.goddard@uha.fr
First published on 13th April 2021
Abstract
Direct metal-free near infra-red photoredox catalysis is applied to organic oxidation, photosensitization and reduction, involving cyanines as photocatalysts. This photocatalyst is competitive with conventional reactions catalyzed under visible light. Kinetic and quenching experiments are also reported. Interestingly, these systems are compatible with water media, opening perspective for various applications.
Introduction
Light as a source of energy for organic transformations has always been an interesting alternative to thermally driven processes. Classically, UV-irradiation is used to activate organic reaction, since most of molecules absorb in this spectral window.1 However, such high energetic light is not innocent and limit the reaction applicability in terms of scope and safety issues. Moving to less energetic wavelengths (i.e. visible light), photoredox catalysis appeared to be a remarkable solution.2 The excitation of a photoredox catalyst (PC to PC*) could give birth to either an oxidant or a reductant, able to promote SET organic transformations. Although extensively studied, the use of visible light has still some limitations like the low penetration of such wavelengths through the solution,3 which limits the set-up to small scales or flow-systems.4 Additionally, the biological window for light is between 650 nm and 950 nm,5 which reduces the impact of visible photoredox catalysis for biological applications. To circumvent these problems, longer wavelengths should be involved, shifting from visible light to near infra-red (NIR) irradiation. Very recently, NIR-photoredox catalysis emerged as a valuable solution resulting in more efficient photochemical processes, due to deeper penetration in various reaction media and the improvement of the irradiation surface. An appropriate photocatalyst able to absorb NIR-light with suitable excited state redox properties is needed. Indirect NIR-photoredox catalysis transformations have been developed based on upconversion phenomena. Two photocatalytic systems are needed, one to convert a NIR-photon into a visible photon and a second, based on conventional photocatalysts, absorbing visible photon to initiate a photoredox transformation (Scheme 1a).6 Recently, direct NIR-photoredox catalysis has been accomplished using Os(II) polypyridyl complexes to promote oxidation/reduction reactions (Scheme 1b).7 Although having remarkable properties, preparation and handling need a glovebox. Moreover, osmium could be an issue in term of toxicity. In parallel, publications have been reported on NIR-polymerization using cyanines as a photoiniator (Scheme 1c).3,8 These NIR-dyes are also well-known as fluorescence probes9 and redox partners for CuAAC, also in a context of polymer science.8,10 Thus, we anticipated that such cyanines could catalyze organic photoredox transformations under NIR light and open a promising field of investigation to push forward the limitation of conventional visible light photoredox catalysis.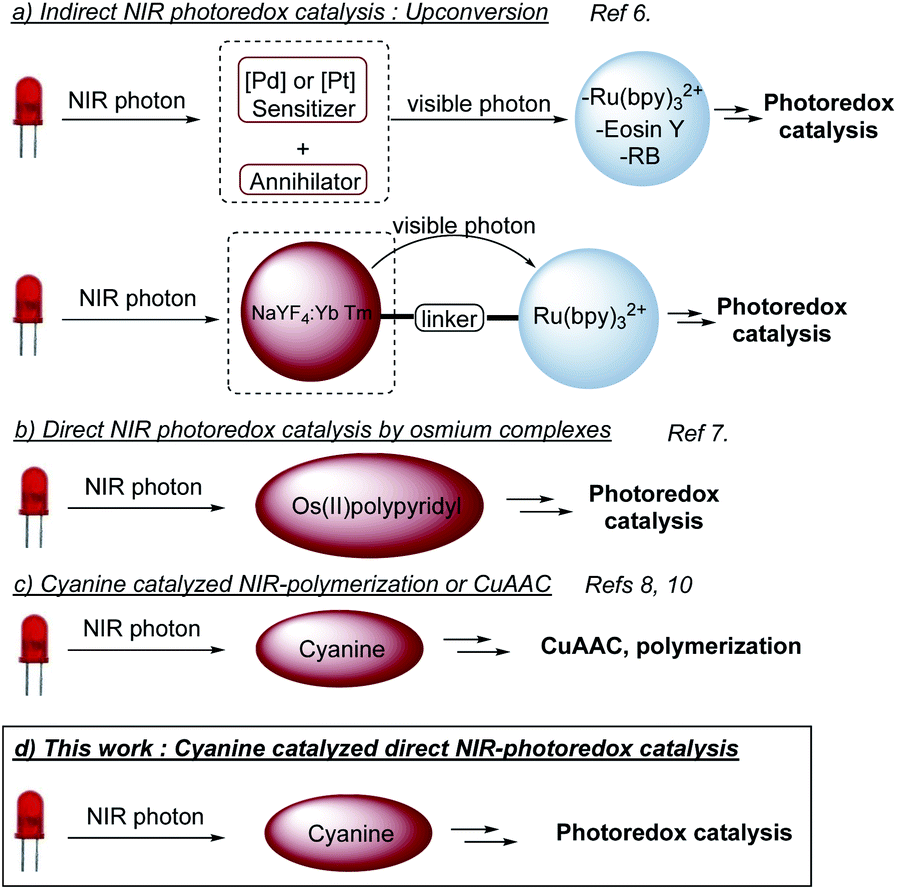 | ||
| Scheme 1 NIR-Photoredox catalysis. | ||
Results and discussion
Among a large variety of cyanines, we selected stable commercial Indocyanine Green (ICG), IR-813, DTTCI, cy746 (Fig. 1), with a structural diversity impacting their photophysical properties (see spectra in ESI §2†). However, this family of dyes owns similar excited state lifetime (∼1 ns)11 and comparable redox properties.12 | ||
| Fig. 1 Selected cyanine photocatalysts. | ||
Then, we began their evaluation with the photoredox oxidation of amine through the aza-Henry reaction (Table 1).13 The optimization has been done with 1a and nitromethane as nucleophile to generate 2a under NIR-light. Gratifyingly, the irradiation (810 nm) of 1a (Eox = 0.62 V vs. SCE)14 with photocatalysts in nitromethane (Table 1, entry 1–4) promoted the formation of 2a in modest to good conversions. Among the tested cyanine photocatalysts, cy746 revealed to be superior (Table 1, entry 4) and we monitored the impact of the irradiation wavelength. As λmax (cy746 in DMSO) = 760 nm, we expected to improve the reaction efficiency with an irradiation at 780 nm but only 14% conversion after 24 h was observed (Table 1, entry 5). No benefit was gained with lower energetic light at 940 nm (Table 1, entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | PC | x (mol%) | Solvent | λ (nm) | Convb. (%) |
| a Run on 0.1 mmol scale. b Conversions are determined on the crude by 1H-NMR. c CH3NO2 is used as solvent. d Run in the dark. e Run under nitrogen atmosphere in the absence of air. | |||||
| 1 | ICG | 10 | CH3NO2c | 810 | 16 |
| 2 | IR-813 | 10 | CH3NO2c | 810 | 28 |
| 3 | DTCCI | 10 | CH3NO2c | 810 | 14 |
| 4 | cy746 | 10 | CH3NO2c | 810 | 57 |
| 5 | cy746 | 10 | CH3NO2c | 780 | 14 |
| 6 | cy746 | 10 | CH3NO2c | 940 | <5 |
| 7 | cy746 | 10 | DCM | 810 | 48 |
| 8 | cy746 | 10 | DMF | 810 | n.r |
| 9 | cy746 | 10 | MeOH | 810 | 28 |
| 10 | cy746 | 10 | DMSO | 810 | 91 |
| 11 | cy746 | 5 | DMSO | 810 | 85 |
| 12 | cy746 | 1 | DMSO | 810 | 67 |
| 13 | cy746 | — | DMSO | 810 | 16 |
| 14d | cy746 | 5 | DMSO | 810 | n.r |
| 15e | cy746 | 5 | DMSO | 810 | 5 |
Finally, variation of solvents and catalyst loadings (Table 1, entry 7–12) revealed that reaction conditions involving 10 equivalents of nitromethane in DMSO under irradiation (810 nm) allowed a very high 91% conversion after 24 h (Table 1, entry 10), which can be maintained at 85% with 5 mol% of cy746 (Table 1, entry 11). When the catalyst was omitted, only 16% conversion was measured (Table 1, entry 13) while no reaction occurred in the dark (Table 1, entry 14). In the absence of oxygen, 5% conversion was observed, indicating that photocatalyst could not be recovered without oxygen (Table 1, entry 15). This clearly indicates the synergistic effect of light, oxygen and photocatalyst to reach high conversion.
These optimal conditions were applied to a set of tetrahydroisoquinolines 1 with various nucleophiles (Scheme 2). An electronic modification of the N-aryl moiety is tolerated given 2a–d in good yields even with more challenging electron poor substrates (i.e., 2d). A more sterically congested nitropropane does not affect the reaction and 2e is obtained in good yield (71%, d.r = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6). Other C–C and C–P bond formation processes have been investigated.14 With dimethyl malonate, 2f is isolated in 74% yield, resulting from the addition of the corresponding enol tautomer onto the iminium intermediate. In the same vein, cyanide anion addition from trimethylsilyl cyanide generates 2g in 79% yield. Diethyl phosphite is also compatible with our optimized conditions and phosphonate 2h is isolated in a satisfactory 75% yield. Our optimized conditions demonstrated a very good tolerance to the catalytic alkynylation of 1a through copper phenylacetylide addition (Scheme 3). The photocatalytic system seems to be undisturbed by this second organometallic catalytic cycle since the corresponding adduct 2i is obtained in 75% yield. In this dually catalyzed transformation, copper(I) intermediate as well as its reactivity, seems to be preserved against redox processes as it was also demonstrated for visible light photoredox catalysis.
:0.6). Other C–C and C–P bond formation processes have been investigated.14 With dimethyl malonate, 2f is isolated in 74% yield, resulting from the addition of the corresponding enol tautomer onto the iminium intermediate. In the same vein, cyanide anion addition from trimethylsilyl cyanide generates 2g in 79% yield. Diethyl phosphite is also compatible with our optimized conditions and phosphonate 2h is isolated in a satisfactory 75% yield. Our optimized conditions demonstrated a very good tolerance to the catalytic alkynylation of 1a through copper phenylacetylide addition (Scheme 3). The photocatalytic system seems to be undisturbed by this second organometallic catalytic cycle since the corresponding adduct 2i is obtained in 75% yield. In this dually catalyzed transformation, copper(I) intermediate as well as its reactivity, seems to be preserved against redox processes as it was also demonstrated for visible light photoredox catalysis.
 | ||
| Scheme 2 Scope of Aza-Henry type reactions. | ||
 | ||
| Scheme 3 Dual catalysis: alkynylation reaction. | ||
To gain insights into the reaction parameters, additional experiments were done. “On/off” experiments demonstrated that the conversion increases during a light-on period and stopped when light is off (Fig. 2a). When the light was switched on after 14 h in the dark, the conversion of 1a was resumed. This demonstrates the stability of the catalyst which was confirmed by the monitoring (1H-NMR) of an irradiated solution of cy746 (DMSO-d6) over 24 h (see ESI §5†).
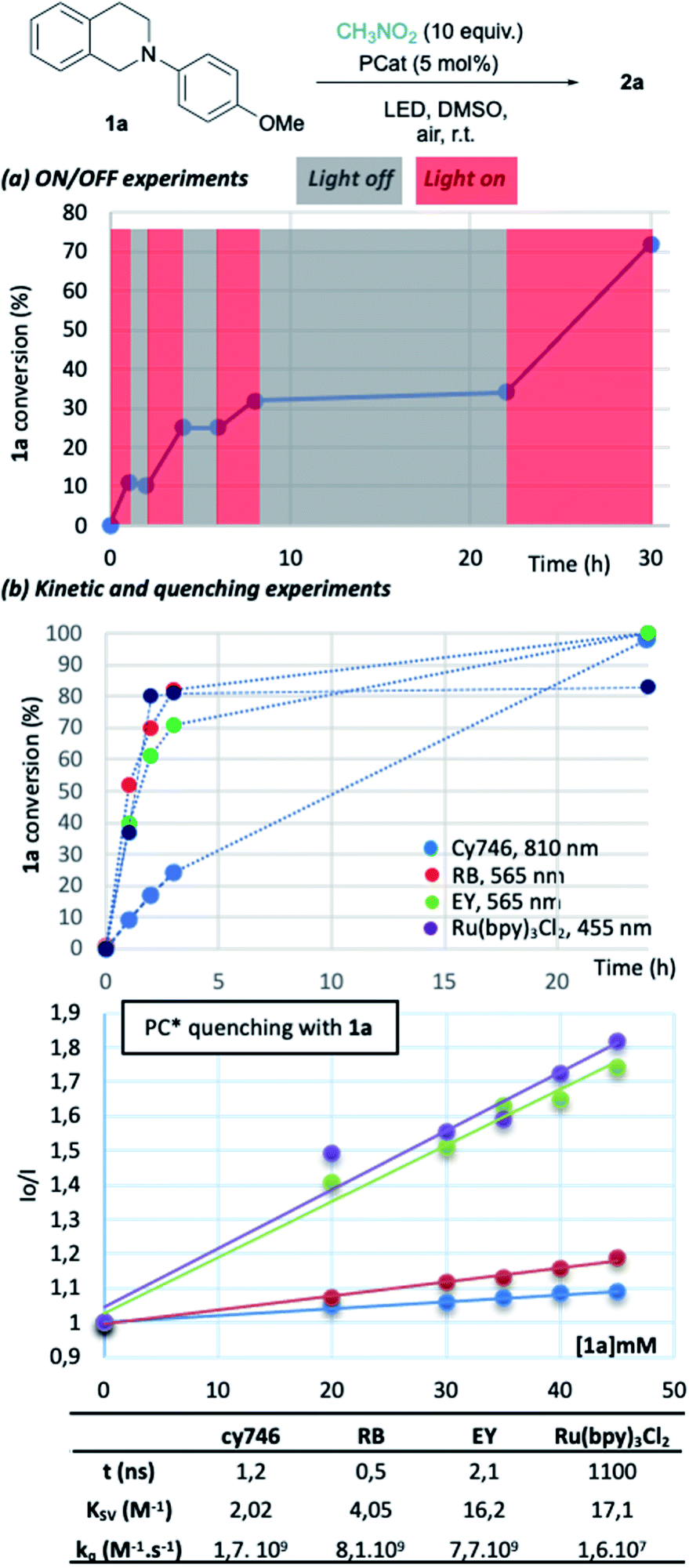 | ||
| Fig. 2 Reaction insights of NIR photoredox process. | ||
We compared the kinetic profile of cy746 (810 nm) with Rose Bengal (565 nm), Eosin Y (565 nm) and Ru(bpy)3Cl2 (455 nm) in their respective optimal conditions for the formation of 2a (Fig. 2b). The visible photocatalysts showed faster formation of 2a to a plateau (∼80%), reached in 4 h. While the reaction with cy746 appeared to be slower (24% conversion in 3 h), full conversion was achieved in 24 h.
In addition, quenching experiments have been done to determine Stern–Volmer constant of the cy746/1a system in comparison with the same three catalysts (Fig. 2b).15 cy746 has comparable excited lifetime11 to other organic dyes (∼1 ns)2d,16 and similar excited state quenching constant (kq = 1.7 109 M−1 s−1) which is 100-fold more than Ru(bpy)3Cl2. Therefore, the slower reactions, observed with cy746 cannot be only explained by those two parameters.
To demonstrate the synthetic potential of cyanines as photoredox catalysts, we extended the scope to heteroatom oxidations, photosensitization and onium salt reduction (Scheme 4). Thus, oxidation of N,N-dimethylaniline 3 (Eox = 0.80 V vs. SCE)17 afforded the corresponding α-aminoalkyl radical which reacted with maleimide followed through a formal [4 + 2] cycloaddition and gave cis-5 in 79% yield as a unique diastereoisomer.18 Thioamide 6 dimerized according to sulfur atom oxidation to provide 1,2,4-thiadiazole 7 in 77%.19 The oxidation of boronic acid 8 was done to generate 9 in 78% yield, as a mild alternative to classical H2O2 oxidation conditions.20
 | ||
| Scheme 4 Selected examples of NIR-triggered reactions. | ||
Cyanines are known to generate 1O2 for dynamic phototherapy.21 Thus, we took advantage of this to apply NIR O2 photosensitization to the oxidation of furfural 10 and tetraphenylcyclopentadienone 12 at 810 nm under O2 atmosphere to get respectively 11 and 13 in 45% and 74% yields.22 The photosensitization of vinyl azide was also successfully applied to 14, producing, after cyclization, the disubstituted pyrrole 15 in 68% yield.23 Interestingly, cy746 is also capable to catalyze reductive processes. The Umemoto's reagent 16 (Ered = −0.75 V vs. Fc/Fc+) was reduced by cy746 under anaerobic conditions to performed trifluoromethylation reactions through the formation of the trifluoromethyl radical intermediate.24,25 Thus, this reactive radical added onto 17 to generate the resulting tertiary bis-benzylic radical that could be further oxidized to provide 18 (50%) in the absence nucleophile. This reaction was improved by using the more electron-rich olefin 19 to form 20 (66%). The compatibility of our NIR-photoredox catalysis with water media has been demonstrated by the formation of the hydroxy trifluoromethylation adduct 21 (46%), coming from the [CF3 radical addition/oxidation/water addition] sequence. It is important to note that no background reaction was observed without light and cy746 (Scheme 4). Further mechanistic considerations as well as two proposed mechanisms are reported in the ESI† section.
Conclusions
In conclusion, we developed an original metal-free NIR-photoredox catalytic system as one of the first to promote organic transformations. This user-friendly method, based on commercial and stable cyanine (cy746), demonstrated its versatility for organic transformations (aza-Henry, heteroatom oxidation, photosensitization and reduction) under mild irradiation (810 nm). Additionally, we compared some photophysical properties with conventional photoredox catalysts. The design and synthesis of new cyanine-based photoredox catalysts are currently in progress in order to improve the reaction parameters.Author contributions
AROK, NS, AB, MC and JPG planned, ran and analyzed the experiments. MC and JPG designed and directed the project and wrote the manuscript with the help of AROK, NS and AB. All the authors contributed to the discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank UHA for providing facilities and funding. JPG and AROK thank IUF for financial support.Notes and references
- A. Albini and M. Fagnoni, Photochemically-Generated Intermediate in Synthesis, Willey-VCH, Weinheim, 2013 Search PubMed.
- (a) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem, 2019, 1, 111–125 CrossRef CAS; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (c) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (e) R. A. Angnes, L. Zhou, C. R. Correia and G. G. Hammond, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC . For mechanistic insight, see L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- A. H. Bonardi, T. M. Grant, G. Noirbent, D. Gigmes, B. H. Lessard, J.-P. Fouassier and J. Lalevée, J. Macromolecules, 2018, 51, 1314–1324 CrossRef CAS.
- E. B. Corcoran, J. P. McMullen, F. Lévesque, M. K. Wismer and J. R. Naber, Angew. Chem., Int. Ed., 2020, 59, 11964–11968 CrossRef CAS PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- (a) F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed; (b) F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CrossRef CAS; (c) L. Huang, W. Wu, K. Huang, W. Lin and G. Han, J. Am. Chem. Soc., 2020, 142, 18460–18470 CrossRef CAS PubMed; (d) B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed; (e) M. Freitag, N. Möller, A. Rühling, C. A. Strassert, B. J. Ravoo and F. Glorius, ChemPhotoChem, 2019, 3, 24–27 CrossRef CAS.
- B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053–2059 CrossRef CAS PubMed.
- (a) B. Strehmel, C. Schmitz, C. Kütahya, Y. Pang, A. Drewitz and H. Mustroph, Beilstein J. Org. Chem., 2020, 16, 415–444 CrossRef CAS PubMed; (b) B. Strehmel, C. Schmitz, K. Cremanns and J. Göttert, Chem. -Eur. J., 2019, 25, 12855–12864 CrossRef CAS PubMed; (c) T. Brömme, C. Schmitz, D. Oprych, A. Wenda, V. Strehmel, M. Grabolle, U. Resh-Genger, S. Ernt, D. Keil, P. Lüs, H. Baumann and B. Strehmel, Chem. Eng. Technol., 2016, 39, 13–25 CrossRef.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- (a) C. Kütahya, Y. Yagci and B. Strehmel, ChemPhotoChem, 2019, 3, 1180–1186 CrossRef; (b) C. Kütahya, C. Schmitz, V. Strehmel, Y. Yagci and B. Strehmel, Angew. Chem., Int. Ed., 2018, 57, 7898–7902 CrossRef PubMed.
- J. K. G. Karlsson, O. Woodford, H. Mustroph and A. Harriman, Photochem. Photobiol. Sci., 2018, 17, 99–106 CrossRef CAS PubMed.
- (a) C. Schmitz, A. Halbhuber, D. Keil and B. Strehmel, Prog. in Org. Coat., 2016, 100, 32–46 CrossRef CAS; (b) B. Strehmel, S. Ernst, K. Reiner, D. Keil, H. Lindauer and H. Baumann, Z. Phys. Chem., 2014, 228, 129–153 CrossRef CAS; (c) For selected data see the ESI† section..
- (a) S. M. Soria- Castro, B. Lebeau, M. Cormier, S. Neunlist, J. Daou and J.-P. Goddard, Eur. J. Org. Chem., 2020, 1572–1578 CrossRef CAS; (b) A. G. Condie, J. C. Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed.
- D. Prasad and B. König, Org. Lett., 2011, 13, 3852–3855 CrossRef PubMed.
- (a) S. P. Pitre, D. C. McTiernan and J. C. Scaino, ACS Omega, 2016, 1, 66–76 CrossRef CAS PubMed; (b) K sv are determined from the curve slopes and kq = Ksv/t (in second)..
- S. Paria and O. Reiser, ChemCatChem, 2014, 6, 2477–2483 CrossRef CAS.
- T. L. Macdonal, W. G. Gutheim, R. B. Martin and F. P. Guengerich, Biochemistry, 1989, 28, 2071–2077 CrossRef PubMed.
- W. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561–3567 CrossRef.
- V. P. Srivastava, A. K. Yadav and L. D. S. Yadav, Synlett, 2013, 24, 0465–0470 CrossRef CAS.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- (a) Q. Zhang, S. Xu, F. Lai, Y. Wang, N. Zhang, M. Nazare and H.-Y. Hu, Org. Lett., 2019, 21, 2121–2125 CrossRef CAS PubMed; (b) L. Jiao, F. Song, J. Cui and X. Peng, Chem. Commun., 2018, 54, 9198–9201 RSC.
- J. M. Carney, R. J. Hammer, M. Hulce, C. M. Lomas and D. Miyashiro, Synthesis, 2012, 44, 2560–2566 CrossRef CAS.
- E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793–797 CrossRef CAS PubMed.
- R. Tomita, Y. Yasu, T. Koike and M. Akita, Beilstein J. Org. Chem., 2014, 10, 1099–1106 CrossRef PubMed.
- T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937–1945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00998b |
| This journal is © The Royal Society of Chemistry 2021 |