
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFLP-type nitrile activation and cyclic ether ring-opening by halo-borane nonagermanide-cluster Lewis acid–base pairs†
Christoph
Wallach‡
,
Felix S.
Geitner‡
and
Thomas F.
Fässler
 *
*
Department Chemie, Technische Universität München, Lichtenbergstraße 4, 85747 Garching b. München, Germany. E-mail: thomas.faessler@lrz.tum.de
First published on 7th April 2021
Abstract
Even though homoatomic nine-atom germanium clusters are known for two decades, their chemical properties are still rarely investigated. We now discovered that Zintl ion main group-element clusters possess a reactive lone pair of electrons, and we show a new pathway to bind ligands with functional groups to the [Ge9] cluster core through Ge–C bond formation. We report on the reactivity of [Ge9{Si(TMS)3}2]2− (TMS = trimethylsilyl) towards a series of Lewis acidic bromo-boranes. The reaction of [Ge9{Si(TMS)3}2]2− and DABo-tol–Br (DAB = 1,3,2-diazaborolidine; o-tol = 2-methylphenyl) resulted, depending on the reaction protocol, either in the formation of [Ge9{Si(TMS)3}2DABo-tol]− (1a) with direct Ge–B interactions, or in [Ge9{Si(TMS)3}2(CH2)4O–DABo-tol]− (2a) featuring a ring-opened thf moiety. Ring opening reactions occur for all bulkier DABR–Br [R: o-xyl (2,6-dimethylphenyl), Mes (2,4,6-trimethylphenyl), Dipp (2,6-diisopropylphenyl)], DAB(II)Dipp–Br and acyclic (iPr2N)2BBr without Ge–B bond formation as shown for the structural characterization of the ring-opened products of thf (3, 4) and trimethylene oxide (5). In contrast to thf, the activation of CH3CN requires the simultaneous presence of Lewis-acid and Lewis-basic reactants allowing the formation of [Ge9{Si(TMS)3}2CH3C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–DABMes]− (6a). Within the presented compounds, 3 and 4 show an unusual substitution pattern of the three ligands at the [Ge9] core in the solid state. The [Ge9] cluster/borane systems correspond to intermolecular frustrated Lewis pairs (FLPs), in which the [Ge9] cluster with several lone pairs represents the Lewis base, and the borane is the Lewis acid.
N–DABMes]− (6a). Within the presented compounds, 3 and 4 show an unusual substitution pattern of the three ligands at the [Ge9] core in the solid state. The [Ge9] cluster/borane systems correspond to intermolecular frustrated Lewis pairs (FLPs), in which the [Ge9] cluster with several lone pairs represents the Lewis base, and the borane is the Lewis acid.
Introduction
Main group element compounds and especially frustrated Lewis pairs (FLPs) have recently emerged in the field of small molecule activation. FLPs rely on a spatial separation of the Lewis-acidic and -basic sites preventing adduct formation and are known to interact with a plethora of different molecules including gasses, unsaturated systems or solvent molecules featuring polarizable bonds.1–4 For the latter, as examples the main group assisted opening of cyclic ethers5–9 or the reaction with nitriles,10–15 which is often accompanied by ring formation, can be quoted.Recently, the idea that transition-metal free [Ge9] clusters might serve as Lewis base in Ge/boron based FLPs was put to front16 and metal-functionalized [Ge9]-Zintl clusters have been employed as homogeneous catalysts, in which the Ge atoms serve as a “support” for the catalytically active transition metal.17 Homoatomic anionic nine-atom germanium clusters offer indeed a framework for a spatial arrangement of various functional groups and possess at least six Ge atoms with lone pairs pointing to the outside of the cage, resembling an overall six-fold Lewis base. Nonagermanide clusters are accessible in high yields and purity via fusion of stoichiometric amounts of K and Ge forming the Zintl phase K4Ge9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18,19 containing discrete, extractable [Ge9]4− anions.20–23 In heterogeneous reactions organic main group element fragments are bound to the [Ge9] core enhancing its solubility and stability. Thus, by treatment of solid K4Ge9 with stoichiometric amounts of an acetonitrile solution of chloro-tris(trimethylsilyl)silane the two- and three-fold silylated clusters [Ge9{Si(TMS)3}2]2−24 and [Ge9{Si(TMS)3}3]−25 are obtained, respectively. The transfer of this method to elusive [Si9] clusters in the phase K12Si17 yielding [Si9{SiR3}2]2−, [Si9{SiR3}3]− (R = TMS, tBu2H),26,27 [Si9H2]2−,28 and [Si9H]3−29,30 shows the huge potential of this synthetic protocol using polar intermetallic compounds (Zintl phases) as precursors.
18,19 containing discrete, extractable [Ge9]4− anions.20–23 In heterogeneous reactions organic main group element fragments are bound to the [Ge9] core enhancing its solubility and stability. Thus, by treatment of solid K4Ge9 with stoichiometric amounts of an acetonitrile solution of chloro-tris(trimethylsilyl)silane the two- and three-fold silylated clusters [Ge9{Si(TMS)3}2]2−24 and [Ge9{Si(TMS)3}3]−25 are obtained, respectively. The transfer of this method to elusive [Si9] clusters in the phase K12Si17 yielding [Si9{SiR3}2]2−, [Si9{SiR3}3]− (R = TMS, tBu2H),26,27 [Si9H2]2−,28 and [Si9H]3−29,30 shows the huge potential of this synthetic protocol using polar intermetallic compounds (Zintl phases) as precursors.
The addition of functional groups such as silyl fragments24,31–35 to bare [Ge9] allowed for a stabilization and enhanced solubility of the cluster. Thus, mixed substituted clusters featuring various group 14 element fragments became accessible.36–41 However, the attachment of phosphanyl-moieties42–45 to [Ge9] showed that the clusters can be decorated with more interesting Lewis basic functional groups. Similarly of interest is the addition of a ligand comprising an electron-deficient boranyl moiety by our group.
The boranyl functionalization of [Ge9] clusters was achieved by the reaction of [Ge9{Si(TMS)3}2]2− with the heterocyclic chloro-boranes DABR–Cl (R: Me, iPr, o-tol; DAB: 1,3,2-diazaborolidine) yielding the anions [Ge9{Si(TMS)3}2DABR]−, thus introducing Lewis acidic moieties to the cluster. In fact, quantum chemical calculations corroborated the anion [Ge9{Si(TMS)3}2BCy2]− (Cy: cyclohexyl) to be an intramolecular frustrated Lewis acid–base pair (FLP), and its reactivity towards small molecules has been anticipated.16 Within the system B/Ge, a bis-amido germylene-based Lewis acid/base-related pair is the only reported example, to the best of our knowledge. Since a Ge–B adduct is formed in the ground state, it does not correspond to a typical FLP, but nevertheless, a cleavage of the CO bond of ketones or isocyanates has been observed.46
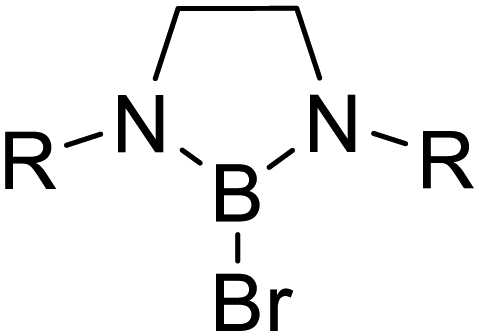
Triggered by the variety of reactions reported for FLP systems and on the basis of our recent achievements in combining boranes and [Ge9] clusters, we now investigated the reactivity of [Ge9{Si(TMS)3}2]2− as an appropriate Lewis base in combination with sterically hindered boron-based electrophiles, focusing on the reactivity towards molecules with polar groups. As boranes we applied the cyclic species DABR–Br (R: iPr, o-tol, o-xyl, Mes, Dipp) and DAB(II)Dipp–Br [(II): unsaturated backbone] as well as acyclic (iPr2N)2BBr, which (according to the acceptor number (AN) determined by the Gutmann–Beckett method) show an increased electrophilicity compared to the chloro-species used for the synthesis of the reported boranyl-functionalized clusters, thus featuring prerequisites for the formation of (reactive) FLPs.16,47–49 Within the presented reactions the transition metal-free Zintl ions can be regarded as an active component in small molecule activation.
Results and discussion
Ring-opening of tetrahydrofuran
The reaction of equimolar amounts of [Ge9{Si(TMS)3}2]2− and DABo-tol–Br in 1,4-dioxane selectively yields anionic [Ge9{Si(TMS)3}2DABo-tol]− (1a). Besides the respective peak at m/z 1398.7 observed in the mass spectrum (Fig. S46†), the NMR data are congruent to the reaction product featuring a Ge–B bond as already reported for the reaction with DABo-tol–Cl (Fig. 1a).16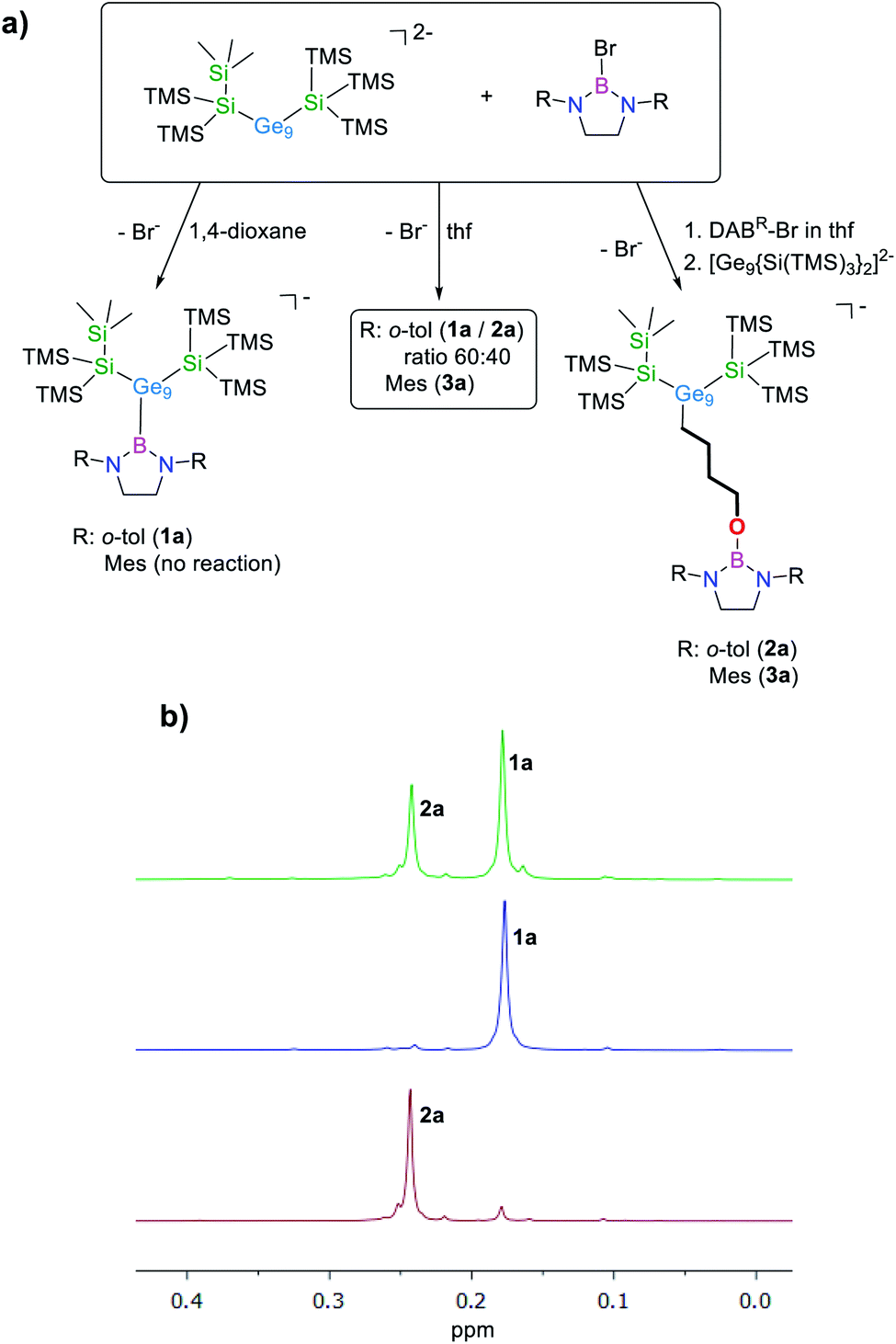 | ||
| Fig. 1 (a) Reactivity of [Ge9{Si(TMS)3}2]2− towards DABR–Br (R: o-tol, Mes) depending on the reaction conditions. (b) Selected areas of the 1H NMR spectra of mixtures of 1a and 2a (top), pure 1a (middle) and pure 2a (bottom). All spectra were acquired in thf-d8. | ||
In thf as solvent for the reaction, the 1H NMR spectrum of the solid residue shows the presence of two distinct species, which are identified as anions 1a and [Ge9{Si(TMS)3}2(CH2)4O–DABo-tol]−, 2a (δ = 0.24 ppm, thf-d8) in a 60:40 ratio (Fig. 1b). This ratio changes to 15:85 performing the reaction at 35 °C. At higher temperature we observe formation of [Ge9{Si(TMS)3}3]−. To selectively yield anion 2a, the bromo-borane is dissolved in thf in a first step and the bis-silylated cluster is added in a subsequent step leading to one single peak at m/z 1470.8 (Fig. S49†) in the ESI MS and the observation of one signal set in the 1H NMR spectrum.
After redissolving the residues of analogous reactions performed in deuterated thf, an ESI mass peak at m/z 1478.8 (2a-d8) is observed, indicative of the formation of the thf ring-opened species in which a –(CD2)4O– moiety is attached to the [Ge9] cluster ion (Fig. S51†). It is noteworthy that pure 1a does not react with thf under ring-opening. Using the sterically more hindered borane DABMes–Br resulted in a complete conversion of the reactants to one single species within 3 h at room temperature in thf, as shown by 1H NMR investigations. Recrystallization of the crude product from toluene at −40 °C yielded red crystals suitable for single crystal X-ray diffraction, which were identified as K[Ge9{Si(TMS)3}2(CH2)4O-DABMes], 3 (Fig. 2a and 3a).
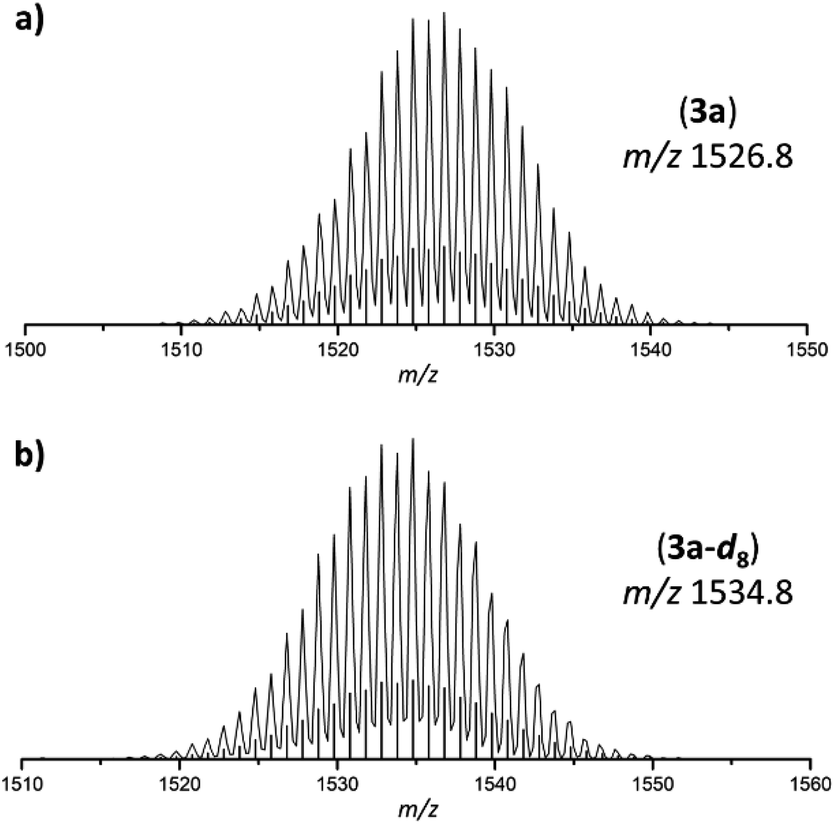 | ||
| Fig. 2 Selected areas of ESI MS spectra (thf, negative-ion mode, 3500 V, 300 °C) of 3a and 3a-d8. (a) Molecule peak of [Ge9{Si(TMS)3}2(CH2)4O–DABMes]− (3a) at m/z 1526.8; (b) molecule peak of [Ge9{Si(TMS)3}2(CD2)4O–DABMes]− (3a-d8) at m/z 1534.8. Calculated isotope patterns are shown as black bars. Overview spectra are provided in the ESI.† | ||
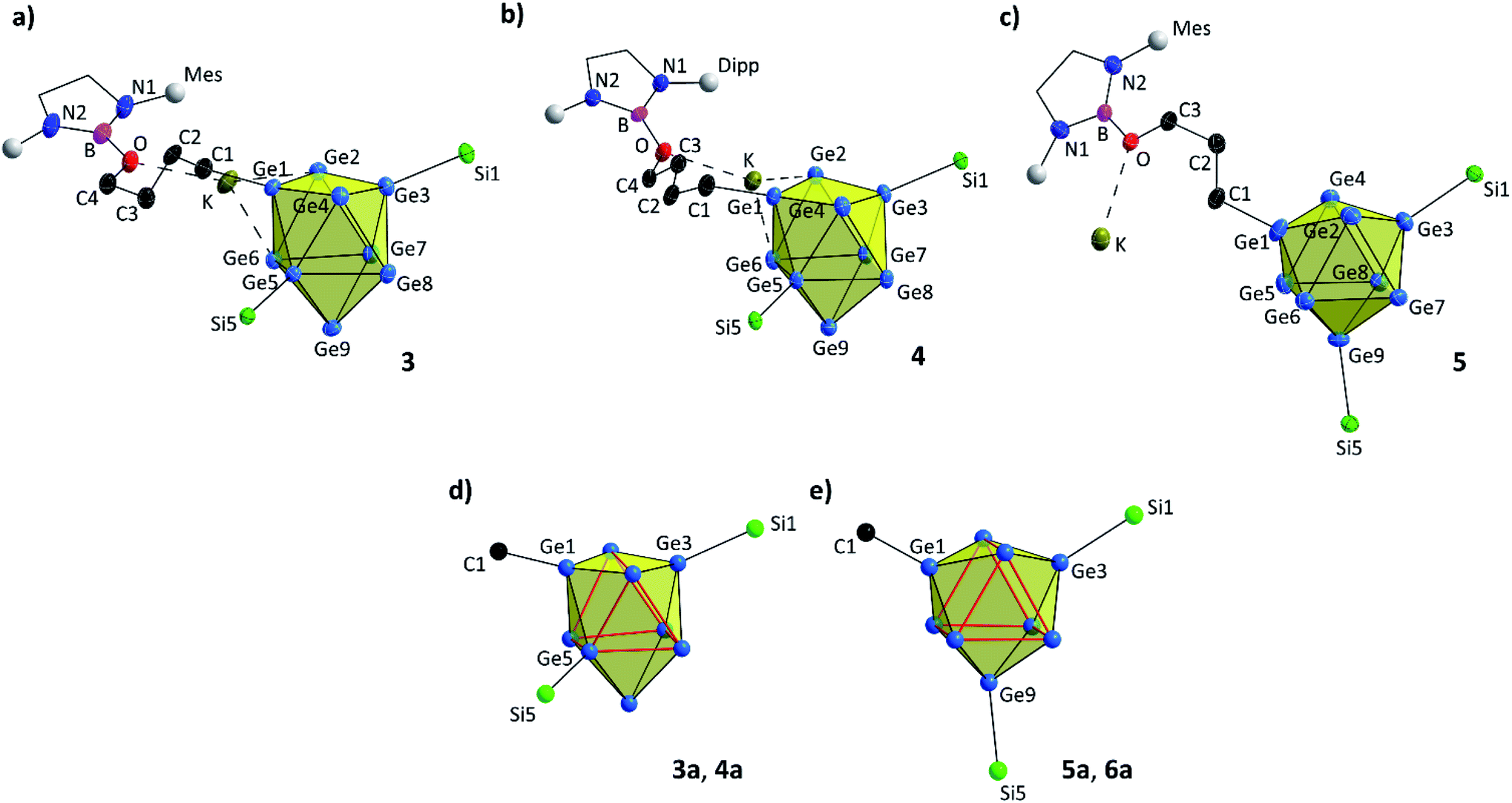 | ||
| Fig. 3 Molecular structures of compounds 3–5 with uncommon (in 3 and 4) and common (in 5) substitution patterns of the [Ge9] core. All ellipsoids are shown at a 50% probability level. For clarity protons, co-crystallized solvent molecules and TMS groups are omitted. Mes and Dipp fragments are shown as grey spheres, and carbon atoms of DAB and DAB(II) moieties are presented as black wire sticks. (a) Molecular structure of 3 with –(CH2)4O– moiety; (b) molecular structure of 4 with –(CH2)4O– moiety; (c) molecular structure of 5 with –(CH2)3O– moiety; emphasis on (d) less common and (e) common substitution isomer of the [Ge9] core in the anions 3a–6a (trigonal prisms are indicated by bold red lines). Selected bond lengths and crystallographic data of 3–5 are presented in the ESI.† | ||
The anion 3a originates from thf ring-opening and insertion between the cluster and the boranyl ligand through Ge–C and B–O bond formation, respectively. The formation is confirmed by ESI MS spectra obtained from the intermediate solid products of the reaction using thf and thf-d8 as solvents, resulting in mass peaks at m/z 1526.8 (3a; Fig. 2a) and m/z 1534.8 (3a-d8, Fig. 2b), respectively.
Similar reactions of [Ge9{Si(TMS)3}2]2− with DAB(II)Dipp–Br yielded the respective anion [Ge9{Si(TMS)3}2(CH2)4O–DAB(II)Dipp]− (4a), with observed molecule peaks at m/z 1608.8 (4a, Fig. S57†) and m/z 1616.8 (4a-d8, Fig. S59†). Full conversion of the reactants occurs upon stirring the reaction mixture for three weeks at 35 °C (at r. t. no reaction is observed at all due to the increased steric demand of the Dipp rest if compared to a Mes group).50 A more convenient access to 4a was found by heating a solution of DAB(II)Dipp–Br in thf to 70 °C for 2 days, followed by the addition of [Ge9{Si(TMS)3}2]2−. Compound 4, K[Ge9{Si(TMS)3}2(CH2)4O-DAB(II)Dipp], was obtained by recrystallization of the crude material from toluene solution at −40 °C.
Compounds 3 and 4 crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In both compounds the bis-silylated clusters carry an additional ligand that comprises a ring-opened thf molecule which is further attached to the boranyl group through the oxygen atom. The [Ge9] cluster cores are best described as distorted tricapped trigonal prisms with three different prims heights (Ge2–Ge4, Ge5–Ge6 and Ge7–Ge8) and thus CS symmetry (Fig. 3a, b and d). All three substituents (one alkyl group and two silyl groups) form classical 2-center-2-electron (2c-2e) exo-bonds to single Ge vertices of the [Ge9] cluster cores, with Ge–Ge, Ge–Si and Ge–C distances being in accordance with reported values (Table 1).24,25,51–53 The alkyl group (–(CH2)4O–) and one of the silyl groups (Si1–Si4) are attached at Ge atoms capping the trigonal prism,25,34,43,44 whereas the second silyl group (Si5–Si8) does not bind to the third capping atom Ge9, but to Ge5 instead, which is an atom of the trigonal prism base. This feature deviates from the typical substitution pattern in [Ge9] clusters presented in Fig. 3e. Isomerization with respect to the ligand arrangement has been observed before.54–56 However, NMR investigations of 3 in solution suggest a symmetrically decorated [Ge9] cage with chemically and magnetically identical silyl groups (Fig. S32†). Even upon cooling of the NMR sample to −90 °C no splitting of the main signal could be observed, nevertheless a minor species (approx. 5%) showing a signal splitting could be monitored reaching its coalescence at 70 °C. Due to the low concentration we believe the latter signal is caused by an impurity. The asymmetric substitution in 3 and 4 could result from packing effects, and thus a rearrangement of the [Ge9] cluster substituents or cluster vertex atoms might occur during crystallization.
. In both compounds the bis-silylated clusters carry an additional ligand that comprises a ring-opened thf molecule which is further attached to the boranyl group through the oxygen atom. The [Ge9] cluster cores are best described as distorted tricapped trigonal prisms with three different prims heights (Ge2–Ge4, Ge5–Ge6 and Ge7–Ge8) and thus CS symmetry (Fig. 3a, b and d). All three substituents (one alkyl group and two silyl groups) form classical 2-center-2-electron (2c-2e) exo-bonds to single Ge vertices of the [Ge9] cluster cores, with Ge–Ge, Ge–Si and Ge–C distances being in accordance with reported values (Table 1).24,25,51–53 The alkyl group (–(CH2)4O–) and one of the silyl groups (Si1–Si4) are attached at Ge atoms capping the trigonal prism,25,34,43,44 whereas the second silyl group (Si5–Si8) does not bind to the third capping atom Ge9, but to Ge5 instead, which is an atom of the trigonal prism base. This feature deviates from the typical substitution pattern in [Ge9] clusters presented in Fig. 3e. Isomerization with respect to the ligand arrangement has been observed before.54–56 However, NMR investigations of 3 in solution suggest a symmetrically decorated [Ge9] cage with chemically and magnetically identical silyl groups (Fig. S32†). Even upon cooling of the NMR sample to −90 °C no splitting of the main signal could be observed, nevertheless a minor species (approx. 5%) showing a signal splitting could be monitored reaching its coalescence at 70 °C. Due to the low concentration we believe the latter signal is caused by an impurity. The asymmetric substitution in 3 and 4 could result from packing effects, and thus a rearrangement of the [Ge9] cluster substituents or cluster vertex atoms might occur during crystallization.
| 3 | 4 | 5 | 6 | |
|---|---|---|---|---|
| h 1 | 3.976(1) | 3.918(1) | 3.429(1) | 3.370(1) |
| h 2 | 2.759(2) | 2.815(2) | 3.525(1) | 3.663(1) |
| h 3 | 2.932(1) | 2.934(1) | 3.295(1) | 3.201(1) |
| h long/hshort | 1.44 | 1.39 | 1.07 | 1.14 |
| Ge–Geshort | 2.5038(8) (Ge1–Ge2) | 2.5046(8) (Ge1–Ge2) | 2.5031(9) (Ge6–Ge9) | 2.5128(7) (Ge3–Ge7) |
| Ge–Gelong | 2.9322(8) (Ge7–Ge8) | 2.9324(8) (Ge7–Ge8) | 2.715(1) (Ge4–Ge8) | 2.7396(7) (Ge4–Ge8) |
| Ge1–C1 | 1.995(5) | 2.007(5) | 1.984(6) | 1.997(4) |
| Ge3–Si1 | 2.396(2) | 2.392(2) | 2.381(2) | 2.369(1) |
| Ge5–Si5 or Ge9–Si5 | 2.395(2) | 2.403(2) | 2.383(2) | 2.381(1) |
The charge balancing potassium cations of 3a and 4a coordinate to two Ge vertices of the [Ge9] unit and to the oxygen atom of the ring-opened ether (Fig. 3a and b). An influence of the K+ cations on the course of the reaction was ruled out, since similar experiments, in which the cations were sequestered by 18-crown-6 or [2.2.2]-cryptand gave the same products. However, the coordination could play a role concerning which possible ligand arrangement at the [Ge9] clusters is found in crystals of 3 and 4.
These species resemble intermolecular frustrated Lewis pairs (FLPs) consisting of the bis-silylated [Ge9] cluster (Lewis base) and the bromo-borane (Lewis acid). The formation of 3 and 4 reveals a new and very promising route to attach further ligands at the silylated [Ge9] cluster. Currently, the formation of [Ge9]–C bonds is limited to vinylation reactions, and a direct alkylation of the [Ge9] cluster using alkyl-halides or acyl-chlorides36,39 suffering either from low yields or limitation to neutral cluster species.38
The cleavage of endocyclic C–O bonds has been reported for boron based Lewis acids57,58 as well as for non-electron-deficient boron subphthalocyanine, proposing the formation of a four-membered ring intermediate and subsequent σ-bond metathesis.59,60 Therefore we tested the reactivity of the boranes DABR–Br towards thf in the absence of any cluster species. Indeed, NMR investigations (1H, 11B; Fig. S5 to S16†) of the residues obtained by stirring DABR–Br (R: o-tol; Mes) or DAB(II)Dipp–Br in thf at room temperature or 70 °C, respectively, revealed thf ring-opening under formation of DABR–O–(CH2)4–Br. As observed for anions 2a to 5a the 11B NMR shifts for the ring-opened species are only slightly up-field shifted if compared to the DAB-reactants, indicating a similar chemical shielding of B. Upon the addition of [Ge9{Si(TMS)3}2]2− species 2a to 4a are formed (Scheme 1), as shown by ESI MS experiments. Reactions with the less hindered DABo-xyl–Br, DABDipp–Br (saturated backbone) and acyclic (iPr2N)2BBr led to the corresponding ring-opened products (Table 2), whereas in reactions with the analogous chloro-boranes DABR–Cl no ring opening of thf was observed (with and without [Ge9{Si(TMS)3}2]2−), which we assign to the lower Lewis acidity47–49 of the chloro-borane, and Cl− being a worse leaving group than Br−.61
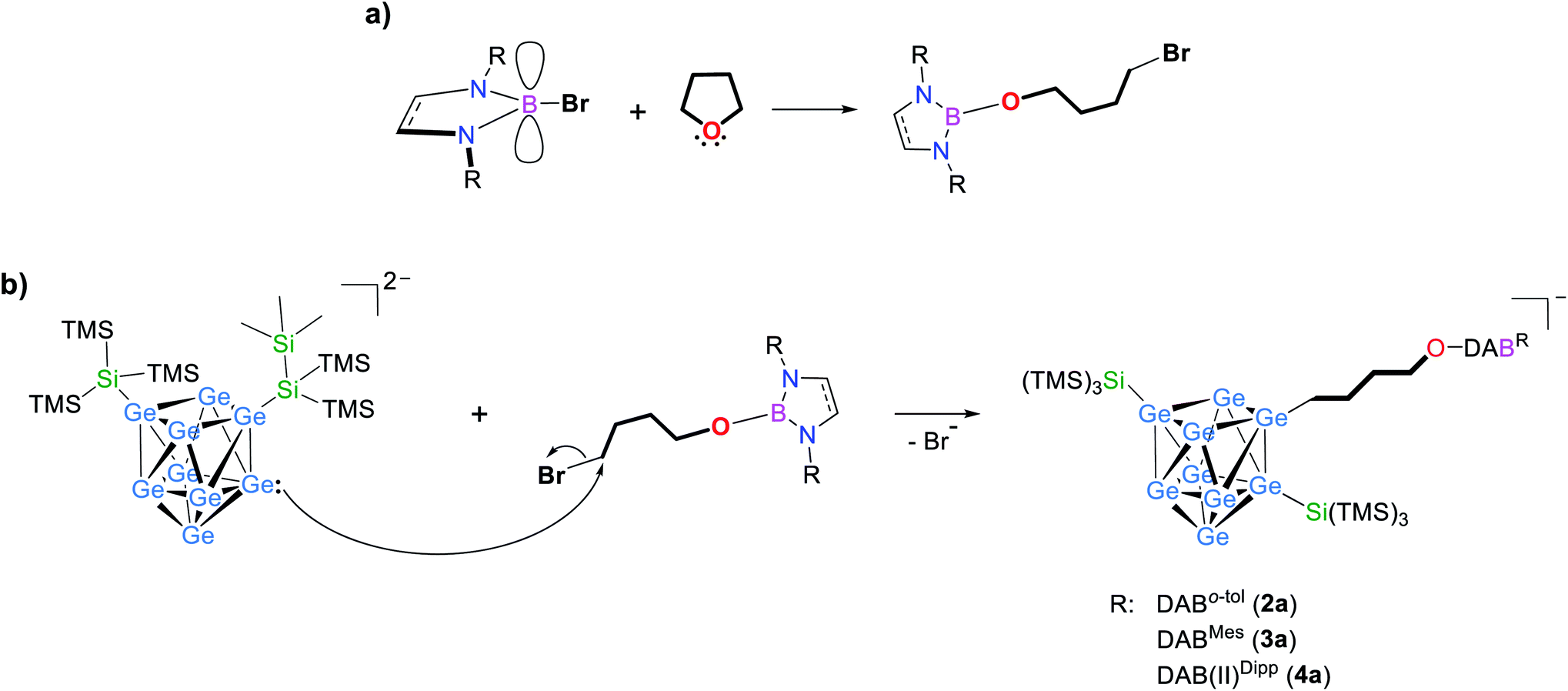 | ||
| Scheme 1 Possible formation path for compounds 2–4 through (a) reaction of DABR–Br (R: o-tol, Mes) and DAB(II)Dipp–Br with thf yielding ring-opened DABR–(CH2)4O–Br and (b) subsequent reaction with [Ge9{Si(TMS)3}2]2−. Detailed reaction parameters are provided in the ESI.† | ||
| Reactant/solvent |
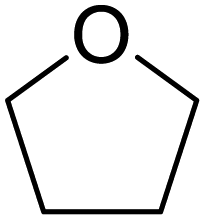
|
|
|
R: o-tol (2) | R: o-xyl |
| o-xyl | Mes (5) | |
| Mes (3) | Dipp | |
| Dipp(4) | ||
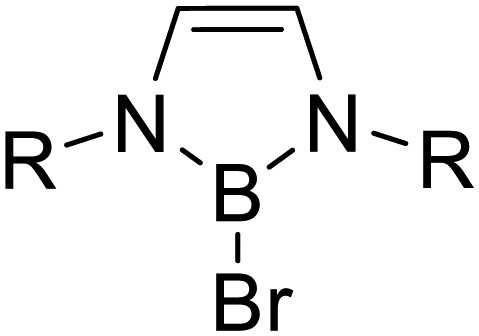
|
R: Dipp | R: Dipp |
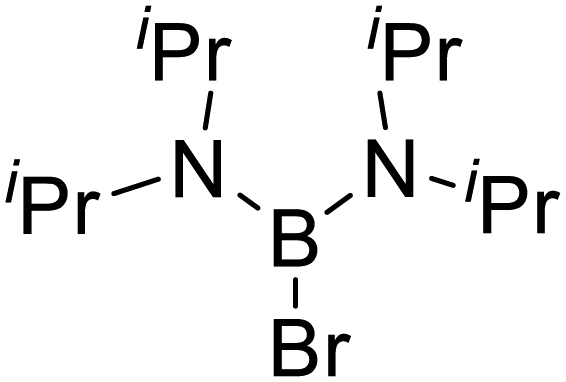
|
Insertion observed | No insertion observed |
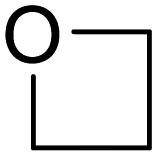
Ring-opening of trimethylene oxide
The reaction of [Ge9{Si(TMS)3}2]2− with DABMes–Br in tmo (trimethylene oxide) accordingly leads to the formation of [Ge9{Si(TMS)3}2(CH2)3O–DABMes]− (5a). Isolation of single crystals allowed the structural characterization and revealed the formation of K[Ge9{Si(TMS)3}2(CH2)3O–DABMes] (5). ESI MS data of 5a (m/z 1512.8, Fig. S61†), and NMR spectroscopic investigations corroborate the structure in solution. Compound 5 crystallizes in the triclinic space group P. The [Ge9] cluster core can be best described as a tricapped trigonal prism with three different prims heights and CS symmetry. All Ge–Ge, Ge–Si and Ge–C distances are in the range of the bond lengths observed in 3 and 4 (Table 1). In contrast to the asymmetric substitution pattern of the [Ge9] cluster in 3 and 4, the three substituents in 5 bind to the capping Ge atoms of the tricapped trigonal prismatic [Ge9] core, as typically observed for tris-substituted [Ge9] species (Fig. 3c and e).25,34,43,44 Apparently, the slightly shorter alkyl chain changes the favoured structure in the solid state, and the potassium counter ion coordinates solely to the oxygen atom of the alkyl substituents and not to Ge vertices of the [Ge9] cluster.
Reactions of [Ge9{Si(TMS)3}2]2− with further bromo-boranes in tmo resulted in the formation of analogous species, which were characterized by ESI MS only (the spectra are shown in the ESI†). However, no selective ring-opening reaction could be observed for the reaction of tmo and various boranes DABR–Br, instead the formation of various unidentified products was observed in the NMR spectra. This indicates that a targeted opening of tmo is only feasible if Lewis basic and acidic moieties are simultaneously present. The results of these experiments show that the insertion of different cyclic ethers between the di-anionic cluster and several bromo-boranes corresponds to an easy route for Ge–C bond formation in [Ge9] cluster chemistry (Table 2). Additional NMR spectra (1H, 11B, 13C, 29Si) of compounds 2 to 5 are provided in the ESI.†
Activation of CH3CN
In contrast to thf the activation of CH3CN requires the simultaneous presence of Lewis-acid and Lewis-basic reactants. A reaction of the nitrile was only observed in the presence of [Ge9{Si(TMS)3}2]2− and DABMes–Br in acetonitrile solution, resulting in the anion [Ge9{Si(TMS)3}2CH3CN–DABMes]− (6a, Fig. 4a). By contrast, stirring acetonitrile solutions of DABMes–Br at r. t. or at 70 °C overnight did not lead to any reaction, as shown by NMR spectroscopy (Fig. S19 and S20†). However, upon the addition of [Ge9{Si(TMS)3}2]2−, compound 6 is rapidly formed, as indicated by a shift of the silyl group protons from 0.16 ppm (bis-silylated [Ge9] cluster) to 0.25 ppm in the 1H NMR spectrum, a 11B shift from 26 ppm to 18 ppm indicating an increased shielding of B and ESI MS investigations (6a at m/z 1495.8, 6a-d3 at m/z 1498.8; Fig. 4b and c).
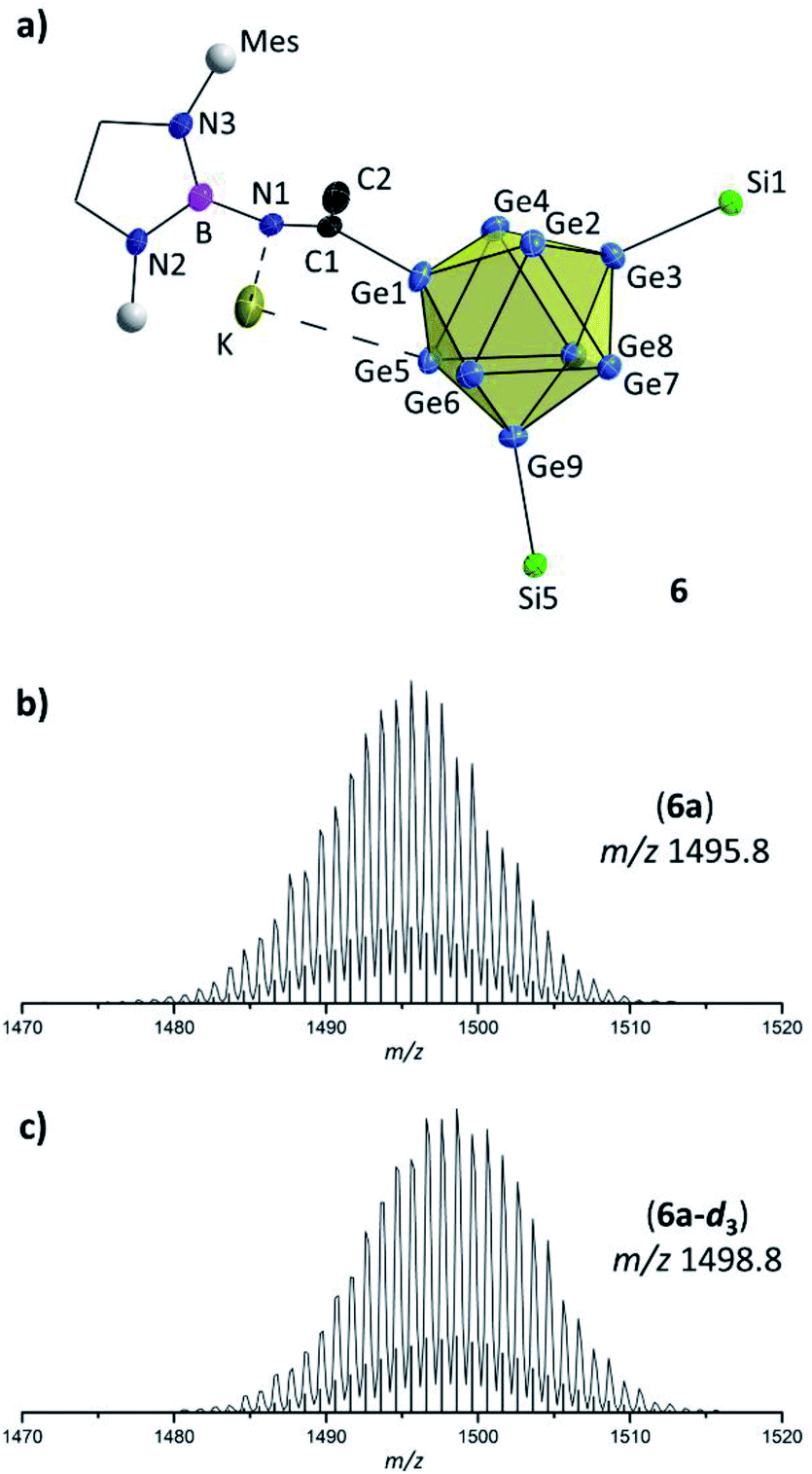 | ||
| Fig. 4 (a) Molecular structure of compound 6 with –(CH3)CN– moiety. All ellipsoids are shown at a 50% probability level. For clarity protons, co-crystallized solvent molecules and TMS groups are omitted. Mes fragments are indicated by grey spheres, and carbon atoms of the DAB moiety are presented as black wire sticks; (b) and (c) selected areas of ESI MS spectra (MeCN, negative-ion mode, 3500 V, 300 °C) of 6a and 6a-d3; (b) molecule peak of [Ge9{Si(TMS)3}2CH3CN–DABMes]− (6a) at m/z 1495.8; (c) molecule peak of [Ge9{Si(TMS)3}2CD3CN–DABMes]− (6a-d3) at m/z 1498.8. Calculated isotope patterns are shown as black bars. Overview spectra are given in the ESI.† | ||
Hence, in contrast to the reactions with the cyclic ether thf, for which a ring opening reaction is already observed in the presence of the respective bromo-borane, MeCN insertion exclusively occurs in the presence of a Lewis acid–base pair (Scheme 2). Recrystallization of the crude product from toluene at room temperature yielded orange needles suitable for single crystal X-ray diffraction of K[Ge9{Si(TMS)3}2CH3CN–DABMes] (6). Compound 6 crystallizes in the monoclinic space group P21. The [Ge9] core has the shape of a tricapped trigonal prism with one prism height significantly elongated compared to the two others, resulting in C2v symmetry (Table 1). As observed in 5, the three substituents (one imine ligand and two silyl groups) bind to the capping Ge atoms of the cluster. The short bond d(C1–N1): 1.273(6) Å and the distorted trigonal planar coordination of C1 by Ge1, N1 and C2 [angles range between 114.2° (Ge1–C1–C2) and 124.4° (N1–C1–C2)] confirm the CN double bond.12 The potassium counter ion is coordinated by the imine nitrogen atom N1 and one Ge vertex atom (Ge5) of the [Ge9] cluster core. Analogous ESI MS experiments for the boranes DABR–Br (R: o-xyl, Dipp), DAB(II)Dipp–Br and (iPr2N)2B–Br indicated insertion of the nitrile moiety between cluster and boranyl ligand only under simultaneous presence of both reactants. The results can be understood in terms of previous publications, which suggest a step-wise reaction path for the cyclo-addition of nitriles or azides to the intramolecular, geminal FLP tBu2P–CH2–BPh2.12,62 According to quantum chemical calculations the nitrile activation is initiated by the coordination of the terminal N atom of MeCN to the electrophilic boron center (intermediate), followed by the nucleophilic attack of the phosphorus' lone pair at the electrophilic C atom of the nitrile. In analogy, we assume that the [Ge9{Si(TMS)3}2]2−/DAB system forms a transition state (Scheme 2), and the reaction occurs according to a frustrated Lewis acid–base-type displacement reaction. These results are further corroborated by the observation that mixtures of [Ge9{Si(TMS)3}2]2− and the acyclic chloro-borane (iPr2N)2B–Cl readily react with MeCN under formation of [Ge9{Si(TMS)3}CH3CN–B(NiPr2)2]− (chloro-boranes are less Lewis acidic than their bromo-congeners), whereas this cluster/borane pair does not open cyclic ethers. This contrasts the findings made for analogous mixtures of [Ge9{Si(TMS)3}2]2− and cyclic DABR–Cl (R: o-tol, Mes), which do not react with MeCN. The difference in the reactivity of the respective cyclic and acyclic chloro-boranes can be explained by the significantly increased Lewis acidity of the acyclic (iPr2N)2B–Cl compared to that of the cyclic DABR–Cl (R: o-tol, Mes) system. This is caused by a decreased electron density donation to the empty B pz-type orbital due to the low barrier of the torsion about the B–N bond in the acyclic molecule.63,64 Therefore, the Lewis acidity can be estimated to follow the sequence DABR–Br > (NiPr2)2–BCl > DABR–Cl.
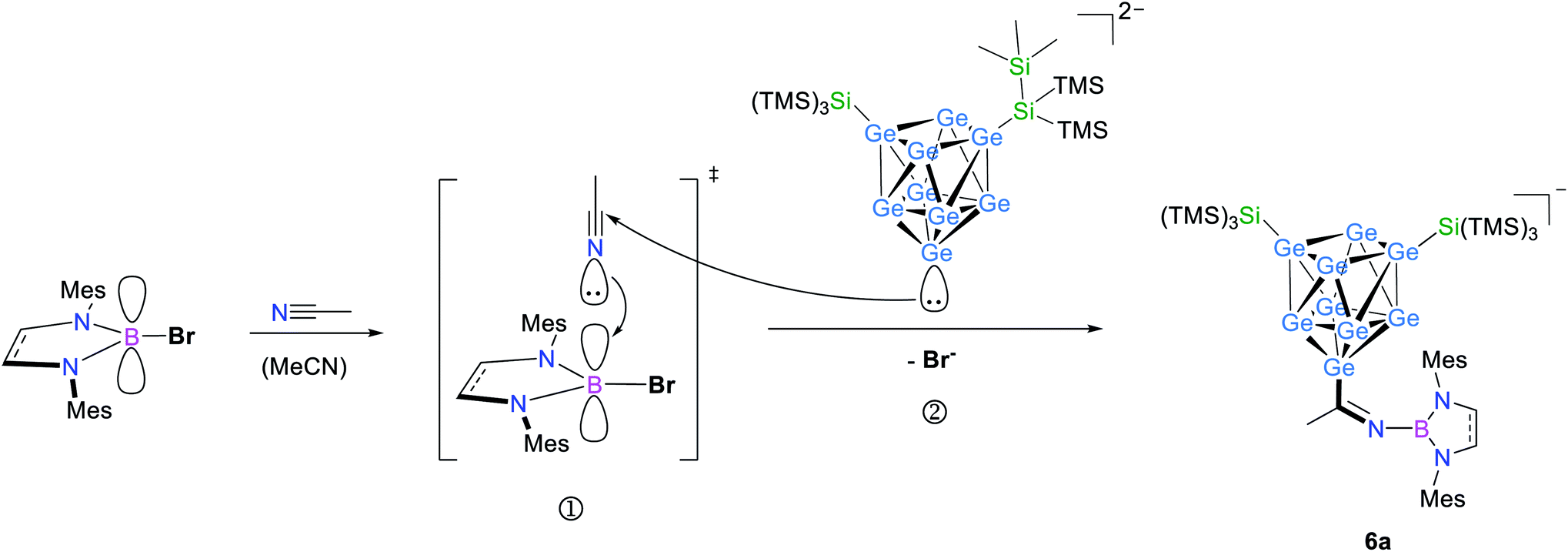 | ||
| Scheme 2 Possible reaction path in analogy to the literature for the formation of 6a through an intermediate activation of acetonitrile by the Lewis acid (➀), subsequent attack of [Ge9{Si(TMS)3}2]2− at the electrophilic C atom of the acetonitrile molecule (②) and product formation under cleavage of Br−.12,62 | ||
Conclusions
We reported on the reactivity of [Ge9{Si(TMS)3}2]2− towards several bromo boranes DABR–Br. The reactions of the bis-silylated [Ge9] cluster with DABo-tol–Br reveal the tunability of this system, yielding either selectively the anion 1a with direct Ge–B bond formation, or a participation of solvent molecules in the reaction. Anion 2a is formed via the intermediate DABo-tol–O(CH2)4–Br, which subsequently reacts with [Ge9{Si(TMS)3}2]2−. Moreover, we found that boranes with sterically demanding ligands such as DABR–Br (R: o-xyl, Mes, Dipp), DAB(II)Dipp–Br or (iPr2N)2B–Br, which cannot bind to [Ge9{Si(TMS)3}2]2−, can also give species analogous to 2a by ring opening of thf or tmo. By contrast, the insertion of CH3CN takes place in analogy to a frustrated Lewis acid–base concept, since a reaction is observed only in the presence of both reactants [Ge9{Si(TMS)3}2]2− (Lewis base) and DABR–Br (R: o-xyl, Mes, Dipp), DAB(II)Dipp–Br or (iPr2N)2B–Br (Lewis acid).The structures of compounds 3 to 6 reveal another interesting aspect of the substitution pattern in spherical homoatomic germanium clusters: compounds 3 and 4 show an unusual substitution pattern of the [Ge9] cores in the solid state (Fig. 5), which is different from that in solution, indicating a rearrangement of the substituents upon crystallization.
 | ||
| Fig. 5 Different substitution isomers of [Ge9]. Common substitution pattern with (a) two exo-bonded silyl groups at Ge atoms capping the trigonal prism;24,36,55 and (b) three ligands capping the trigonal prism.25,34,35,43,44,65 Novel substitution isomers with (c) two silyl groups55 and (d) three ligands binding to different cluster vertices (this manuscript). Trigonal prisms are indicated by red lines. NHCiPr: 1,3-bis(isopropyl)imidazolylidene; NHCMes: 1,3-bis(2,4,6-trimethylphenyl)imidazolylidene. | ||
To the best of our knowledge these systems are the first examples for intermolecular Ge/B frustrated Lewis acid–base pairs comprising nine-atom germanide clusters capable of reacting with small molecules. Currently, further investigations on the activation of small molecules by these germanide cluster/bromo-borane systems are carried out in our laboratories.
Author contributions
The experimental work was performed equally by C. W. and F. S. G. The manuscript was authored by C. W., F. S. G. and T. F. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. W. thanks the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for funding (project number 245845833) within the International Research Training Group IRTG 2022 – Alberta Technical University of Munich School for Functional Hybrid Materials (ATUMS). Support within TUM IGSSE is greatly appreciated. C. W. also thanks the Studienstiftung des Deutschen Volkes for granting a PhD scholarship. This work was financially supported by the Bavarian Ministry of Economic Affairs, Regional Development and Energy “SolarTechnologies go Hybrid”. The authors acknowledge Kevin Frankiewicz for the acquisition of variable temperature NMR data.References
- P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed.
- B. Birkmann, T. Voss, S. J. Geier, M. Ullrich, G. Kehr, G. Erker and D. W. Stephan, Organometallics, 2010, 29, 5310 CrossRef CAS.
- S. Fantasia, J. M. Welch and A. Togni, J. Org. Chem., 2010, 75, 1779 CrossRef CAS PubMed.
- T. Krachko, E. Nicolas, A. W. Ehlers, M. Nieger and J. C. Slootweg, Chem.–Eur. J., 2018, 24, 12669 CrossRef CAS PubMed.
- G. C. Welch, J. D. Masuda and D. W. Stephan, Inorg. Chem., 2006, 45, 478 CrossRef CAS PubMed.
- G. Wittig and A. Rückert, Justus Liebigs Ann. Chem., 1950, 566, 101 CrossRef CAS.
- K. Samigullin, I. Georg, M. Bolte, H. W. Lerner and M. Wagner, Chem.–Eur. J., 2016, 22, 3478 CrossRef CAS PubMed.
- L. M. Elmer, G. Kehr, C. G. Daniliuc, M. Siedow, H. Eckert, M. Tesch, A. Studer, K. Williams, T. H. Warren and G. Erker, Chem.–Eur. J., 2017, 23, 6056 CrossRef CAS PubMed.
- E. R. Habraken, L. C. Mens, M. Nieger, M. Lutz, A. W. Ehlers and J. C. Slootweg, Dalton Trans., 2017, 46, 12284 RSC.
- Z. Jian, G. Kehr, C. G. Daniliuc, B. Wibbeling and G. Erker, Dalton Trans., 2017, 46, 11715 RSC.
- L. Keweloh, N. Aders, A. Hepp, D. Pleschka, E.-U. Würthwein and W. Uhl, Dalton Trans., 2018, 47, 8402 RSC.
- J. Li, C. Mück-Lichtenfeld, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2020, 59, 12477 CrossRef CAS PubMed.
- C. Wallach, F. S. Geitner, A. J. Karttunen and T. F. Fässler, Angew. Chem., Int. Ed., 2021, 60, 2648 CrossRef CAS PubMed.
- O. P. Townrow, C. Chung, S. A. Macgregor, A. S. Weller and J. M. Goicoechea, J. Am. Chem. Soc., 2020, 142, 18330 CrossRef CAS PubMed.
- S. Ponou and T. F. Fässler, Z. Anorg. Allg. Chem., 2007, 633, 393 CrossRef CAS.
- H. Von Schnering, M. Baitinger, U. Bolle, W. Carrillo-Cabrera, J. Curda, Y. Grin, F. Heinemann, J. Llanos, K. Peters and A. Schmeding, Z. Anorg. Allg. Chem., 1997, 623, 1037 CrossRef CAS.
- W. Carrillo-Cabrera, U. Aydemir, M. Somer, A. Kircali, T. F. Fässler and S. D. Hoffmann, Z. Anorg. Allg. Chem., 2007, 633, 1575 CrossRef CAS.
- C. Downie, J.-G. Mao and A. M. Guloy, Inorg. Chem., 2001, 40, 4721 CrossRef CAS PubMed.
- M. Somer, W. Carrillo-Cabrera, E. M. Peters, K. Peters and H. G. v. Schnering, Z. Anorg. Allg. Chem., 1998, 624, 1915 CrossRef CAS.
- C. Suchentrunk, J. Daniels, M. Somer, W. Carrillo-Cabrera and N. Korber, Z. Naturforsch., B: J. Chem. Sci., 2005, 60, 277 CrossRef CAS.
- O. Kysliak and A. Schnepf, Dalton Trans., 2016, 45, 2404 RSC.
- F. Li and S. C. Sevov, Inorg. Chem., 2012, 51, 2706 CrossRef CAS PubMed.
- L. J. Schiegerl, A. J. Karttunen, W. Klein and T. F. Fässler, Chem.–Eur. J., 2018, 24, 19171 CrossRef CAS PubMed.
- L. J. Schiegerl, A. J. Karttunen, W. Klein and T. F. Fässler, Chem. Sci., 2019, 10, 9130 RSC.
- L. J. Schiegerl, A. J. Karttunen, J. Tillmann, S. Geier, G. Raudaschl-Sieber, M. Waibel and T. F. Fässler, Angew. Chem., Int. Ed., 2018, 57, 12950 CrossRef CAS PubMed.
- C. Lorenz, F. Hastreiter, J. Hioe, N. Lokesh, S. Gärtner, N. Korber and R. M. Gschwind, Angew. Chem., Int. Ed., 2018, 57, 12956 CrossRef CAS PubMed.
- T. Henneberger, W. Klein and T. F. Fässler, Z. Anorg. Allg. Chem., 2018, 644, 1018 CrossRef CAS.
- K. Mayer, L. J. Schiegerl and T. F. Fässler, Chem.–Eur. J., 2016, 22, 18794 CrossRef CAS PubMed.
- O. Kysliak, C. Schrenk and A. Schnepf, Inorg. Chem., 2015, 54, 7083 CrossRef CAS PubMed.
- L. J. Schiegerl, F. S. Geitner, C. Fischer, W. Klein and T. F. Fässler, Z. Anorg. Allg. Chem., 2016, 642, 1419 CrossRef CAS.
- O. Kysliak, T. Kunz and A. Schnepf, Eur. J. Inorg. Chem., 2017, 2017, 805 CrossRef CAS.
- K. Mayer, L. Schiegerl, T. Kratky, S. Günther and T. Fässler, Chem. Commun., 2017, 53, 11798 RSC.
- S. Frischhut, W. Klein and T. F. Fässler, C. R. Chim., 2018, 21, 932 CrossRef CAS.
- S. Frischhut and T. F. Fässler, Dalton Trans., 2018, 47, 3223 RSC.
- F. Li and S. C. Sevov, J. Am. Chem. Soc., 2014, 136, 12056 CrossRef CAS PubMed.
- S. Frischhut, W. Klein, M. Drees and T. F. Fässler, Chem.–Eur. J., 2018, 24, 9009 CrossRef CAS PubMed.
- F. Li, A. Muñoz-Castro and S. C. Sevov, Angew. Chem., Int. Ed., 2012, 51, 8581 CrossRef CAS PubMed.
- O. Kysliak and A. Schnepf, Z. Anorg. Allg. Chem., 2019, 645, 335 CrossRef CAS.
- F. S. Geitner, W. Klein and T. F. Fässler, Angew. Chem., Int. Ed., 2018, 57, 14509 CrossRef CAS PubMed.
- F. S. Geitner, J. V. Dums and T. F. Fässler, J. Am. Chem. Soc., 2017, 139, 11933 CrossRef CAS PubMed.
- F. S. Geitner, C. Wallach and T. F. Fässler, Chem.–Eur. J., 2018, 24, 4103 CrossRef CAS PubMed.
- C. Wallach, F. S. Geitner, W. Klein and T. F. Fässler, Chem.–Eur. J., 2019, 25, 12349 CrossRef CAS PubMed.
- J. Li, B. Li, R. Liu, L. Jiang, H. Zhu, H. W. Roesky, S. Dutta, D. Koley, W. Liu and Q. Ye, Chem.–Eur. J., 2016, 22, 14499 CrossRef CAS PubMed.
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629 CrossRef CAS.
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270, 75 CrossRef.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC.
- A. Schnepf, Angew. Chem., Int. Ed., 2003, 42, 2624 CrossRef CAS PubMed.
- M. W. Hull and S. C. Sevov, J. Am. Chem. Soc., 2009, 131, 9026 CrossRef CAS PubMed.
- M. W. Hull, A. Ugrinov, I. Petrov and S. C. Sevov, Inorg. Chem., 2007, 46, 2704 CrossRef CAS PubMed.
- S. Frischhut, F. Kaiser, W. Klein, M. Drees, F. E. Kühn and T. F. Fässler, Organometallics, 2018, 37, 4560 CrossRef CAS.
- F. S. Geitner and T. F. Fässler, Inorg. Chem., 2020, 59, 15218 CrossRef CAS PubMed.
- F. Li, A. Muñoz-Castro and S. C. Sevov, Angew. Chem., Int. Ed., 2016, 55, 8630 CrossRef CAS PubMed.
- H. C. Brown, P. V. Ramachandran and J. Chandrasekharan, Heteroat. Chem., 1995, 6, 117 CrossRef CAS.
- J. B. Heilmann, Y. Qin, F. Jäkle, H.-W. Lerner and M. Wagner, Inorg. Chim. Acta, 2006, 359, 4802 CrossRef CAS.
- C. Bonnier and T. P. Bender, Molecules, 2015, 20, 18237 CrossRef CAS PubMed.
- J. Guilleme, L. Martinez-Fernandez, D. Gonzalez-Rodriguez, I. Corral, M. Yanez and T. Torres, J. Am. Chem. Soc., 2014, 136, 14289 CrossRef CAS PubMed.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Hoboken, New Jersey, Vol. 8, 2020, pp. 778–779 Search PubMed.
- D. H. Boom, A. R. Jupp, M. Nieger, A. W. Ehlers and J. C. Slootweg, Chem.–Eur. J., 2019, 25, 13299 CrossRef CAS PubMed.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed., 1996, 35, 1121 CrossRef CAS.
- L. M. Slaughter, ACS Catal., 2012, 2, 1802 CrossRef CAS.
- O. Kysliak, C. Schrenk and A. Schnepf, Inorg. Chem., 2017, 56, 9693 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data of compounds 3–6; selected bond lengths of compounds 3–6; NMR spectra of 1–6 (1H, 13C, 11B, 29Si; including VT NMR spectra of 3); synthesis procedure for DABR–O–(CH2)4–Br and (iPr2N)2B–O–(CH2)4–Br together with corresponding 1H and 11B NMR spectra; details on ESI MS sample preparation and spectra. CCDC 1993875–1993878. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00811k |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |