DOI:
10.1039/D1RA02403E
(Paper)
RSC Adv., 2021,
11, 17734-17739
Pd-catalyzed synthesis of 1-(hetero)aryl-2,2,2-trichloroethanols using chloral hydrate and (hetero)arylboroxines†
Received
26th March 2021
, Accepted 11th May 2021
First published on 18th May 2021
Abstract
1-(Hetero)aryl-2,2,2-trichloroethanols are useful key intermediates for the synthesis of various bioactive compounds. Herein, we describe N-heterocyclic carbene (NHC)-coordinated cyclometallated palladium complex (CYP)-catalyzed (hetero)aryl addition of chloral hydrate using (hetero)arylboroxines, providing a new approach to 1-(hetero)aryl-2,2,2-trichloroethanols. Notably, PhS-IPent-CYP which coordinated the bulky yet flexible 2,6-di(pentan-3-yl)aniline (IPent)-based NHC showed good catalytic activities and promoted the transformation in 24–97% yields.
Introduction
1-(Hetero)aryl-2,2,2-trichloroethanols are one of the most useful building blocks for the synthesis of bioactive compounds,1 because the carbinol moiety is easily transformed to various α-substituted carboxylic acid derivatives.2–6 So far, 1-(hetero)aryl-2,2,2-trichloroethanols have two kinds of possible synthetic routes as depicted in Scheme 1. One is an addition of the trichloromethyl anion to carbonyl compounds such as aldehydes or ketones (i).7 This way has generally needed the use of toxic trichloromethyl anion sources such as chloroform and trichloroacetic acid. The other is an addition of moisture-sensitive organometallic reagents such as organomagnesium compounds to dehydrated chloral (ii).8
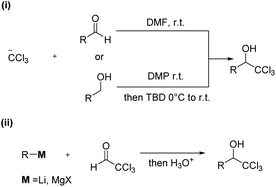 |
| | Scheme 1 Previous synthesis of trichloromethylcarbinols. | |
The transition metal-catalyzed 1,2-addition of organoboronic acids and their derivatives to carbonyl compounds is a convenient method compared to the Grignard reaction, because this could be conducted in the presence of water.9 Although several research groups have reported the Rh-catalyzed 1,2-addition of arylboronic acids to trifluoromethyl ketones,10 the transition metal-catalyzed addition of arylboron compounds to trichloromethyl carbonyl compounds such as chloral have not been examined yet. It is well known that N-heterocyclic carbenes (NHC) coordinated palladium complexes are useful for various applications such as anticancer drugs, OLEDs and catalysts.11 Recently, we have developed the NHC coordinated cyclometallated palladium complexes (CYPs) that catalyzed the 1,2-addition of organoboron compounds to a wide range of carbonyl compounds including hemiacetals such as aqueous formaldehyde and glyoxylate hemiacetals (Scheme 2).12 Therefore, we envisaged that the NHC-CYPs exhibit a good catalytic activity of the addition of arylboron compounds to chloral hydrate without a dehydration process. Here, we report the direct aryl addition to chloral hydrate with triarylboroxines using NHC-CYPs as a catalyst.
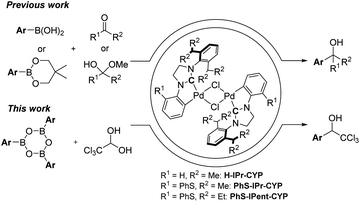 |
| | Scheme 2 NHC-CYPs-catalyzed 1,2-addition of arylboron compounds and carbonyl compounds. | |
Results and discussion
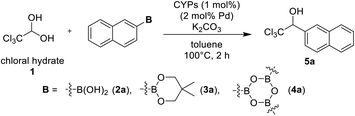
At first, we examined CYPs-catalyzed 1,2-addition of chloral hydrate 1 and 2-naphthylboron compounds (Table 1). PhS-IPr-CYP have catalyzed the addition of arylboronic acids to an excess amount of aqueous formaldehyde to provide the corresponding benzylic alcohols in satisfactory yields,12a,d although PhS-IPr-CYP catalyzed reaction of 2-naphthaleneboronic acid 2a and 4 equivalent of chloral hydrate afforded the desired product 5a in 39% yield (entry 1). In this case, the yield of 5a was improved by the use of an excess of 2a relative to chloral hydrate (entry 2). Then, using 2-naphthaleneboronate 3a instead of 2a increased slightly the yield of 3a to 70% (entry 3). We have confirmed the efficacy of arylboroxines in the arylation of trifluoroacetaldehyde hemiacetal from a preliminary investigation.12c When this reaction was performed using tri(naphthalene-2-yl)boroxine 4a, the yield of 5a was improved considerably to 82% (entry 4). Dehydrated chloral was usable as well as chloral hydrate for this addition reaction (entry 5). H-IPr-CYP has shown more catalytic activity than PhS-IPr-CYP in the CYPs-catalyzed arylation of glyoxylate hemiacetals,12b but it was not suitable for this reaction (entry 6). PhS-IPent-CYP having sterically bulky alkyl group had more active towards the addition than PhS-IPr-CYP (entry 7).
Table 1 Optimization of reaction conditions of CYPs-catalyzed 1,2-addition of chloral hydrate 1 and 2-naphtalenelboron compounds
|
| Entry |
1 (mmol) |
B (mmol) |
K2CO3 (mmol) |
CYPs |
Yielda (%) |
| Yields were determined by 1H-NMR using triphenylmethane as an internal standard. 3a was used instead of 2a. 4a was used instead of 2a. Dehydrated chloral was used instead of chloral hydrate. Isolated yield. |
| 1 |
2.0 |
0.5 |
0.5 |
PhS-IPr-CYP |
39 |
| 2 |
0.5 |
1.5 |
1.5 |
PhS-IPr-CYP |
66 |
| 3b |
0.5 |
1.5 |
1.5 |
PhS-IPr-CYP |
70 |
| 4c |
0.5 |
0.5 |
1.5 |
PhS-IPr-CYP |
82 |
| 5c,d |
0.5 |
0.5 |
1.5 |
PhS-IPr-CYP |
81 |
| 6c |
0.5 |
0.5 |
1.5 |
H-IPr-CYP |
73 |
| 7c |
0.5 |
0.5 |
1.5 |
PhS-IPent-CYP |
95 (95)e |
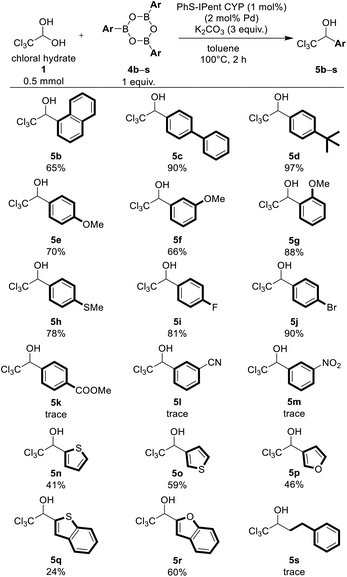
Under the optimized conditions, we synthesized various functionalized trichloromethyl carbinols using PhS-IPent-CYP catalyzed reaction (Table 2). Substrates bearing sterically hindered 1-naphthyl group was also converted to the corresponding alcohol 5b in moderate yield of 65%. Arylboroxines bearing electron-donating groups like tert-butyl, phenyl, methoxy and methylthio groups furnished the corresponding products 5c–5h in satisfactory yields of 66–97%. Interestingly, sterically bulky 2-methoxyphenylboroxine reacted more smoothly than 3-methoxyphenyl and 4-methoxyphenylboroxines. 4-Fluorophenyl and 4-bromophenylboroxines provided the corresponding products 5i and 5j in excellent yields, but the reaction using arylboroxines having strong electron-withdrawing groups such as nitrile, nitro or methoxycarbonyl group have not afforded the products 5k–5m. Remarkably, the bromo group on the aromatic ring remained intact, and the Suzuki–Miyaura cross-coupling product did not observe under this reaction condition. This catalytic reaction was also applicable to heteroarylboroxines containing oxygen or sulfur atom and provided the products 5n–5r in low to moderate yields, but was not applicable to an aliphatic boroxine such as 2-phenylethylboroxine 4s.
Table 2 PhS-IPent-CYP-catalyzed 1,2-addition of arylboroxines 4 to chloral hydrate 1a
| Reaction conditions: 1 (1 equiv., 0.5 mmol), 4 (1.0 equiv., 0.5 mmol), K2CO3 (3.0 equiv., 1.5 mmol), PhS-Ipent-CYP (0.005 mmol, 1 mol%) and toluene (1 mL) at 100 °C for 2 h in a sealed tube. Isolated yield. |
 |
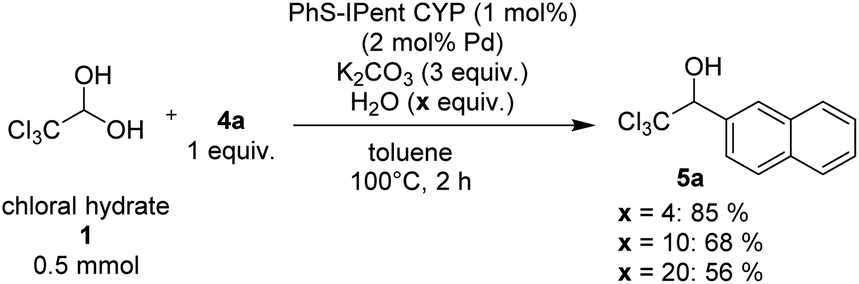
Since arylboroxines are more suitable for this reaction than arylboronic acids, we examined an experiment under the reaction conditions with H2O (Scheme 3). Because arylboroxines is known to rapidly absorb H2O and transform to boronic acids, and adding water is expected to reduce the dehydration performance of boroxine. Practically, the yield declined as the amount of H2O added increased, indicating that arylboroxines may be involved in the dehydration step of chloral hydrate. So, we proposed a plausible catalytic cycle which is described in Scheme 4. Initially, dehydrated chloral and arylboronic acids are generated from the hydrolysis of arylboroxines by chloral hydrate. Then arylpalladium intermediate 6 is formed from a base-promoted transmetallation between an arylboronic acid and PhS-IPent-CYP, alkoxypalladium 7 is generated from an insertion of the aryl group on 6 to chloral. Finally, a transmetallation of complex 7 between an arylboronic acid results in the formation of 1-(hetero)aryl-2,2,2-trichloroethanol and the regeneration of complex 6.
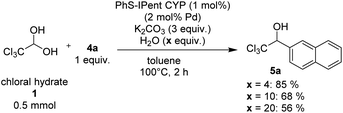 |
| | Scheme 3 The effect of the used amount of H2O on the catalytic addition reaction. | |
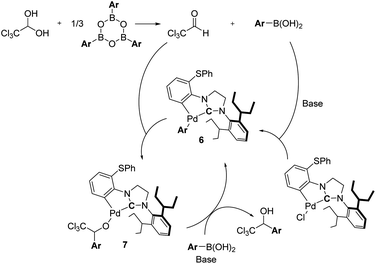 |
| | Scheme 4 Proposed reaction mechanism. | |
Conclusions
We have achieved a nucleophilic arylation to chloral hydrate using PhS-IPent-CYP as a catalyst. The use of arylboroxine is critical for this reaction, and arylboroxines have acted not only as an arylcarbanion source but also as a dehydrating agent for chloral hydrate.
Experimental
General
All reactions were carried out under an argon atmosphere. 1H, 13C, and 19F NMR spectra were recorded on an AVANCE III 400 spectrometer (400.15 MHz) at ambient temperature. Melting points were recorded on Yanako MP-S3. HRMS were recorded on a Thermo Fisher Scientific Exactive (Orbitrap) using ESI or APCI. Commercially available organic and inorganic compounds were used without purification. PhS-IPr-CYP,12a H-IPr-CYP,12a PhS-IPent-CYP12d and arylboroxines 413 were prepared according to the literature procedures.
Preparation and characterizations of compounds
2,2,2-Trichloro-1-(naphthalen-2-yl)ethan-1-ol7b 5a. Chloral hydrate (83 mg, 0.50 mmol), 2-naphtyl boroxine (231 mg, 0.500 mmol), PhS-IPent-CYP (6.1 mg, 0.0050 mmol) and potassium carbonate (207 mg, 1.50 mmol) were charged in 10 mL test tube sealed with a rubber septum. The test tube was evacuated and backfilled with argon. This sequence was repeated three times. Then dehydrated toluene (1 mL) was added via the rubber septum with syringe. In an argon flow, the rubber septum was replaced with a Teflon liner screw cap. The sealed test tube was placed into an oil bath preheated 100 °C. After the reaction was stirred for 2 h and cooled to room temperature, the obtained crude was purified by passing it though a silica gel column with a hexane/ethyl acetate to give 131 mg (0.475 mmol, 95%) of product 5a as a pale yellow solid, mp 93–94 °C (lit.7b 93–94 °C). 1H NMR (400 MHz, CDCl3, ppm): δ 8.09 (s, 1H, ArH), 7.85–7.90 (m, 3H, ArH), 7.88 (dd, J1 = 7.3 Hz, J2 = 9.8 Hz, 1H, ArH), 7.52 (t, J = 4.1 Hz, 2H, ArH), 5.40 (d, J = 3.4 Hz, 1H, CH(OH)CCl3), 3.39 (d, J = 3.4 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 133.8 (Ar), 132.5 (Ar), 132.3 (Ar), 129.3 (Ar), 128.4 (Ar), 127.7 (Ar), 127.4 (Ar), 126.9 (Ar), 126.4 (Ar), 126.2 (Ar), 103.3 (CCl3), 84.7 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C12H9OCl4: 308.9413. Found: 308.9424.
2,2,2-Trichloro-1-(naphthalen-1-yl)ethan-1-ol3d 5b. Product 5b was prepared by utilizing the general procedure using 1-naphtyl boroxine (231 mg, 0.500 mmol) and was isolated as a pale yellow liquid (91 mg, 0.33 mmol, 66%). 1H NMR (400 MHz, CDCl3, ppm): δ 8.28 (d, J = 8.5 Hz, 1H, ArH), 8.09 (d, J = 8.5 Hz, 1H, ArH), 7.91–7.97 (m, 2H, ArH), 7.51–7.62 (m, 3H, ArH), 6.23 (d, J = 4.2 Hz, 1H, CH(OH)CCl3), 3.45 (d, J = 4.2 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 133.5 (Ar), 132.0 (Ar), 131.4 (Ar), 130.3 (Ar), 129.0 (Ar), 127.2 (Ar), 126.4 (Ar), 125.6 (Ar), 124.9 (Ar), 123.7 (Ar), 103.5 (CCl3), 79.0 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C12H9OCl4: 308.9413. Found: 308.9422.
1-([1,1′-Biphenyl]-4-yl)-2,2,2-trichloroethan-ol 5c. Product 5c was prepared by utilizing the general procedure using 4-biphenyl boroxine (270 mg, 0.500 mmol) and was isolated as a pale yellow solid (136 mg, 0.451 mmol, 90%), mp 119–120 °C. 1H NMR (400 MHz, CDCl3, ppm): δ 7.70 (s, 1H, Ar), 7.67 (s, 1H, Ar), 7.59–7.62 (m, 4H, Ar), 7.45 (t, J = 7.5 Hz, 2H, Ar), 7.36 (t, J = 7.5 Hz, 1H, Ar), 5.26 (s, 1H, CH(OH)CCl3), 3.32 (s, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 142.4 (Ar), 140.4 (Ar), 133.8 (Ar), 129.7 (Ar), 128.9 (Ar), 127.7 (Ar), 127.2 (Ar), 126.6 (Ar), 103.2 (CCl3), 84.4 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C14H11OCl4: 334.9569. Found: 334.9583.
1-(4-(tert-Buthyl)phenyl)-2,2,2-trichloroethan-1-ol7a 5d. Product 5d was prepared by utilizing the general procedure using 4-tert-buthylpheny boroxine (240 mg, 0.500 mmol) and was isolated as a pale yellow solid (95 mg, 0.34 mmol, 67%), mp 77–78 °C. 1H NMR (400 MHz, CDCl3, ppm): δ 7.52 (d, J = 8.4 Hz, 2H, ArH), 7.39 (d, J = 8.4 Hz, 2H, ArH), 5.16 (d, J = 4.1 Hz, 1H, CH(OH)CCl3), 3.33 (d, J = 4.1 Hz, 1H, OH) 1.32 (s, 9H, C(CH3)3); 13C NMR (100 MHz, CDCl3, ppm): δ 152.6 (Ar), 132.0 (Ar), 128.9 (Ar), 124.8 (Ar), 103.3 (CCl3), 84.4 (CH(OH)CCl3), 34.7 (C(CH3)3), 31.3 (C(CH3)3); HRMS (EI) m/z: [M + Cl]− calcd for C12H15OCl4: 314.9882. Found: 314.9894.
2,2,2-Trichloro-1-(4-methoxyphenyl)ethan-1-ol7b 5e. Product 5e was prepared by utilizing the general procedure using 4-methoxyphenyl boroxine (201 mg, 0.500 mmol) and was isolated as a pale yellow liquid (90 mg, 0.35 mmol, 70%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.52 (d, J = 8.7 Hz, 2H, ArH), 6.90 (d, J = 8.7 Hz, 2H, ArH), 5.15 (d, J = 2.3 Hz, 1H, CH(OH)CCl3), 3.81 (s, 3H, OCH3), 3.35 (d, J = 2.3 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 160.4 (Ar), 130.4 (Ar), 127.0 (Ar), 113.2 (Ar), 103.5 (CCl3), 84.2 (CH(OH)CCl3), 55.3 (OCH3); HRMS (EI) m/z: [M + Cl]− calcd for C9H9O2Cl4: 288.9362. Found: 288.9374.
2,2,2-Trichloro-1-(3-methoxyphenyl)ethan-1-ol 5f. Product 5f was prepared by utilizing the general procedure using 3-methoxyphenyl boroxine (201 mg, 0.500 mmol) and was isolated as a colourless liquid (85 mg, 0.33 mmol, 66%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.28 (dd, J1 = 8.0 Hz, J2 = 8.0 Hz, 1H, ArH), 7.16–7.17 (m, 2H, ArH), 6.94 (d, J = 8.0 Hz, 1H, ArH), 5.16 (d, J = 4 Hz, 1H, CH(OH)CCl3), 3.80 (s, 3H, OCH3), 3.43 (d, J = 12 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 159.0 (Ar), 136.4 (Ar), 128.8 (Ar), 121.8 (Ar), 115.0 (Ar), 114.9 (Ar), 103.0 (CCl3), 84.4 (CH(OH)CCl3), 55.3 (OCH3); HRMS (EI) m/z: [M + Cl]− calcd for C9H9O2Cl4: 288.9362. Found: 288.9372.
2,2,2-Trichloro-1-(2-methoxyphenyl)ethan-1-ol 5g. Product 5g was prepared by utilizing the general procedure using 2-methoxyphenyl boroxine (201 mg, 0.500 mmol) and was isolated as a colourless liquid (113 mg, 0.44 mmol, 88%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.60 (dd, J1 = 1.7 Hz, J2 = 7.7 Hz, 1H, ArH), 7.35 (m, 1H, ArH), 6.91–7.01 (m, 1H, ArH), 5.59 (d, J = 6.9 Hz, 1H, CH(OH)CCl3), 4.25 (d, J = 6.9 Hz, 1H, OH), 3.84 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3, ppm): δ 157.7 (Ar), 130.6 (Ar), 130.4 (Ar), 123.5 (Ar), 120.5 (Ar), 111.2 (Ar), 103.5 (CCl3), 80.3 (CH(OH)CCl3), 55.6 (OCH3); HRMS (EI) m/z: [M + Cl]− calcd for C9H9O2Cl4: 288.9362. Found: 288.9371.
2,2,2-Trichloro-1-(4-(methylthio)phenyl)ethan-1-ol 5h. Product 5h was prepared by utilizing the general procedure using 4-(methylthio)phenyl boroxine (225 mg, 0.500 mmol) and was isolated as a pale yellow solid (107 mg, 0.394 mmol, 78%), mp 89.5–90.0 °C. 1H NMR (400 MHz, CDCl3, ppm): δ 7.50 (d, J = 8.4 Hz, 2H, ArH), 7.23 (d, J = 8.4 Hz, 2H, ArH), 5.14 (d, J = 3.0 Hz, 1H, CH(OH)CCl3), 3.46 (d, J = 3.0 Hz, 1H, OH), 2.48 (s, 3H, SCH3); 13C NMR (100 MHz, CDCl3, ppm): δ 140.4 (Ar), 131.4 (Ar), 129.6 (Ar), 125.3 (Ar), 103.1 (CCl3), 84.2 (CH(OH)CCl3), 15.2 (SCH3); HRMS (EI) m/z: [M + Cl]− calcd for C9H9OCl4S: 304.9134. Found: 304.9146.
2,2,2-Trichloro-1-(4-fluorophenyl)ethan-1-ol1c 5i. Product 5i was prepared by utilizing the general procedure using 4-fluorophenyl boroxine (183 mg, 0.500 mmol) and was isolated as a pale yellow liquid (99 mg, 0.41 mmol, 81%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.58–7.61 (m, 2H, ArH), 7.07 (t, J = 8.7 Hz, 2H, ArH), 5.20 (d, J = 3.1 Hz, 1H, CH(OH)CCl3), 3.40 (d, J = 3.1 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 163.4 (d, 1JC–F = 247 Hz, Ar), 131.1 (d, 2JC–F = 8.6 Hz, Ar), 130.6 (d, 3JC–F = 3.0 Hz, Ar), 114.9 (d, 4JC–F = 21.4 Hz, Ar), 103.1 (d, 5JC–F = 2.4 Hz, CCl3), 83.8 (CH(OH)CCl3); 19F (377 MHz, CDCl3, ppm): δ −117.8(s, 1F, ArF); HRMS (EI) m/z: [M + Cl]− calcd for C8H6OCl4F: 276.9162. Found: 276.9170.
1-(4-Bromophenyl)-2,2,2-trichloroethan-1-ol7c 5j. Product 5j was prepared by utilizing the general procedure using 4-bromophenyl boroxine (274 mg, 0.500 mmol) and was isolated as a pale yellow liquid (138 mg, 0.45 mmol, 90%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.49–7.56 (m, 4H, ArH), 5.18 (s, 1H, CH(OH)CCl3), 3.59 (s, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 133.8 (Ar), 131.1 (Ar), 130.9 (Ar), 123.9 (Ar), 102.7 (CCl3), 83.9 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C8H6OBrCl4: 336.8362. Found: 336.8374.
2,2,2-Trichloro-1-(thiophen-2-yl)ethan-1-ol7b 5n. Product 5n was prepared by utilizing the general procedure using 2-thiophene boroxine (165 mg, 0.500 mmol) and was isolated as a pale yellow liquid (47 mg, 0.20 mmol, 41%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.40 (dd, J1 = 1.2 Hz, J2 = 5.1 Hz, 1H, ArH), 7.31 (m, 1H, ArH), 7.04 (dd, J1 = 3.6 Hz, J2 = 5.1 Hz, 1H, ArH), 5.48 (d, J = 4.4 Hz, 1H, CH(OH)CCl3), 3.40 (d, J = 4.4 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 137.3 (Ar), 129.2 (Ar), 127.1 (Ar), 126.3 (Ar), 102.5 (CCl3), 81.6 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C6H5OCl4S: 264.8821. Found: 264.8833.
2,2,2-Trichloro-1-(thiophen-3-yl)ethan-1-ol14 5o. Product 5o was prepared by utilizing the general procedure using 3-thiophene boroxine (165 mg, 0.500 mmol) and was isolated as a pale yellow liquid (68 mg, 0.29 mmol, 59%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.55 (t, J = 2.4 Hz, 1H, ArH), 7.32 (m, 2H, ArH), 5.32 (d, J = 4.4 Hz, 1H, CH(OH)CCl3), 3.29 (d, J = 4.4 Hz, 1H, CCl3); 13C NMR (100 MHz, CDCl3, ppm): δ 136.0 (Ar), 127.6 (Ar), 126.2 (Ar), 125.2 (Ar), 102.8 (CCl3), 81.3 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C6H5OCl4S: 264.8821. Found: 264.8831.
2,2,2-Trichloro-1-(furan-3-yl)ethan-1-ol 5p. Product 5p was prepared by utilizing the general procedure using 3-furan boroxine (141 mg, 0.500 mmol) and was isolated as a pale yellow liquid (50 mg, 0.23 mmol, 46%). 1H NMR (400 MHz, CDCl3, ppm): δ 7.66 (t, J = 0.7 Hz, 1H, ArH), 7.45 (t, J = 1.7 Hz, 1H, ArH), 6.66 (dd, J1 = 0.7 Hz, J2 = 1.7 Hz, 1H, ArH), 5.21 (d, J = 4.7 Hz, 1H, CH(OH)CCl3), 3.28 (d, J = 4.7 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 142.8 (Ar), 142.6 (Ar), 120.8 (Ar), 110.0 (Ar), 102.8 (CCl3), 79.0 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C6H5O2Cl4: 248.9049. Found: 248.9056.
1-(Benzo[b]thiophene-2-yl)-2,2,2-trichloroethan-1-ol8a 5q. Product 5q was prepared by utilizing the general procedure using 2-benzo[b]thiophene boroxine (240 mg, 0.500 mmol) and was isolated as a pale yellow solid (34 mg, 0.12 mmol, 24%), mp 109–110 °C(lit.8a 109–110 °C). 1H NMR (400 MHz, CDCl3, ppm): δ 7.82–7.89 (m, 2H, ArH), 7.59 (s, 1H, ArH), 7.39–7.41 (m, 2H, ArH), 5.57 (d, J = 4.4 Hz, 1H, CH(OH)CCl3), 3.48 (d, J = 4.4 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 140.1 (Ar), 138.4 (Ar), 138.0 (Ar), 126.2 (Ar), 125.2 (Ar), 124.5 (Ar), 124.1 (Ar), 122.3 (Ar), 102.1 (CCl3), 82.0 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd for C10H7OCl4S: 314.8977. Found: 314.8992.
1-(Benzofuran-2-yl)-2,2,2-trichloroethan-1-ol 5r. Product 5r was prepared by utilizing the general procedure using 2-benzofuran boroxine (207 mg, 0.500 mmol) and was isolated as a pale yellow solid (80 mg, 0.30 mmol, 60%), mp 71–72 °C. 1H NMR (400 MHz, CDCl3, ppm): δ 7.60 (m, 1H, ArH), 7.51 (m, 1H, ArH), 7.24–7.36 (m, 2H, ArH), 6.98 (s, 1H, ArH), 5.35 (d, J = 7.2 Hz, 1H, CH(OH)CCl3), 3.59 (d, J = 7.2 Hz, 1H, OH); 13C NMR (100 MHz, CDCl3, ppm): δ 154.7 (Ar), 150.7 (Ar), 127.4 (Ar), 125.3 (Ar), 123.3 (Ar), 121.6 (Ar), 111.6 (Ar), 107.9 (Ar), 100.9 (CCl3), 79.7 (CH(OH)CCl3); HRMS (EI) m/z: [M + Cl]− calcd. for C10H7O2Cl4: 298.9206. Found: 298.9216.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Prof. Mino, Chiba University, for the HRMS measurements. This work was supported in part by the Research Institute for Science and Technology, Tokyo Denki University Grant Number Q19E-06 and Q18E-04.
Notes and references
-
(a) I. Hwang, J. Kim, H. Kim and S. Kyung, Bull. Korean Chem. Soc., 2009, 30, 1475 CrossRef CAS;
(b) A. Schäfer, A. Wellner, M. Strauss, A. Schäfer, G. Wolber and R. Gust, J. Med. Chem., 2012, 55, 9607 CrossRef PubMed;
(c) X. Zhu, C. T. Hu, J. Yang, L. A. Joyce, M. Qiu, M. D. Ward and B. Kahr, J. Am. Chem. Soc., 2019, 141, 16858 CrossRef CAS PubMed.
-
(a) E. J. Corey and J. O. Link, J. Am. Chem. Soc., 1992, 114, 1906 CrossRef CAS;
(b) R. O. Duthaler, Tetrahedron, 1994, 50, 1539 CrossRef CAS;
(c) C. Mellin-Morliére, D. J. Aitken, S. D. Bull, S. G. Davis and H. Husson, Tetrahedron: Asymmetry, 2001, 12, 149 CrossRef;
(d) R. L. Tennyson, G. S. Cortez, H. J. Galicia, C. R. Kreiman, C. M. Thompson and D. Romo, Org. Lett., 2002, 4, 533 CrossRef CAS PubMed;
(e) A. Scaffidi, B. W. Skelton, R. V. Stick and A. H. White, Aust. J. Chem., 2004, 57, 723 CrossRef CAS;
(f) M. S. Perryman, M. E. Harris, J. L. Foster, A. Joshi, G. J. Clarkson and D. J. Fox, Chem. Commun., 2013, 49, 10022 RSC;
(g) C. Hobson, M. S. Perryman, G. Kirby, G. J. Clarkson and D. J. Fox, Tetrahedron Lett., 2018, 59, 3965 CrossRef CAS.
-
(a) E. J. Corey and J. O. Link, Tetrahedron Lett., 1992, 33, 3431 CrossRef;
(b) K. Funabiki, N. Honma, W. Hashimoto and M. Matsui, Org. Lett., 2003, 5, 2059 CrossRef CAS PubMed;
(c) H. Yu, Y. Fang, Y. Xia and J. Wu, Synth. Commun., 2006, 36, 2421 CrossRef CAS;
(d) S. Federico, T. Khan, N. Relitti, G. Chemi, M. Brindisi, S. Brogi, E. Novellino, D. M. Zisterer, G. Campiani, S. Gemma and S. Butini, Tetrahedron Lett., 2018, 59, 4466 CrossRef CAS.
-
(a) D. A. Dudley, A. M. Bunker, L. Chi, W. L. Cody, D. R. Holland, D. P. Ignasiak, N. Janiczek-Dolphin, T. B. McClanahan, T. E. Mertz, L. S. Narasimhan, S. T. Rapundalo, J. A. Trautschold, C. A. V. Huis and J. J. Edmunds, J. Med. Chem., 2000, 43, 4063 CrossRef CAS PubMed;
(b) J. Blanchet and J. Zhu, Tetrahedron Lett., 2004, 45, 4449 CrossRef CAS;
(c) M. S. Perryman, M. W. M. Earl, S. Greatorex, G. J. Clarkson and D. J. Fox, Org. Biomol. Chem., 2015, 13, 2360 RSC.
-
(a) J. L. Shamshina and T. S. Snowden, Org. Lett., 2006, 8, 5881 CrossRef CAS PubMed;
(b) A. Scaffidi, B. W. Skelton, R. V. Stick and A. H. White, Aust. J. Chem., 2006, 59, 426 CrossRef CAS.
- T. S. Snowden, ARKIVOC, 2012, 24 Search PubMed , (ii), .
-
(a) E. Y. Ko, C. H. Lim and K. Chung, Bull. Korean Chem. Soc., 2006, 27, 432 CrossRef CAS;
(b) M. K. Gupta, Z. Li and T. S. Snowden, J. Org. Chem., 2012, 77, 4854 CrossRef CAS PubMed;
(c) A. B. Jensen and A. T. Lindhardt, J. Org. Chem., 2014, 79, 1174 CrossRef CAS PubMed;
(d) M. K. Gupta, Z. Li and T. S. Snowden, Org. Lett., 2014, 16, 1602 CrossRef CAS PubMed;
(e) R. N. Ram and V. K. Soni, J. Org. Chem., 2015, 80, 8922 CrossRef CAS PubMed.
-
(a) R. P. Gajewski, J. L. Jackson, N. D. Jones, J. K. Swartzendruber and J. B. Deeter, J. Org. Chem., 1989, 54, 3311 CrossRef CAS;
(b) S. Dohi, K. Moriyama and H. Togo, Eur. J. Org. Chem., 2013, 34, 7815 CrossRef.
-
(a) D. V. Partyka, Chem. Rev., 2011, 111, 1529 CrossRef CAS PubMed;
(b) C. S. Marques and A. J. Burke, Catalytic Arylation Methods, Wiley-VCH, 2014, p. 329 Search PubMed.
-
(a) S. L. X. Martina, B. C. J. Richard, J. G. de Vries, F. L. Feringa and A. J. Minnard, Chem. Commun., 2006, 39, 4093 RSC;
(b) J. R. White, G. J. Price, P. K. Plucinsky and C. G. Frost, Tetrahedron Lett., 2009, 50, 7365 CrossRef CAS;
(c) V. R. Jumde, S. Facchetti and A. Iuliano, Tetrahedron: Asymmetry, 2010, 21, 2775 CrossRef CAS;
(d) R. Luo, K. Li, Y. Hu and W. Tang, Adv. Synth. Catal., 2013, 355, 1297 CrossRef CAS;
(e) V. Valdivia, I. Fernández and N. Khiar, Org. Biomol. Chem., 2014, 12, 1211 RSC;
(f) L. S. Dobson and G. Pattison, Chem. Commun., 2016, 52, 11116 RSC;
(g) L. G. Borrego, R. Recio, M. Alcarranza, N. Khiar and I. Fernández, Adv. Synth. Catal., 2018, 360, 1273 CrossRef CAS.
- For selected recent examples:
(a) M. Sreenivasulu, K. S. Kumar, P. R. Kumar, K. B. Chandrasekhar and M. Pal, Org. Biomol. Chem., 2012, 10, 1670 RSC;
(b) A. F. Henwood, M. Lesieur, A. K. Bansal, V. Lemaur, D. Beljonne, D. G. Thompson, D. Graham, A. M. Z. Slawin, I. D. W. Samuel, C. S. J. Cazin and E. Zysman-Colman, Chem. Sci., 2015, 6, 3248 RSC;
(c) T. T. H. Fong, C. N. Lok, C. Y. S. Chung, Y. M. E. Fung, P. K. Chow, P. K. Wan and C. M. Che, Angew. Chem., Int. Ed., 2016, 55, 11935 CrossRef CAS PubMed;
(d) F. Schroeter, J. Soellner and T. Strassner, Organometallics, 2018, 37, 4267 CrossRef CAS;
(e) S. Y. Hussaini, R. A. Haque, T. Fatima, M. T. Agha, A. M. S. A. Majid and M. R. Razali, J. Coord. Chem., 2018, 71, 2787 CrossRef CAS;
(f) Q. Deng, Q. Zheng, B. Zuo and T. Tu, Green Synth. Catal., 2020, 1, 75 CrossRef.
-
(a) T. Yamamoto, A. Zhumagazin, T. Furusawa, R. Tanaka, T. Yamakawa, Y. Oe and T. Ohta, Adv. Synth. Catal., 2014, 356, 3525 CrossRef CAS;
(b) M. Sugaya, T. Yamamoto and H. Shinozaki, Tetrahedron Lett., 2017, 58, 2495 CrossRef CAS;
(c) M. S. T. Yagihashi, T. Yamamoto and H. Shinozaki, The 8th Tokyo Conference on Advanced Catalytic Science and Technology, Japan, August 6, 2018, p. P1200 Search PubMed;
(d) R. Akiyama, M. Sugaya, H. Shinozaki and T. Yamamoto, Synth. Commun., 2019, 49, 1193 CrossRef CAS;
(e) Y. Okuda, M. Nagaoka and T. Yamamoto, ChemCatChem, 2020, 12, 6291 CrossRef CAS.
- Q. Xiao, L. Tian, R. Tan, Y. Xia, D. Qiu, Y. Zhang and J. Wang, Org. Lett., 2012, 14, 4230 CrossRef CAS PubMed.
- A. I. Ayi, R. Condom, T. N. Wade and R. Guedj, J. Fluorine Chem., 1979, 14, 437 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02403e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc