
A self-healing and recyclable polyurethane-urea Diels–Alder adduct synthesized from carbon dioxide and furfuryl amine†
Peixuan
Wu
abc,
Haiyang
Cheng
 *ac,
Xinchao
Wang
abc,
Ruhui
Shi
abc,
Chao
Zhang
ac,
Masahiko
Arai
ac and
Fengyu
Zhao
*abc
*ac,
Xinchao
Wang
abc,
Ruhui
Shi
abc,
Chao
Zhang
ac,
Masahiko
Arai
ac and
Fengyu
Zhao
*abc
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, No. 5625 Renmin Rd., Changchun, 130022, China. E-mail: zhaofy@ciac.ac.cn; hycyl@ciac.ac.cn
bUniversity of Science and Technology of China, No. 96 Jinzhai Rd., Hefei, 230026, China
cJilin Province Key Laboratory of Green Chemistry and Process, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, No. 5625 Renmin Rd., Changchun, 130022, China
First published on 28th November 2020
Abstract
The fabrication of self-healing and recyclable CO2-based polymeric materials meets the requirement of sustainable development. By introducing reversible hydrogen bonds and heat-responsive Diels–Alder bonds, a new, environmentally friendly, self-healing and recyclable polyurethane-urea Diels–Alder adduct (PUUa-DA) was successfully synthesized using CO2 and furfuryl amine as renewable feedstocks. PUUa-DA was semi-crystalline and exhibited high thermal stability, 5% weight loss temperature being above 220 °C. Moreover, PUUa-DA with a cross-linked structure showed a tensile stress of 18.5 MPa, a modulus of 151 MPa and an elongation at break of 136%. The dynamic Diels–Alder bonds and hydrogen bonds provide the self-healing response and contribute to 94% recovery of the tensile strength upon heating. Therefore, PUUa-DA will have potential applications as a functional material in a wide range of practical fields. The synthesis of PUUa-DA using CO2 and bio-based furfuryl amine will be of importance in green and sustainable chemistry.
Introduction
The progress achieved in polymer materials has benefited human life in recent decades. The development of self-healing and recyclable materials is of significance and a major challenge in polymer science.1 Thus, much effort has been devoted so far and some progress has been made in preparing self-healing and recyclable polymeric materials such as polyolefins, polyesters, polyamides, and polyurethanes.2,3 It is a prerequisite to easily build, break and rebuild dynamic covalent bonds and/or noncovalent supramolecular interactions in the polymeric structure for obtaining the desired self-healing and recycling properties. Dynamic covalent bonds such as disulfide,4,5 B–O bonds,6,7 imine bonds,8,9 and Diels–Alder bonds10–14 and noncovalent supramolecular interactions such as hydrogen bonding,15,16 metal–ligand interaction,17,18 ionic interaction,19,20 and π–π stacking interactions21,22 have been frequently used in the design of self-healing and recyclable polymers.Polyurethanes and polyureas are among the most important polymeric materials that have versatile properties, so they have been used in huge volumes in many industrial and commodity products. The traditional synthesis of polyurethanes and polyureas usually invokes isocyanate chemistry, which uses toxic multifunctional isocyanates. Therefore, the use of CO2 as a nontoxic and renewable feedstock for the synthesis of polymers has attracted growing attention from both academic and industrial communities.23 In recent years, various CO2-based polyurethanes and polyureas have been synthesized through direct and indirect routes, in which the former uses CO2 as a monomer and the latter uses CO2-derived building blocks.23 CO2 is reacted with appropriate reagents and catalysts to produce polyureas.24–36 CO2-derived monomers such as cyclic carbonate,37–40 carbamate,41,42 and urea43–45 are used to synthesize polyurethanes and polyureas. To date, a few types of self-healing and recyclable CO2-based polymeric materials have been reported.10,46–48 In 2016, Caillol and coworkers developed thermo-responsive CO2-based polyurethane by converting furfuryl glycidyl ether into a cyclic carbonate derivative and mixing it with dimaleimide-terminated polypropylene glycol and an appropriate diamine. The resulting polyurethane can return from a solid state to a liquid state due to the breaking of Diels–Alder adducts, which is desired for many thermo-reversible material applications.49 In 2019, Wu and coworkers reported CO2-based healable thermoplastic elastomers via a tandem synthetic strategy, and they exhibit characteristic properties of thermoplastic elastomers with a Young's modulus of up to 10.1 MPa. Moreover, a robust self-healing ability could be achieved via the addition of a small amount of boronic ester as a dynamic cross-linker.46 In our recent work, a self-healing CO2-based polyurea was fabricated via reasonable regulation of the strength, arrangement and density of hydrogen bonds. The synthesized CO2-based polyurea is self-healable at room temperature and 95% of the superior mechanical properties of tensile strength (6.6 MPa) and toughness (28.7 MJ m−3) can be recovered.47 In the present work, we have further attempted to design a self-healing and recyclable CO2-based polymer with high strength properties by introducing stronger cross-linking bonds via the Diels–Alder reaction, in which stronger dynamic covalent bonds would provide superior mechanical strength and stability. The Diels–Alder reaction is one of the most promising reactions in self-healing polymer materials as reported in the literature.13,50–54 Sun and coworkers have reported recyclable polyurethane networks with an excellent mechanical performance. The reprocessed specimens via hot-pressing and solution casting show no decrease in the strength, stiffness and toughness.55 Cheng and coworkers have synthesized recyclable, shape-memory, and self-healing soy oil-based polyurethane networks by the thermoreversible Diels–Alder reaction between 1,5-bis(maleimido)-2-methylpentane and furfuryl alcohol.11
Herein, the authors report the synthesis of a self-healing and recyclable CO2-based polyurethane-urea Diels–Alder adduct from CO2 and furfuryl amine. Compared to the previously reported CO2 route,56,57 this new route is greener and more eco-friendly, because no isocyanates are used, but CO2 is used as a comonomer with a bio-based furfuryl amine. Linear polyurethane-urea was prepared by the polyaddition reaction between the furfuryl amine cyclocarbonate derivative and oligourea. Then polyurethane-urea was cross-linked by the Diels–Alder reaction between furfuryl and 4,4-bismaleimidodiphenylmethane (BIM). The mechanical properties (modulus, tensile stress and elongation at break) of the polyurethane-urea Diels–Alder adduct are excellent due to the combination of ordered and disordered hydrogen bonding interactions and the appropriate amounts of crosslinked Diels–Alder groups. Meanwhile, the polyurethane-urea Diels–Alder adduct showed excellent self-healing capability and recyclability. The present green route is effective at building strong and tough self-healing and recyclable polymers.
Experimental
Materials
Furfuryl amine (FA, 98%), 4,7,10-trioxa-1,13-tridecanediamine (TTD, 98%), epichlorohydrin (ECH, 99%), tetrabutylammonium bromide (TBAB, 99%), lithium bromide (LiBr, 99%), and 4,4-bismaleimidodiphenylmethane (BIM, 96%) were purchased from Tokyo Chem. Ind. and used as received. Poly(propylene glycol) bis(2-aminopropyl ether) (PEA, number-average molecular weight (Mn) 1542 g mol−1 based on matrix-assisted laser desorption and ionization time-of-flight mass spectra), N,N-dimethylformamide (DMF, 99.5%), chloroform-d (99.8%), sodium hydroxide (96%), and magnesium sulfate (99.5%) were purchased from Aladdin and used as received. Carbon dioxide (99.999%) and nitrogen (99.999%) were purchased from Changchun Juyang Gas Company.Synthesis of oligourea (OUa)
4,7,10-Trioxa-1,13-tridecanediamine (40 g, 0.182 mol) was introduced into a 50 mL autoclave and it was sealed and purged with N2 three times to remove air. Then, the autoclave was heated to 180 °C and CO2 was introduced up to 11.5 MPa. The reaction was run at a stirring speed of 400 rpm for 6 h. After the reaction was finished, the autoclave was cooled to ambient temperature and CO2 was vented slowly. The product was dried in a N2 flow at 140 °C for 3 h and then under vacuum at 80 °C for 24 h (38.5 g, 86% yield). The Mn of oligourea (OUa) was 1550 g mol−1 based on the 1H NMR data.58Synthesis of polyurea (PUa)
Oligourea (25 g, 0.0162 mol) was introduced into a 250 mL three-necked flask with a mechanical stirrer. At a constant CO2 flow rate (50 mL min−1) and at a constant stirring speed (170 rpm), the reaction was carried out at 200 °C for 1 h. Then the temperature was increased to 250 °C, and the reaction was conducted at this temperature for 1.16 h. After the reaction, the product was cooled to room temperature, collected, and stored (20.5 g, 82% yield). The Mn of polyurea (PUa) was 4952 g mol−1 based on the 1H NMR data.36Synthesis of polyurethane-urea (PUUa)
Oligourea (31 g, 20 mmol), bis(cyclic carbonate) furfuryl amine (5.95 g, 20 mmol) and DMF (30 mL) were introduced into a 100 mL round-bottom flask. Then the reaction mixture was heated to 150 °C and stirred for 2 h. After the reaction, DMF was removed by distillation and the product of polyurethane-urea (PUUa) was collected and dried under vacuum at 60 °C for 24 h (36.5 g, 98% yield).Synthesis of cross-linked polyurethane-urea (PUUa-DA)
Polyurethane-urea (18.5 g, 10 mmol) and 4,4-bismaleimidodiphenylmethane (1.79 g, 5 mmol) were dissolved in 50 mL DMF. The resulting solution was poured into a Teflon mold and dried in air at 60 °C for 24 h. The polyurethane-urea Diels–Alder adduct (PUUa-DA) was obtained by further drying under vacuum at 60 °C for 24 h (19.3 g, 95% yield).Synthesis of cross-linked polyurethane-urea from TTD and PEA
4,7,10-Trioxa-1,13-tridecanediamine (6.6 g, 30 mmol), bis(cyclic carbonate) furfuryl amine (8.91 g, 30 mmol) and DMF (30 mL) were introduced into a 100 mL round-bottom flask. Then the reaction mixture was heated to 150 °C and stirred for 2 h. After the reaction, DMF was removed by distillation and the product was collected and dried under vacuum at 60 °C for 24 h. Then, the product (12.18 g, 23.5 mmol) and 4,4-bismaleimidodiphenylmethane (4.22 g, 11.7 mmol) were dissolved in 50 mL DMF. The resulting solution was poured into a Teflon mold and dried in air and vacuum at 60 °C for 24 h each. The product was denoted as TTD-DA. Similarly, PEA-DA was prepared from PEA by a similar process.Characterization
1H NMR (400 MHz) analysis was conducted on a Bruker AV-400 at room temperature with CDCl3 as a solvent and tetramethylsilane as an internal reference.Fourier-transform infrared (FTIR) spectra were obtained using a Thermo Scientific Nicolet iS50 spectrometer with a DTGS detector in the region of 4000–400 cm−1. Temperature-dependent Fourier-transform infrared (TD-FTIR) spectra were obtained on a Bruker INVENIO spectrometer equipped with a diamond ATR crystal in the range between 4000 and 600 cm−1. The sample was heated from 30 to 120 °C at 3 °C min−1 and FTIR spectra were collected at the desired temperatures.
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed on TA-Q50 and TA-Q100 instruments, respectively. Samples (about 5–10 mg) were heated from 50 to 500 °C for TGA and from −50 to 150 °C for DSC at a heating rate of 10 °C min−1 under a nitrogen atmosphere.
A Leica DM2500P polarizing microscope was used to examine the change of a polymer sample during self-healing. The sample was prepared by casting a polymer onto a glass slide with a thickness of 0.9 mm. It was scratched with a razor, and then heated from 30 to 120 °C at a heating rate of 5 °C min−1.
Mechanical tensile measurements were performed on an Instron 1121 electronic universal testing machine according to a standard method (GB/T 1040 – 2006 Type 5A). Dumbbell shaped specimens were prepared by the melt-press method (the size of the test section was 20 mm × 4 mm × 0.9 mm). The samples were measured at a strain rate of 20 mm min−1. Young's modulus was determined from the initial slope of the stress–strain curves. Toughness is defined as the area surrounded by the tensile stress (σ)–strain (ε) curve and calculated using the following eqn (1):
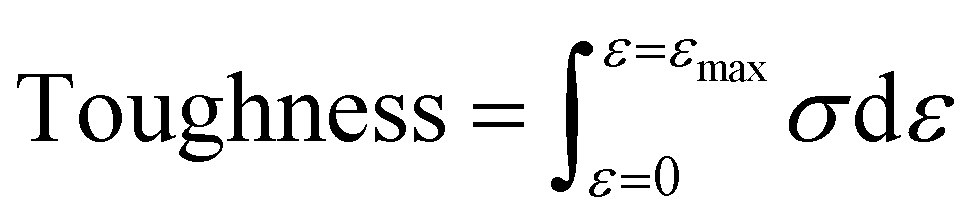 | (1) |
Abbreviations of chemicals, products and polymeric materials are used in the following and these are listed and explained in Table S1.†
Results and discussion
Synthesis and characterization of polyurethane-urea (PUUa)
PUUa was prepared from bis(cyclic carbonate) furfuryl amine (BCCFA) and oligourea (OUa) by a polyaddition reaction, as illustrated in Scheme 1. Either BCCFA or OUa was synthesized with CO2. Bio-based furfurylamine was first converted into the corresponding diglycidyl furfurylamine (DGFA) by reaction with epichlorohydrin, and then DGFA was converted into the corresponding difunctional cyclic carbonate of BCCFA by reacting with CO2. The 1H NMR spectra of DGFA and BCCFA are in accordance with the ones given in the literature (Fig. S1†).49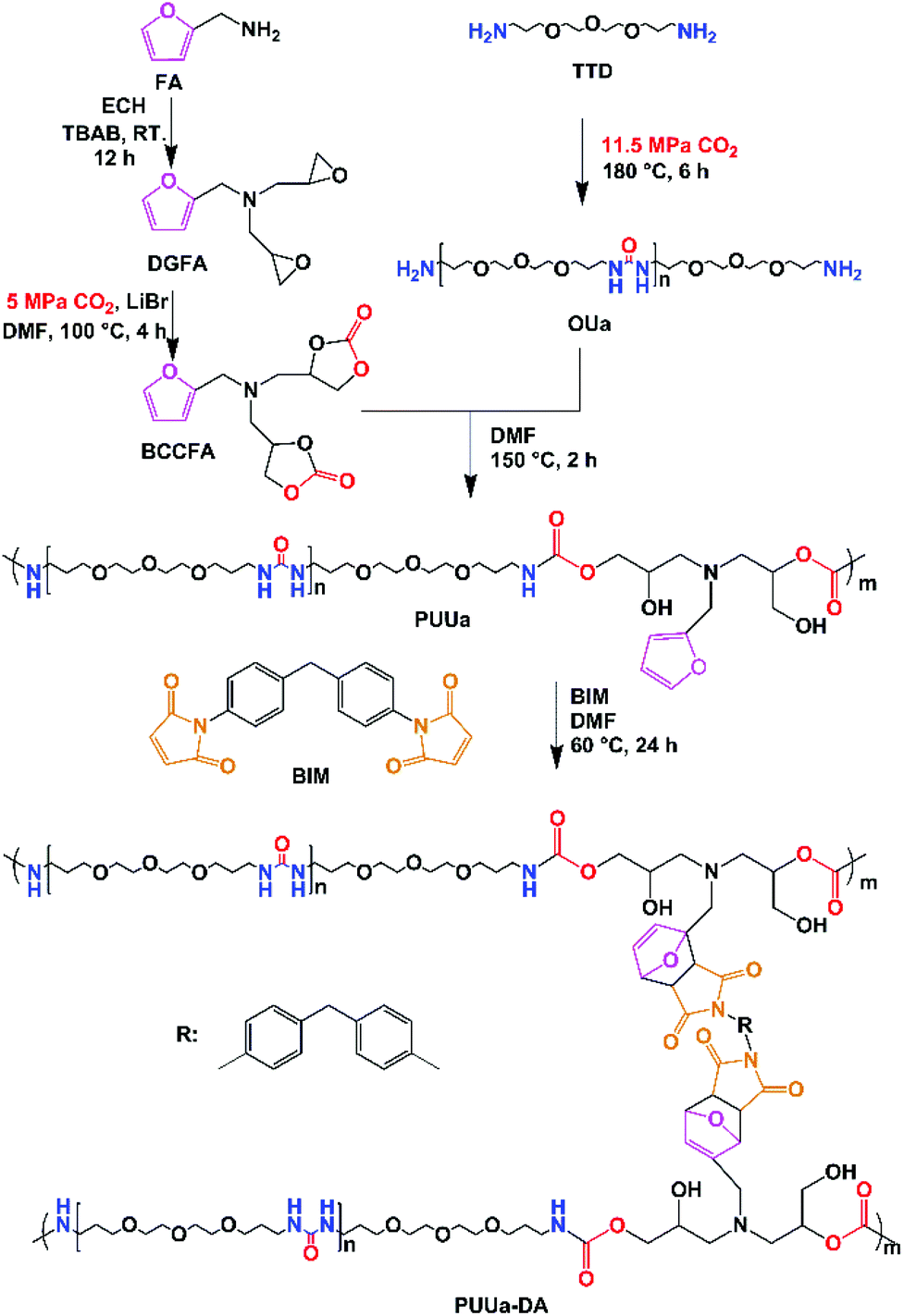 | ||
| Scheme 1 Synthesis of non-isocyanate polyurethane-urea (PUUa) from CO2, and sequential transformation of PUUa with 4,4-bismaleimidodiphenylmethane to the polyurethane-urea Diels–Alder adduct (PUUa-DA). | ||
OUa was synthesized from 4,7,10-trioxa-1,13-tridecanediamine (TTD) and CO2 by the polycondensation reaction.59 The structure of OUa prepared was confirmed by FTIR and 1H NMR. As shown in Fig. 1a, the formation of urea linkages was confirmed by the absorption bands of the –C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)– stretching vibration at 1612 cm−1 and –NH bending vibration at 1590 cm−1, in addition to the absorption band of the –NH stretching vibration at 3321 cm−1 (Fig. 1a).601H NMR showed that a new signal at 3.2 ppm appeared in OUa, as compared with TTD, which was the characteristic signal of –CH2– protons linked to the urea group (Fig. S2†). Moreover, the molecular weight of OUa was calculated by 1H NMR to be about 1550 g mol−1.58 To compare the mechanical properties, polyurea (PUa) was prepared by the further reaction of OUa and CO2 at a high temperature of 250 °C to get a reference polymer with a high molecular weight (4952 g mol−1) (Scheme S1†).36 The FTIR spectrum of PUa was similar to that of OUa; the absorption bands of the CO stretching vibration at 1610 cm−1 and the N–H bending vibration at 1587 cm−1 were ascribed to the strong and ordered hydrogen bonded urea groups of PUa (Fig. S3†).
O)– stretching vibration at 1612 cm−1 and –NH bending vibration at 1590 cm−1, in addition to the absorption band of the –NH stretching vibration at 3321 cm−1 (Fig. 1a).601H NMR showed that a new signal at 3.2 ppm appeared in OUa, as compared with TTD, which was the characteristic signal of –CH2– protons linked to the urea group (Fig. S2†). Moreover, the molecular weight of OUa was calculated by 1H NMR to be about 1550 g mol−1.58 To compare the mechanical properties, polyurea (PUa) was prepared by the further reaction of OUa and CO2 at a high temperature of 250 °C to get a reference polymer with a high molecular weight (4952 g mol−1) (Scheme S1†).36 The FTIR spectrum of PUa was similar to that of OUa; the absorption bands of the CO stretching vibration at 1610 cm−1 and the N–H bending vibration at 1587 cm−1 were ascribed to the strong and ordered hydrogen bonded urea groups of PUa (Fig. S3†).
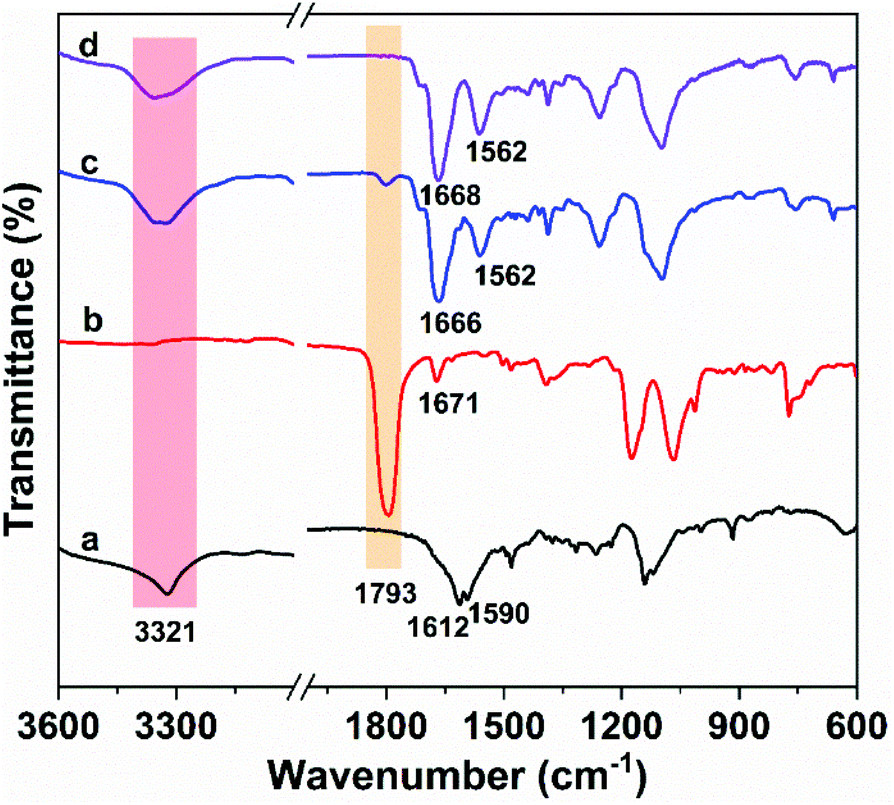 | ||
| Fig. 1 FTIR spectra of (a) oligourea (OUa), (b) bis(cyclic carbonate) furfuryl amine (BCCFA), and the reaction mixture of BCCFA and OUa reacted at 150 °C in (c) 1 h, and (d) 2 h. | ||
OUa cannot be pressed into the shape of a bar suitable to measure the mechanical properties, but its amino groups at both ends are reactive and OUa can dissolve in DMF to react with BCCFA. PUa could not dissolve in DMF.36 Therefore, OUa was used to react with BCCFA to synthesize PUUa. The reaction was detected by FTIR (Fig. 1). A strong absorption band at 1793 cm−1 ascribed to the CO stretching vibration in the cyclic carbonate groups of BCCFA weakened in 1 h and disappeared in 2 h, meaning that the cyclic carbonate groups fully reacted with the –NH2 groups of OUa in 2 h. Meanwhile, two new absorption bands appeared at 1668 and 1562 cm−1, which were assigned to the CO stretching vibration and N–H bending vibration in the urethane groups formed. The CO stretching vibration and the N–H bending vibration of the urea groups in the OUa segment were overlapped by the CO stretching vibration and the N–H bending vibration of the urethane groups in PUUa. In addition, 1H NMR was also used to characterize the chemical structure of PUUa (Fig. 2). The appearance of signals at 3.3 and 4.1 ppm was associated with the formation of urethane and a peak at 3.7–4.0 ppm indicated the hydroxyl group formed by the cyclic carbonate/amine reaction.
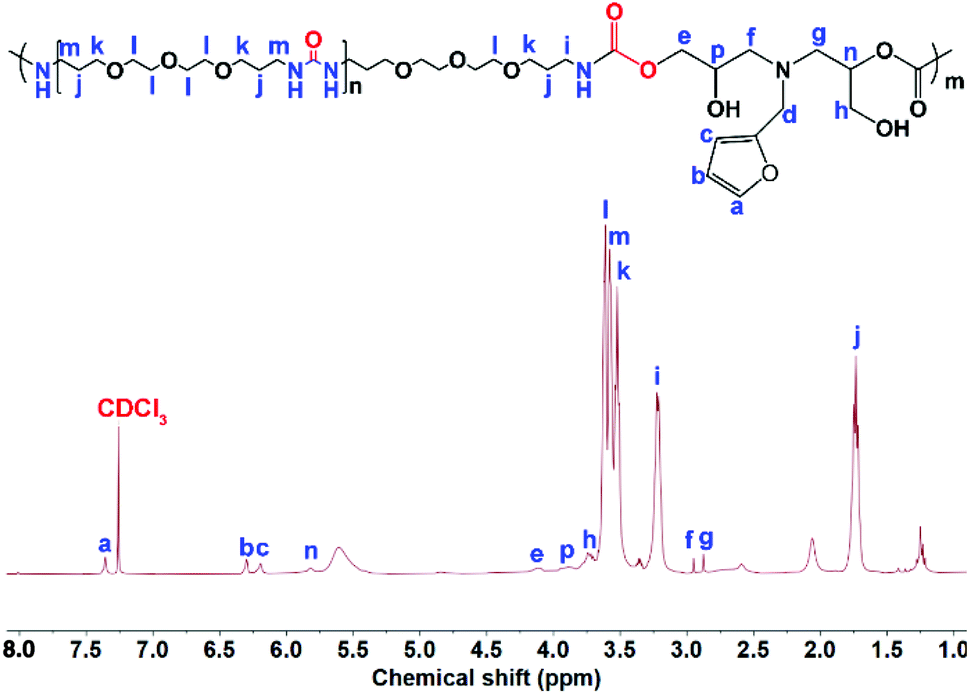 | ||
| Fig. 2 1H NMR spectrum of polyurethane-urea (PUUa). | ||
Synthesis and characterization of the polyurethane-urea Diels–Alder adduct (PUUa-DA)
The Diels–Alder reaction between furan and maleimide groups could build dynamic cross-linked thermoplastics with thermo-reversible properties.61 PUUa has a furan group, so self-healing materials can be constructed by the Diels–Alder reaction. The PUUa Diels–Alder adduct (PUUa-DA) was synthesized from PUUa and BIM. Fig. 3 shows two new characteristic absorption bands at 1710 and 1773 cm−1 ascribed to the Diels–Alder adduct bonds (Fig. 3vs.Fig. 1d),10 which means that the cross-linked PUUa-DA was successfully synthesized. The weight ratio of CO in the urea group of PUUa-DA is 11.8 wt%; i.e., the consumption of CO2 in PUUa-DA is 16.1 wt%. The amount of bio-based furylamine in PUUa-DA is 4.7 wt%. With the increase in the temperature from 30 to 120 °C, the absorption bands of the CO stretching vibration at 1609 cm−1 and the N–H bending vibration at 1589 cm−1 in the urea groups were gradually weakened. Meanwhile, the intensity of the N–H stretching vibration band at 3317 cm−1 gradually decreased, while the absorption bands of the “free” CO stretching vibration at 1658 cm−1 and the “free” N–H bending vibration at 1542 cm−1 gradually became stronger. These results indicated that the strong and ordered hydrogen bonding of urea groups changed to the weak and disordered one with increasing temperature. After cooling to 30 °C for 24 h, the FTIR spectrum became similar to that of the starting material of PUUa-DA, indicating that strong and ordered hydrogen bonding was reformed (Fig. S4†). Furthermore, the characteristic peak of maleimide at 690 cm−1 did not appear at 30 °C, indicating that maleimide reacted with the furan ring through the Diels–Alder reaction. When PUUa-DA was heated to 70 °C, the absorption band at 690 cm−1 ascribed to maleimide was observed, indicating that the retro-Diels–Alder reaction occurred. The reaction was also verified by differential scanning calorimetry (DSC) in the first heating scan (Fig. S5†). PUUa-DA showed a significant broad endothermic temperature at round 108 °C, which was due to the retro-Diels–Alder reaction.
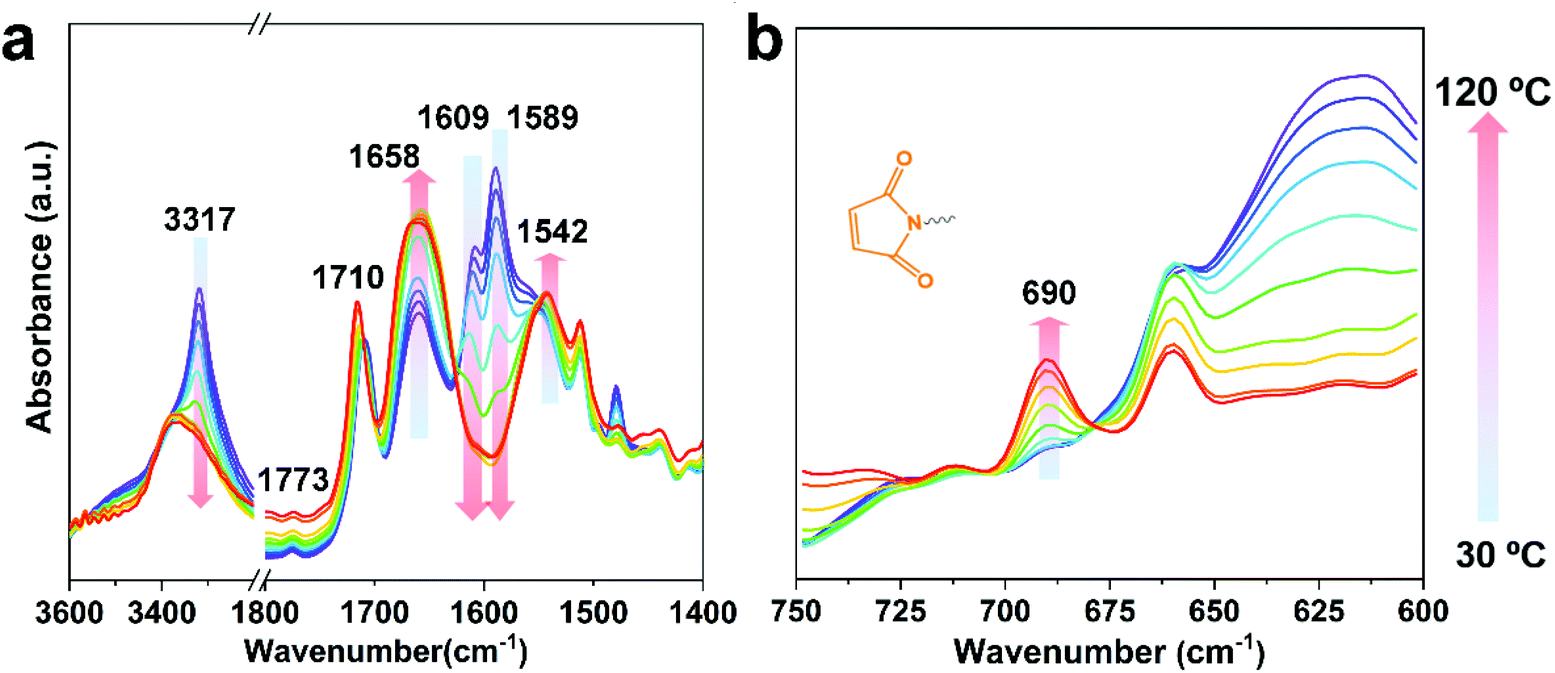 | ||
| Fig. 3 Temperature-dependent FTIR spectra of the polyurethane-urea Diels–Alder adduct (PUUa-DA) from 30, 40, 50, 60, 70, 80, 90, 100, 110 to 120 °C. (a) The region is from 3600 to 1400 cm−1 and (b) the region is from 750 to 600 cm−1. | ||
Non-isocyanate polyurethane is usually obtained by the reaction of diamine with difunctional cyclic carbonate. For comparison, OUa was replaced by TTD or PEA to react with BCCFA, the obtained product was further reacted with BIM, and the corresponding polymer (TTD-DA or PEA-DA) was prepared (Scheme S2†). From FTIR spectra, compared with those of PUUa-DA, there is no strong and ordered hydrogen bonding in TTD-DA and PEA-DA, because TTD-DA and PEA-DA have only the carbamate group but no urea group (Fig. S3†).
Thermal properties
From the TGA curves measured, all samples exhibited high thermal stability with an initial decomposition temperature (5% weight loss, Td,5%) of >220 °C except for TTD-DA (Fig. 4). From the DSC results obtained, the glass transition temperature (Tg) and the melting temperature (Tm) of OUa are lower than those of PUa (Fig. 5, Table S2†). For OUa, PUUa and PUUa-DA, Tg gradually increased from −26 °C to −14 and −8 °C after the reaction of OUa with BCCFA and the following Diels–Alder reaction. Meanwhile, the melting peaks gradually decreased and disappeared for PUUa-DA, indicating that the introduction of furan with an irregular structure and Diels–Alder reaction groups decreased the crystallinity. These results were in agreement with those of FTIR (Fig. 1a, d and Fig. S3†) and XRD (Fig. S6†); the weak and disordered hydrogen bonding increased and the XRD pattern became weakened and broader in the order of PUa → PUUa → PUUa-DA. It is worth noting that two distinct melting peaks appeared in the first heating curve of PUUa-DA, but no melting peak was observed in the second heating curve. This may be due to insufficient crystallization of the OUa segment in PUUa-DA during the cooling process at a rate of 10 °C min−1. According to DSC and XRD (Fig. 5 and Fig. S6†), OUa, PUa, PUUa, and PUUa-DA were all semi-crystalline materials. For TTD-DA, Td,5% is too low, so DSC measurement is impossible. For PEA-DA, Tg may be lower than −50 °C. The crystallization temperatures (Tc) of OUa and PUa were similar, and Tc was not clearly seen for PUUa, PUUa-DA, and PEA-DA (Fig. S7†).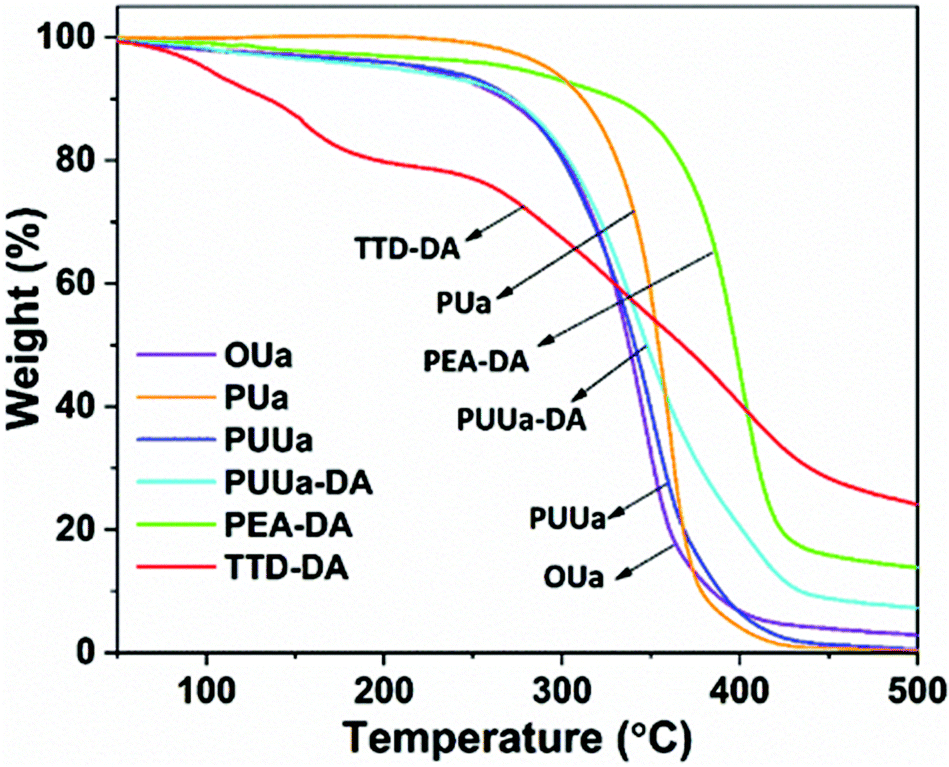 | ||
| Fig. 4 The TGA curves of oligourea (OUa), polyurea (PUa), polyurethane-urea (PUUa), the polyurethane-urea Diels–Alder adduct (PUUa-DA), the poly(propylene glycol) bis(2-aminopropyl ether) Diels–Alder adduct (PEA-DA) and the 4,7,10-trioxa-1,13-tridecanediamine Diels–Alder adduct (TTD-DA). | ||
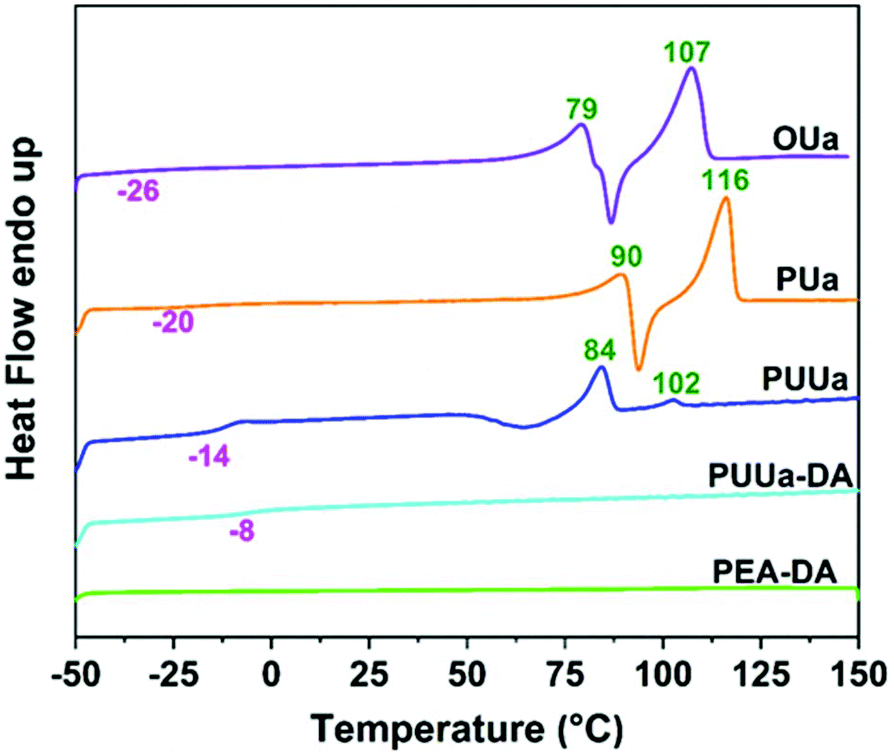 | ||
| Fig. 5 DSC curves of oligourea (OUa), polyurea (PUa), polyurethane-urea (PUUa), the polyurethane-urea Diels–Alder adduct (PUUa-DA) and the poly(propylene glycol) bis(2-aminopropyl ether) Diels–Alder adduct (PEA-DA) from the second heating scan. | ||
Mechanical properties
The mechanical properties were characterized using a uniaxial tensile test at a strain rate of 20 mm min−1 (Fig. 6, Table S3†). OUa obtained from TTD and CO2 was too fragile to be characterized due to its low molecular weight (1550 g mol−1). When OUa was further reacted with CO2, the obtained high molecular weight PUa (4952 g mol−1) was still typical of a stiff polymer with a high Young's modulus of 527 MPa and an elongation at break of 3.2%. Both OUa and PUa are semi-crystalline and have strong, ordered and 3D network crosslinked intermolecular hydrogen bonds (Fig. 1a, 5 and Fig. S4†).25,36 When TTD was reacted with BCCFA, the viscous liquid product obtained was further reacted with BIM and the TTD-DA so produced was a fragile gel, as a result, its mechanical properties cannot be measured, in which short TTD segments led to a very high crosslinking density of the DA adduct. To get a tough and strong material, we prepared PUUa. First, either BCCFA or OUa was prepared from CO2. Then BCCFA was reacted with OUa. The PUUa so formed was found to be a tough thermoplastic with a tensile stress of 15.9 MPa, an elongation at break of 46% and a Young's modulus of 206 MPa. Compared to PUa, the enhanced toughness of PUUa was caused by the introduction of the irregular structure of furan and the formation of the carbamate group, reducing the crystallization due to an increase in the disordered hydrogen bonding interactions. To further enhance the toughness and get a self-healing action, PUUa was further reacted with BIM through the reversible Diels–Alder reaction, and a cross-linking PUUa-DA material was then obtained. PUUa-DA served as a tough and self-healing thermosetting material; compared with PUUa, the Young's modulus decreased from 206 to 151 MPa, but the tensile stress increased from 15.9 to 18.5 MPa and the elongation at break increased from 46% to 136% due to the formation of a cross-linking structure. Compared with OUa, PUa and TTD-DA, PUUa-DA showed significantly improved mechanical properties. In PUUa-DA, on the one hand, the long molecular chains in OUa were favorable for the formation of appropriate amounts of crosslinked Diels–Alder groups, and these crosslinked structures reduced the degree of crystallization of PUUa-DA. On the other hand, OUa segments provided the strong and ordered hydrogen bonding interaction, while the carbamate groups provided the weak and disordered hydrogen bonding interaction. The combination of the ordered and disordered hydrogen bonding interactions and the appropriate amounts of crosslinked Diels–Alder groups were favorable for the improvement of mechanical properties. In order to further examine the influence of the strong hydrogen bonding on the mechanical properties, a polyetheramine of PEA with a Mn similar to that of OUa was selected to replace OUa. The PEA-DA prepared exhibited a tensile stress of 0.085 MPa, an elongation at break of 58% and a Young's modulus of 0.21 MPa, which were much smaller than those of PUUa-DA (18.5 MPa, 136%, 151 MPa, respectively). The Mn values of PEA and OUa were similar, so the amounts of crosslinked DA groups should be similar. Thus, the difference in the properties of PEA-DA and PUUa-DA was due to the fact that PEA could not provide strong and ordered hydrogen bonding for the former (Fig. S3†).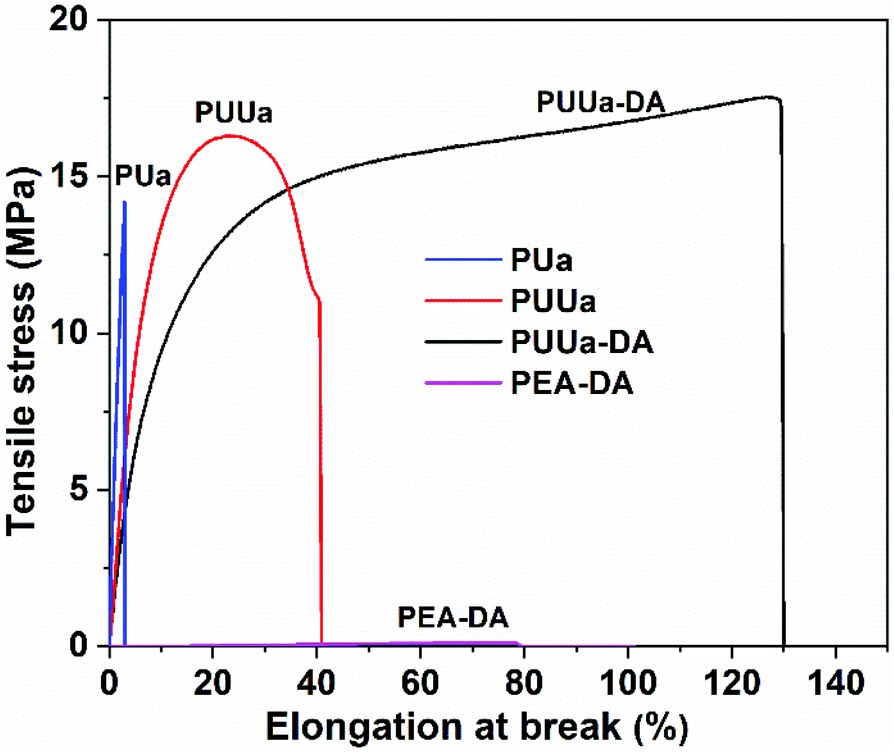 | ||
| Fig. 6 Stress–strain curves of polyurea (PUa), polyurethane-urea (PUUa), the polyurethane-urea Diels–Alder adduct (PUUa-DA) and the poly(propylene glycol) bis(2-aminopropyl ether) Diels–Alder adduct (PEA-DA). | ||
Self-healing and recyclability of PUUa-DA
To examine the macroscopic self-healing behavior, a rectangular specimen of PUUa-DA was cut into two pieces and the fresh surfaces were brought in contact immediately (Fig. 7). Then, it was processed by thermal treatment at 120 °C for 10 min and 60 °C for 24 h to allow the retro-Diels–Alder and Diels–Alder reactions, respectively. After this, the healed specimen can be twisted and bent without breaking. At 120 °C, the retro-Diels–Alder reaction occurred very quickly and PUUa-DA was transformed to PUUa, which is a thermoplastic, then PUUa melted because its melting temperature (84 or 102 °C) is lower. Then the broken hydrogen bond in PUUa was recombined, and the crosslinked Diels–Alder groups were reformed by the Diels–Alder reactions at 60 °C for 24 h. However, at a healing temperature lower than 108 °C, the retro-Diels–Alder reaction is inefficient; although the melted OUa segment makes the molecular chains move easily, PUUa-DA is still a thermosetting material without melting, so PUUa-DA cannot be healed effectively at a temperature below 108 °C. In addition, the self-healing process was also recorded using a polarizing optical microscope (Fig. 8). A crack was made on the surface of PUUa-DA using a blade. The cracked specimen was put on a heating stage and heated from 30 to 120 °C with a heating speed of 5 °C min−1. It was observed that the crack was slightly closed during heating at temperatures lower than 100 °C, but the crack healed fast at 120 °C and no crack was seen on the surface after healing for 2 min. Furthermore, the self-healing process of PUUa-DA was quantitatively evaluated by tensile measurement (Fig. 9), and the healing efficiency was calculated using the ratio of the toughness of the healed specimen against the original one. The healing efficiency of the specimen became as high as 94% when it was healed at 120 °C for 10 min and then healed at 60 °C for 24 h. However, when PUUa-DA was cut and healed at a low temperature of 60 °C for 24 h or healed at 120 °C for 10 min, the healing efficiency was only 39% and 23%, respectively (Fig. 9, Table S4†). | ||
| Fig. 7 Pictures of the healing test of the polyurethane-urea Diels–Alder adduct (PUUa-DA). | ||
 | ||
| Fig. 8 Polarizing optical microscopy images of the self-healing process of a crack on the polyurethane-urea Diels–Alder adduct (PUUa-DA). | ||
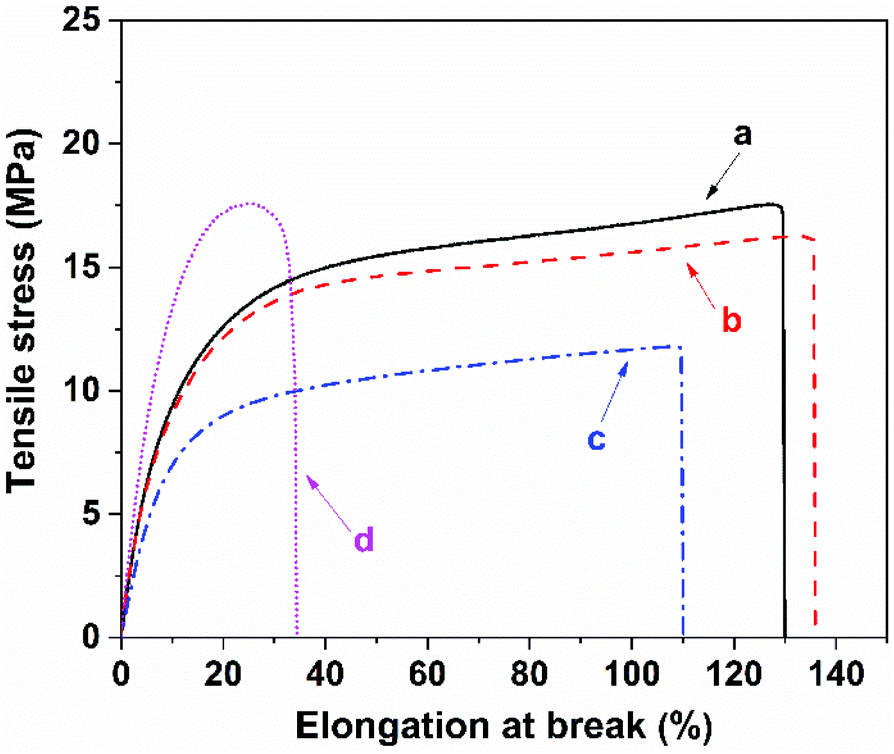 | ||
| Fig. 9 Stress–strain curves of the polyurethane-urea Diels–Alder adduct (PUUa-DA) before and after cutting and healing at different temperatures and times. (a) Original specimen; (b) healed at 120 °C for 10 min and then healed at 60 °C for 24 h; (c) healed at 60 °C for 24 h; (d) healed at 120 °C for 10 min. | ||
The self-healing mechanism of PUUa-DA is depicted in Scheme 2. At the beginning, micro cracks were caused by some damage. The subsequent heating at 120 °C for 10 min broke the molecular chains into small molecular weight fragments via a retro-Diels–Alder reaction and hydrogen bond dissociation (Fig. 3). Meanwhile, the cracks can be filled with the decomposed small chains because the molecular chains have a good mobility at 120 °C (Fig. 5). Finally, after the cracked PUUa-DA is closely connected and kept at 60 °C for 24 h, the dissociative maleimide and furan parts will be connected again via the Diels–Alder reaction. At the same time, the hydrogen bond between urea and carbamate groups will also be reformed and, therefore, the cracked sample is healed and 94% healing efficiency is achieved. However, PUUa-DA showed a lower healing efficiency of 39% at 60 °C for 24 h, because the broken hydrogen bond could be recombined, but the broken DA adduct could not be healed at 60 °C. There are still a large number of undissociated DA bonds in the healed PUUa-DA; the healing temperature of 60 °C is higher than the Tg of −8 °C; molecular chains could move and some undissociated DA bonds moved to the broken domain; the healed PUUa-DA was still a thermosetting material. When PUUa-DA was only healed at 120 °C for 10 min, the retro-Diels–Alder reaction occurred while the Diels–Alder reaction could not occur. Therefore, the DA adduct could not be formed and the healed PUUa-DA at 120 °C for 10 min presented to be a thermoplastic material, similar to that of PUUa. Regarding the reversible nature of the Diels–Alder bond and hydrogen bond, the crosslinked PUUa-DA could be recycled by the conventional hot-pressing molding method. When the dumbbell specimen of PUUa-DA used for mechanical tensile measurements was cut into small fragments and remolded, the remolded sample could retain similar mechanical properties to the original one, indicating that the PUUa-DA thermosetting material can be reused and recycled successfully (Fig. S8†).
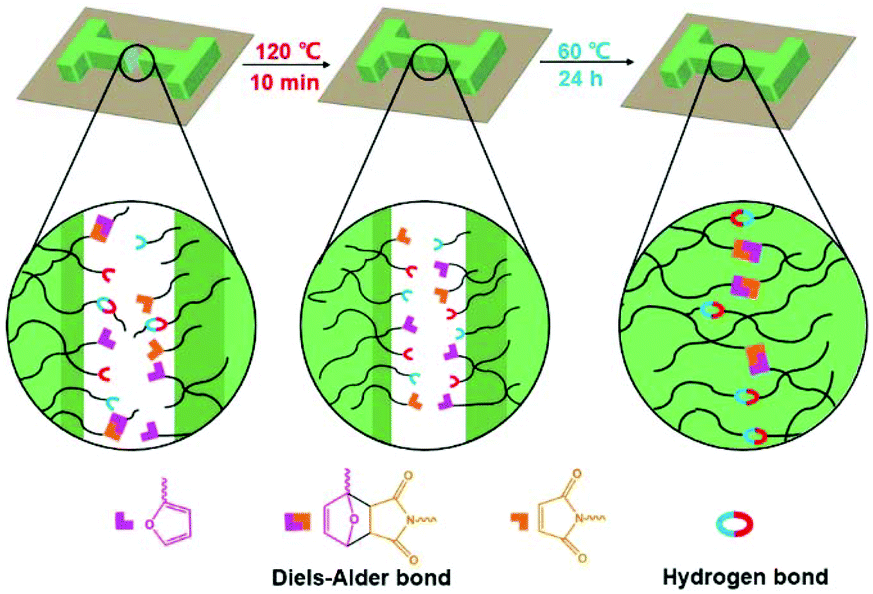 | ||
| Scheme 2 Schematic illustration of the self-healing mechanism of the polyurethane-urea Diels–Alder adduct (PUUa-DA). | ||
Conclusions
In this work, we have reported a green route to synthesize a bio-based non-isocyanate polyurethane-urea Diels–Alder adduct using CO2 as a nontoxic and renewable feedstock. It was used as a co-monomer with a bio-based furfuryl amine. First, bio-based furfurylamine was converted into diglycidyl furfurylamine, and then diglycidyl furfurylamine was converted into bis(cyclic carbonate) furfuryl amine by reacting with CO2. Second, oligourea was synthesized from 4,7,10-trioxa-1,13-tridecanediamine and CO2 by the polycondensation reaction. Oligourea was a fragile polymer due to the strong, ordered and 3D network crosslinked intermolecular hydrogen bonds. Third, polyurethane-urea was prepared by the reaction of oligourea and bis(cyclic carbonate) furfuryl amine. Polyurethane-urea was a tough thermoplastic with a tensile stress of 15.9 MPa, an elongation at break of 46% and a Young's modulus of 206 MPa. The larger toughness, compared to the oligourea, resulted from the irregular structure of furan and the formation of the carbamate group reduced the crystallization and ordered hydrogen bonding interactions. Finally, polyurethane-urea was further reacted with 4,4-bismaleimidodiphenylmethane through the reversible Diels–Alder reaction, and a cross-linking polyurethane-urea Diels–Alder adduct was obtained. The polyurethane-urea Diels–Alder adduct is a tough and self-healing thermosetting material, compared with polyurethane-urea; the Young's modulus decreased to 151 MPa, but the tensile stress increased to 18.5 MPa and the elongation at break increased to 136%. The combination of the ordered and disordered hydrogen bonding interactions and the appropriate amounts of crosslinked Diels–Alder groups in the polyurethane-urea Diels–Alder adduct make it exhibit excellent mechanical and self-healing properties. The healing efficiency is 94% when it was healed at 120 °C for 10 min and then healed at 60 °C for 24 h. In addition, the polyurethane-urea Diels–Alder adduct showed good recyclability and could be remolded via hot-pressing. The successful synthesis of the polyurethane-urea Diels–Alder adduct with great performance would further expand the potential application of environmentally friendly non-isocyanate polyurethane.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the National Key Research and Development Program of China (2016YFA0602900), the National Natural Science Foundation of China (NSFC 21672204), the Youth Innovation Promotion Association CAS (2016206), and the Chinese Academy of Sciences President's International Fellowship Initiative (2018VCA0012).References
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- B. Willocq, J. Odent, P. Dubois and J.-M. Raquez, RSC Adv., 2020, 10, 13766–13782 RSC.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS.
- L. Ling, J. H. Li, G. P. Zhang, R. Sun and C. P. Wong, Macromol. Res., 2018, 26, 365–373 CrossRef CAS.
- C. Bao, Z. Guo, H. Sun and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 9478–9486 CrossRef CAS.
- W. A. Ogden and Z. Guan, J. Am. Chem. Soc., 2018, 140, 6217–6220 CrossRef CAS.
- K. Huang, S. Ma, S. Wang, Q. Li, Z. Wu, J. Liu, R. Liu and J. Zhu, Green Chem., 2019, 21, 4964–4970 RSC.
- S. Dhers, G. Vantomme and L. Averous, Green Chem., 2019, 21, 1596–1601 RSC.
- Y. Fang, X. Du, Y. Jiang, Z. Du, P. Pan, X. Cheng and H. Wang, ACS Sustainable Chem. Eng., 2018, 6, 14490–14500 CrossRef CAS.
- K. Zheng, Y. Tian, M. Fan, J. Zhang and J. Cheng, J. Appl. Polym. Sci., 2018, 135, 1–10 Search PubMed.
- J. Xu, S. Ye and J. Fu, J. Mater. Chem. A, 2018, 6, 24291–24297 RSC.
- D. Wang, S. Chen, J. Zhao and Z. Zhang, Mater. Today Commun., 2020, 23, 101138 CrossRef CAS.
- K.-K. Tremblay-Parrado, C. Bordin, S. Nicholls, B. Heinrich, B. Donnio and L. Avérous, Macromolecules, 2020, 53, 5869–5880 CrossRef CAS.
- P. Song and H. Wang, Adv. Mater., 2019, 32, 1901244 CrossRef.
- L. Zhang, J. B. Bailey, R. H. Subramanian, A. Groisman and F. A. Tezcan, Nature, 2018, 557, 86–91 CrossRef CAS.
- Y. Y. Han, X. D. Wu, X. X. Zhang and C. H. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 20106–20114 CrossRef CAS.
- Y. L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y. C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. Gu, C. Wang, M. He, J. W. Chung and Z. Bao, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS.
- S. So and R. C. Hayward, ACS Appl. Mater. Interfaces, 2017, 9, 15785–15790 CrossRef CAS.
- G. Z. Zhang, L. Lv, Y. H. Deng and C. Y. Wang, Macromol. Rapid Commun., 2017, 38, 1700018 CrossRef.
- S. Burattini, B. W. Greenland, W. Hayes, M. E. Mackay, S. J. Rowan and H. M. Colquhoun, Chem. Mater., 2011, 23, 6–8 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS.
- B. Grignard, S. Gennen, C. Jerome, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- N. Yamazaki, F. Higashi and T. Iguchi, J. Polym. Sci., Part C: Polym. Lett., 1974, 12, 517–521 CAS.
- C. Wu, J. Wang, P. Chang, H. Cheng, Y. Yu, Z. Wu, D. Dong and F. Zhao, Phys. Chem. Chem. Phys., 2012, 14, 464–468 RSC.
- J. Shang, S. Liu, X. Ma, L. Lu and Y. Deng, Green Chem., 2012, 14, 2899–2906 RSC.
- L. Hao, Y. Zhao, B. Yu, H. Zhang, H. Xu, J. Xu and Z. Liu, J. Colloid Interface Sci., 2014, 424, 44–48 CrossRef CAS.
- P. Wang, X. Ma, Q. Li, B. Yang, J. Shang and Y. Deng, RSC Adv., 2016, 6, 54013–54019 RSC.
- Z. Ying, Y. Dong, J. Wang, Y. Yu, Y. Zhou, Y. Sun, C. Zhang, H. Cheng and F. Zhao, Green Chem., 2016, 18, 2528–2533 RSC.
- Z. Ying, C. Zhang, S. Jiang, Q. Wu, B. Zhang, Y. Yu, M. Lan, H. Cheng and F. Zhao, J. CO2 Util., 2016, 15, 131–135 CrossRef CAS.
- Z. Liu, Z. Yang, L. Hao, X. Liu, H. Zhang, B. Yu and Z. Liu, New J. Chem., 2017, 41, 51–55 RSC.
- S. Jiang, R. Shi, H. Cheng, C. Zhang and F. Zhao, Green Energy Environ., 2017, 2, 370–376 CrossRef.
- Z. Ying, C. Wu, C. Zhang, S. Jiang, R. Shi, H. Cheng, B. Zhang, Y. Li and F. Zhao, J. CO2 Util., 2017, 19, 209–213 CrossRef CAS.
- P. Wang, Y. Fei and Y. Deng, New J. Chem., 2018, 42, 1202–1207 RSC.
- P. Wang, Y. Fei, Y. Long and Y. Deng, J. CO2 Util., 2018, 28, 403–407 CrossRef CAS.
- S. Jiang, H.-Y. Cheng, R.-H. Shi, P.-X. Wu, W.-W. Lin, C. Zhang, M. Arai and F.-Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47413–47421 CrossRef CAS.
- N. Yadav, F. Seidi, D. Crespy and V. D'Elia, ChemSusChem, 2019, 12, 724–754 CrossRef CAS.
- B. S. Ritter and R. Mülhaupt, Macromol. Mater. Eng., 2017, 302, 1600338 CrossRef.
- L. Longwitz, J. Steinbauer, A. Spannenberg and T. Werner, ACS Catal., 2017, 8, 665–672 CrossRef.
- S.-E. Dechent, A. W. Kleij and G. A. Luinstra, Green Chem., 2020, 22, 969–978 RSC.
- H. Bindu and A. Palanisamy, ChemistrySelect, 2019, 4, 11052–11060 CrossRef CAS.
- H. Y. Chen, W. C. Pan, C. H. Lin, C. Y. Huang and S. A. Dai, J. Polym. Res., 2012, 19, 114 Search PubMed.
- J. M. Dennis, L. I. Steinberg, A. M. Pekkanen, J. Maiz, M. Hegde, A. J. Muller and T. E. Long, Green Chem., 2018, 20, 243–249 RSC.
- J. M. Sirrine, S. A. Schexnayder, J. M. Dennis and T. E. Long, Polymer, 2018, 154, 225–232 CrossRef CAS.
- B. T. White, J. M. Migliore, E. U. Mapesa, J. D. Wolfgang, J. Sangoro and T. E. Long, RSC Adv., 2020, 10, 18760–18768 RSC.
- G. W. Yang and G. P. Wu, ACS Sustainable Chem. Eng., 2019, 7, 1372–1380 CrossRef CAS.
- P. Wu, H. Cheng, Y. Wang, R. Shi, Z. Wu, M. Arai and F. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 12677–12685 CrossRef CAS.
- G. W. Yang, Y. Y. Zhang, Y. Wang, G. P. Wu, Z. K. Xu and D. J. Darensbourg, Macromolecules, 2018, 51, 1308–1313 CrossRef CAS.
- E. Dolci, V. Froidevaux, G. Michaud, F. Simon, R. Auvergne, S. Fouquay and S. Caillol, J. Appl. Polym. Sci., 2017, 134, 44408 CrossRef.
- P. A. Pratama, M. Sharifi, A. M. Peterson and G. R. Palmese, ACS Appl. Mater. Interfaces, 2013, 5, 12425–12431 CrossRef CAS.
- C. Y. Shao, M. Wang, H. L. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- S. Schafer and G. Kickelbick, Macromolecules, 2018, 51, 6099–6110 CrossRef.
- Y. Peng, Y. J. Hou, Q. Q. Shen, H. Wang, G. Li, G. S. Huang and J. R. Wu, Acta Polym. Sin., 2020, 51, 158–165 Search PubMed.
- K. Urdl, S. Weiss, P. Christofl, A. Kandelbauer, U. Muller and W. Kern, Eur. Polym. J., 2020, 127, 109601 CrossRef.
- S. Yu, R. C. Zhang, Q. Wu, T. H. Chen and P. C. Sun, Adv. Mater., 2013, 25, 4912–4917 CrossRef CAS.
- Z. Ying, C. Y. Wu, S. Jiang, R. H. Shi, B. Zhang, C. Zhang and F. Y. Zhao, Green Chem., 2016, 18, 3614–3619 RSC.
- J. Qin, J. Jiang, S. Ye, J. Liang, Y. Tao, S. Wang, D. Han and M. Xiao, Adv. Ind. Eng. Poly. Res., 2019, 2, 186–195 Search PubMed.
- P. X. Wu, H. Y. Cheng, R. H. Shi, S. Jiang, Q. F. Wu, C. Zhang, M. Arai and F. Y. Zhao, Adv. Synth. Catal., 2019, 361, 317–325 CrossRef CAS.
- Z. Ying, L. J. Zhao, C. Zhang, Y. C. Yu, T. Liu, H. Y. Cheng and F. Y. Zhao, RSC Adv., 2015, 5, 42095–42100 RSC.
- S. Li, Z. Sang, J. Zhao, Z. Zhang, J. Zhang and W. Yang, Ind. Eng. Chem. Res., 2016, 55, 1902–1911 CrossRef CAS.
- K. Yasuda, K. Sugane and M. Shibata, J. Polym. Res., 2020, 27, 18 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S4, Schemes 1, 2 and Fig. S1–S8. See DOI: 10.1039/d0gc03695a |
| This journal is © The Royal Society of Chemistry 2021 |