
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphynes: indispensable nanoporous architectures in carbon flatland
Anto James
,
Chris John
,
Cheriyacheruvakkara Owais
,
Stephen Nagaraju Myakala
,
Sarap Chandra Shekar†
,
Jyoti Roy Choudhuri‡
and
Rotti Srinivasamurthy Swathi
 *
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram (IISER-TVM), Vithura, Kerala, India-695551. E-mail: swathi@iisertvm.ac.in
First published on 22nd June 2018
Abstract
Theoretical design and experimental realization of novel nanoporous architectures in carbon membranes has been a success story in recent times. Research on graphynes, an interesting class of materials in carbon flatland, has contributed immensely to this success story. Graphyne frameworks possessing sp and sp2 hybridized carbon atoms offer a variety of uniformly distributed nanoporous architectures for applications ranging from water desalination, gas separation, and energy storage to catalysis. Theory has played a pivotal role in research on graphynes, starting from the prediction of various structural forms to the emergence of their remarkable applications. Herein, we attempt to provide an up-to-date account of research on graphynes, highlighting contributions from numerous theoretical investigations that have led to the current status of graphynes as indispensable materials in carbon flatland. Despite unsolved challenges in large-scale synthesis, the future appears bright for graphynes in present theoretical and experimental research scenarios.
 Anto James | Anto James is currently pursuing his doctoral studies under the supervision of Dr R. S. Swathi at the School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram (IISER-TVM), India. His research is aimed at designing nano-oscillators by investigating the permeation of fullerenes through the pores of higher order graphynes. |
 Chris John | Chris John is an Integrated PhD student pursuing her doctoral studies under the supervision of Dr R. S. Swathi at IISER-TVM. Her research focuses on developing optimal parameters for model potentials governing the interactions of atoms and molecules with graphene, carbon nanotubes, as well as boron nitride sheets and tubes. |
 Cheriyacheruvakkara Owais | Cheriyacheruvakkara Owais is an Integrated PhD student pursuing his doctoral studies under the supervision of Dr R. S. Swathi at IISER-TVM. His research focuses on employing continuum approaches for modeling carbon-based nanostructures such as graphene, carbon nanotubes and graphynes. |
 Stephen Nagaraju Myakala | Stephen Nagaraju Myakala is a BS-MS student at IISER-TVM working under the supervision of Dr R. S. Swathi. He is currently a recipient of the INSPIRE fellowship. |
 Sarap Chandra Shekar | S. Chandra Shekar completed his PhD under the supervision of Dr. R. S. Swathi at IISER-TVM. He is currently pursuing his postdoctoral studies at the Institute for Computational Physics, University of Stuttgart, Germany. His research is mainly focused on the two-dimensional carbon networks such as graphene, graphyne and graphdiyne and their utility in the context of lithium ion batteries, DNA sensing and photoswitching. |
 Jyoti Roy Choudhuri | Jyoti Roy Choudhuri completed his PhD under the supervision of Prof. Amalendu Chandra at IIT, Kanpur. Subsequently, he had a postdoctoral stint at the Virginia Commonwealth University under the supervision of Prof. Alenka Luzar. He followed this with a short stint in the research group of Dr R. S. Swathi at IISER-TVM. Jyoti is currently an Assistant Professor at the Presidency University, Bangalore, India. |
 Rotti Srinivasamurthy Swathi | R S Swathi obtained PhD in theoretical chemistry from Indian Institute of Science, Bangalore. Subsequently, she joined as an Assistant Professor in School of Chemistry, IISER-TVM. Her research group employs analytical and computational approaches for modeling chemical phenomena involving carbon-based and metal-based nanostructures. She is a recipient of the Young Scientist Awards from Indian National Science Academy, National Academy of Sciences, Indian Academy of Sciences, Kerala State Council for Science, Technology and Environment and Distinguished Lectureship Award from the Chemical Society of Japan. She is currently on the Editorial Advisory Board of ACS Applied Materials & Interfaces. |
1 Introduction to the family of graphynes
“Carbon seems the most favourable case, because it has the richest chemistry” is a remark made by Stephen Hawking about the chemical basis of alien life in the universe.1 Carbon is the second most abundant element in the human body, due to its catenation property, although it is the fifteenth most abundant element in the Earth’s crust. Carbon has many allotropic forms: the natural ones are diamond and graphite, and the important man-made allotropes are fullerenes, carbon nanotubes (CNTs), graphene and carbon nanofoams. Interestingly, most of these materials essentially consist of carbon atoms in the sp2 state of hybridization. They have expanded the scope of available zero-, one-, and two-dimensional materials (0D, 1D, and 2D, respectively) with their wide range of mechanical, electronic, optical, chemical and electrochemical properties leading to many advanced applications in photovoltaic cells, organic light emitting devices, field-effect transistors and chemical sensors.2–4 Among these, one-atom-thick graphene is known to be one of the most important allotropes exhibiting a variety of intriguing electronic, thermal,5 optical,6,7 and mechanical properties.8 It has potential applications in electronic and optoelectronic devices,9–11 chemical sensors,12 nanocomposites13,14 and energy storage.15–20 First scientific study of graphene was carried out by Wallace in 1947,21 although it was first observed by Boehm et al.22 in 1962 and isolated by Geim and Novoselov23 in 2004. Wallace theoretically predicted the band structure of graphene considering it as the monolayer model structure for graphite. Other forms of carbon networks such as graphene oxide, graphane, graphone and fluoro-graphane can be derived by chemical functionalization of graphene. Hydrogenation and fluorination of graphene yield graphane and fluorographene, respectively. Partial hydrogenation (on alternate carbons) of graphene gives graphone. The introduction of epoxy, carboxyl and hydroxyl linkages on graphene leads to the formation of graphene oxide. Of these range of systems, one can notice that graphene has sp2 hybridized carbons, graphane and fluorographene have sp3 hybridized carbons and graphone and graphene oxide have both sp2 and sp3 hybridized carbons in their networks.In the last two decades, new members of the carbon family referred to as graphynes, one-atom-thick two-dimensional networks similar to graphene have gained much importance.24,25 Graphynes (GYs) were first theoretically proposed by Baughman and co-workers in 1987. Indeed, theory preceded and guided the synthesis and fabrication of most of the new forms of carbon-based materials known today. For instance, graphane and graphone were also first predicted theoretically and subsequently realized in experiments.26–29 Theoretically, GYs are constructed by replacing some carbon–carbon sp2 bonds in graphene with acetylenic (i.e., consisting of single- and triple-bonded) linkages.30 Various types of GYs are proposed based on the variation of positions at which the acetylenic linkers are added. Depending on the structures, they are classified into various members known as α-GY, β-GY, γ-GY, δ-GY, rhombic-GY, 6,6,12-GY etc.,31 as shown in Fig. 1. GYs possess intrinsically porous structures and the porosity can be tuned by changing the number of acetylenic linkers in the architectures. According to the number of acetylenic linker groups (N), they are named as GY-N with N equal to zero yielding the graphene. GY-2 of γ-GY (known as graphdiyne, GDY) and rhombic-GY (known as carbon ene–yne, CEY) are the only structures reported to be successfully synthesised in large quantities.32,33 Thin films of GDY have been synthesised on top of copper34 and silver substrates.35 A recent report shows that thin films of rhombic-GY-2 can also be synthesised on copper substrates.36 However, small annulenic units of various GYs are prepared from the benzannulated and perethynylated dehydroannulene-derived substructures. A recent review on GDY gives a detailed picture of various significant applications as well as importance of GDY.37 Unique structural and electronic properties of various GYs make them even more imperative. Alike graphene, GYs also show some unique chemical, electronic, mechanical and structural properties38–41 that have important applications ranging from energy storage to nanoelectronics.42 Nanoporous membranes of graphene and its analogues have been employed for achieving selective permeation of gas molecules, ions, hydrocarbons, biomolecules etc. However, attaining uniform porosity in graphene is a difficult task and hence carbon membranes like GYs which possess intrinsic pores are of great interest.43 Theoretical studies have predicted the chemical inertness of free-standing graphyne layers and their stability under ambient temperature.44–46 γ-GYs exhibit non-zero band gap in contrast to the zero band gap of graphene. The distortion in the Dirac cone of 6,6,12-GY arising due to the presence of sp and sp2 carbons results in the directionality of electric conductance.47 Graphene is one of the stiffest materials known to exist after carbyne and diamond. Addition of acetylenic linkers in graphene results in reduction of stiffness. From γ-GY-1 to γ-GY-5, increase in sp carbon atom content results in 10 to 50% reduction in Young’s modulus compared to graphene.39 The decrease in carbon atom density in GYs compared to graphene also results in lower values of elastic modulus,48 as shown in Table 1. The velocity of sound waves in γ-GY is predicted to vary with external stress, indicating the application of GYs as surface acoustic sensors.49 Researchers suggest that these materials can surpass graphene in the near future.47
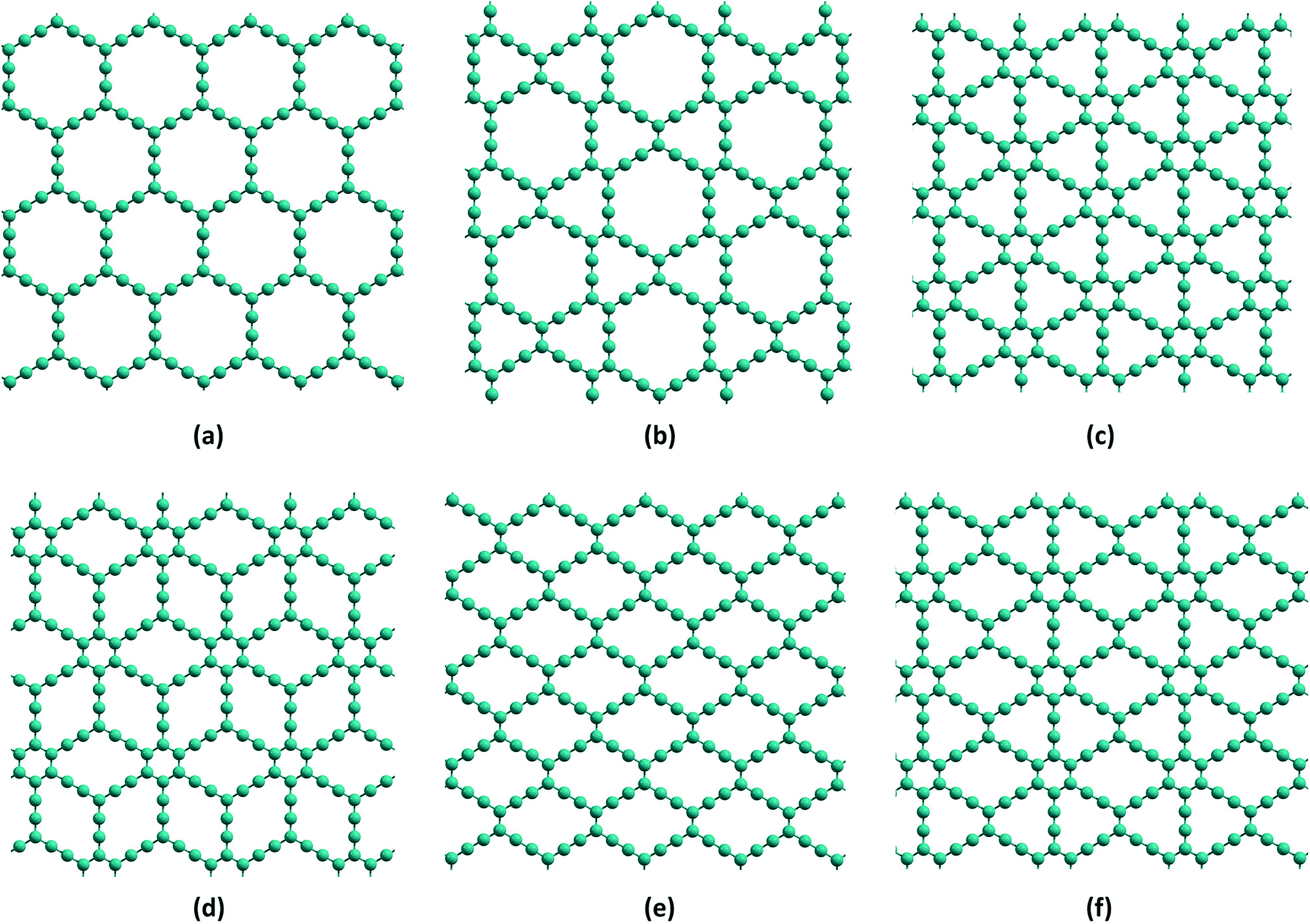 | ||
| Fig. 1 Representative structural models for (a) α-GY (b) β-GY (c) γ-GY (d) δ-GY (e) rhombic-GY and (f) 6,6,12-GY. | ||
| Model system | Percentage of acetylenic linkages | Atom density (atoms per nm2) | Young’s Modulus (GPa) |
|---|---|---|---|
| α-GY | 100 | 18.92 | 120 |
| β-GY | 66.67 | 23.13 | 261 |
| γ-GY | 33.33 | 29.61 | 505 |
| 6,6,12-GY | 41.67 | 28.02 | 445 |
| Graphene | 0 | 39.95 | 995 |
Similar to graphene which can be rolled into CNTs, GYs also form nanotubes. Graphyne nanotubes (GNTs) were proposed in 2003 by Coluci and co-workers.50 The key difference between a CNT and a GNT is that the latter has porous walls, thus allowing material transport through the sidewalls. Coluci et al. classified the GNTs into three families, α-GNT, β-GNT and γ-GNT, each derived from their corresponding parent graphyne sheets, respectively. Furthermore, the metallic or semiconducting nature of GNTs is governed by the same rule as in case of CNTs. (n,0) and (n,n) α-GNTs are referred to as zigzag and armchair, respectively, whereas, (2m,m) and (n,0) β-GNTs and γ-GNTs are referred to as zigzag and armchair, respectively.50 Comparing the band structures of the three, it was found that the band gap has an oscillatory behaviour for α-GNT and β-GNT as a function of the GNT diameter, while γ-GNT has diameter-independent band gap. Contrary to this, Wang et al. have found that γ-GNTs also show oscillatory behaviour51 (Fig. 2a and b). The key issues at present are the stability of the different forms of GNTs and the feasibility of the different synthetic methods. Although several reports predict that γ-GNTs are likely the first ones to be synthesised because of their lowest energy (and hence the most stable amongst the three forms), there have been successful attempts at the synthesis of other types of GNTs. Li and co-workers successfully demonstrated synthesis of high-quality GDY nanotubes with a wall thickness of 15 nm and nanotube length of about 40 microns obtained after annealing of the GNTs at 650 °C.52 Alaei et al. studied the interaction of transition metals with γ-GNTs. Complexes of γ-GNTs with Co and Fe yielded net non-zero magnetic moments, opening up several applications in electronics and spintronics.53 In addition to being nonmagnetic, pristine γ-GNTs possess zero electric dipole moment as well. Deb et al. have shown that functionalization of H2O molecule on γ-GNTs can cause an increase in dipole moment.54 Thus, γ-GNTs act as electron acceptors and therefore can be used as n-type semiconductors. Hu et al. found that γ-GNTs have remarkably low thermal conductivity (10 W mK−1), about two orders of magnitude lower when compared to any of the pristine or chemically functionalized CNTs.55 Direct proportionality of thermal conductivity was observed to the length of the tube in the range of 0.1 to 1.5 μm, beyond which it showed negligible change. Likewise, there was steep increase of thermal conductivity for tubes with a diameter less than 2 nm but was independent for all diameters greater than 2 nm (Fig. 2c and d). In addition to their array of distinct properties, de Sousa et al. have found that γ-GNTs also exhibit superplasticity.56 Studies of the newly synthesised GNTs and nanowires with their field emission properties, high conductivity and mobility open numerous opportunities in the fabrication of graphyne nanodevices.52,57 Graphyne nanoscrolls are structures obtained by rolling nanosheets in a papyrus-like topology. The nanoscroll diameter can be easily tuned when compared to the GNT diameters. Solis et al. have predicted that their stability depends on the critical value of the ratio between the length and the height of the GY sheets used.58
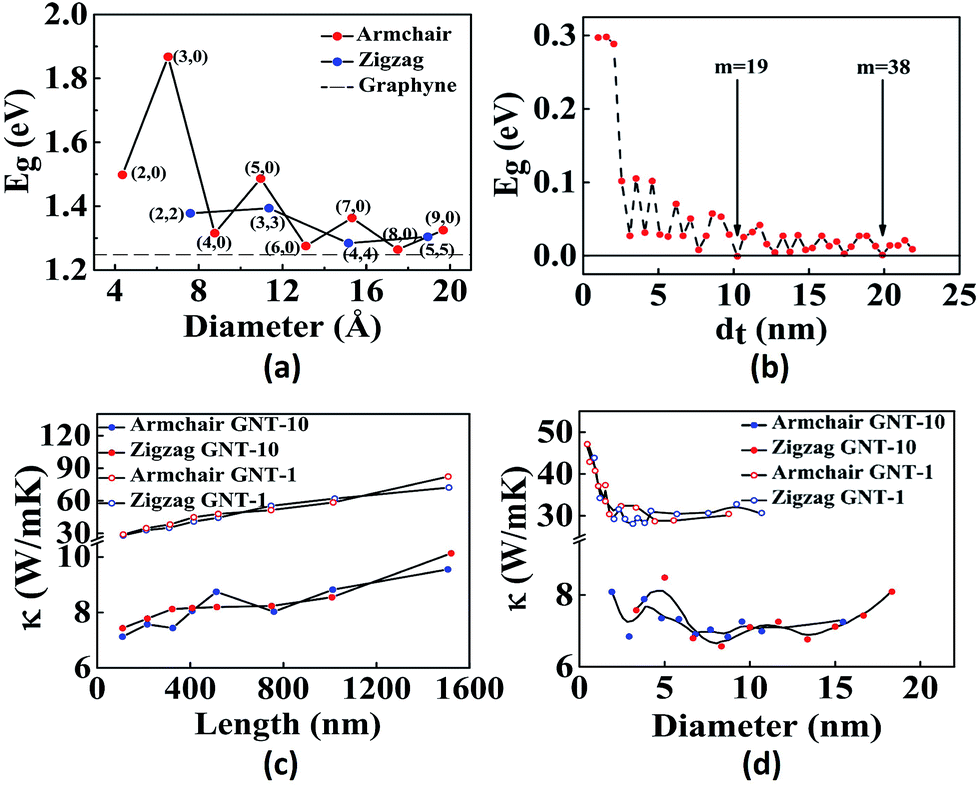 | ||
| Fig. 2 Representative properties of GNTs: effect of tube diameter on the band gap of (a) γ-GNTs and (b) zigzag β-GNTs. Dependence of the thermal conductivity on (c) tube length and (d) tube diameter of various GNTs. Part a is reproduced from ref. 51 with permission from the American Chemical Society. Part (b) is reproduced from ref. 59 and parts (c) and (d) are reproduced from ref. 55 with permissions from the American Physical Society. | ||
In this review, we describe some of the potential chemical applications of GYs on the basis of the latest theoretical and experimental results. We have attempted to launch an up-to-date account of most topics relevant in this field. This review is organised as follows. In Section 2, we have discussed the different approaches that have been used for the synthesis of GYs. This is followed by Sections 3 to 8, where various potential applications of GYs ranging from water desalination, gas separation, gas sensing, energy storage, catalysis to optoelectronics are discussed in detail. Finally, the review is concluded by giving a brief exposition to the future of GYs in Section 9.
2 Approaches for the synthesis of graphynes
Ever since they were theoretically predicted in 1987, GYs in various forms and structures have been modelled. Extensive studies of their unique properties have been carried out, indicating a plethora of applications. However, the central challenge of devising efficient industrially realizable synthetic methods still remains. Recently, Li et al. synthesised large area of GDY films via the homocoupling of hexaethynylbenzene on a Cu foil in the presence of pyridine.61 As predicted by Baughman and co-workers, the obtained films were highly crystalline in nature, as confirmed by their XRD patterns. Qian et al. synthesised GDY films on ZnO nanorod arrays by using a self-catalysed vapor–liquid–solid growth process. GDY films were fabricated by vapour deposition on ZnO nanorod arrays from the vapour source of GDY powder. These GDY films exhibit very low turn on field of 4.2 V μm−1.62 Despite the large area and high crystallinity obtained in the early methods of GDY synthesis, these methods involve use of hazardous solvents and strong reaction conditions. However, Nishihara and co-workers overcame this limitation by synthesising few layers of GDY of higher quality using a bottom-up approach (Fig. 3a).60 They carried out catalytic polymerization of hexaethynylbenzene in a mixture of dichloromethane and toluene with Cu as a catalyst. GDY sheets thus synthesised are characterized by transmission electron microscope and atomic force microscope (Fig. 3b and c).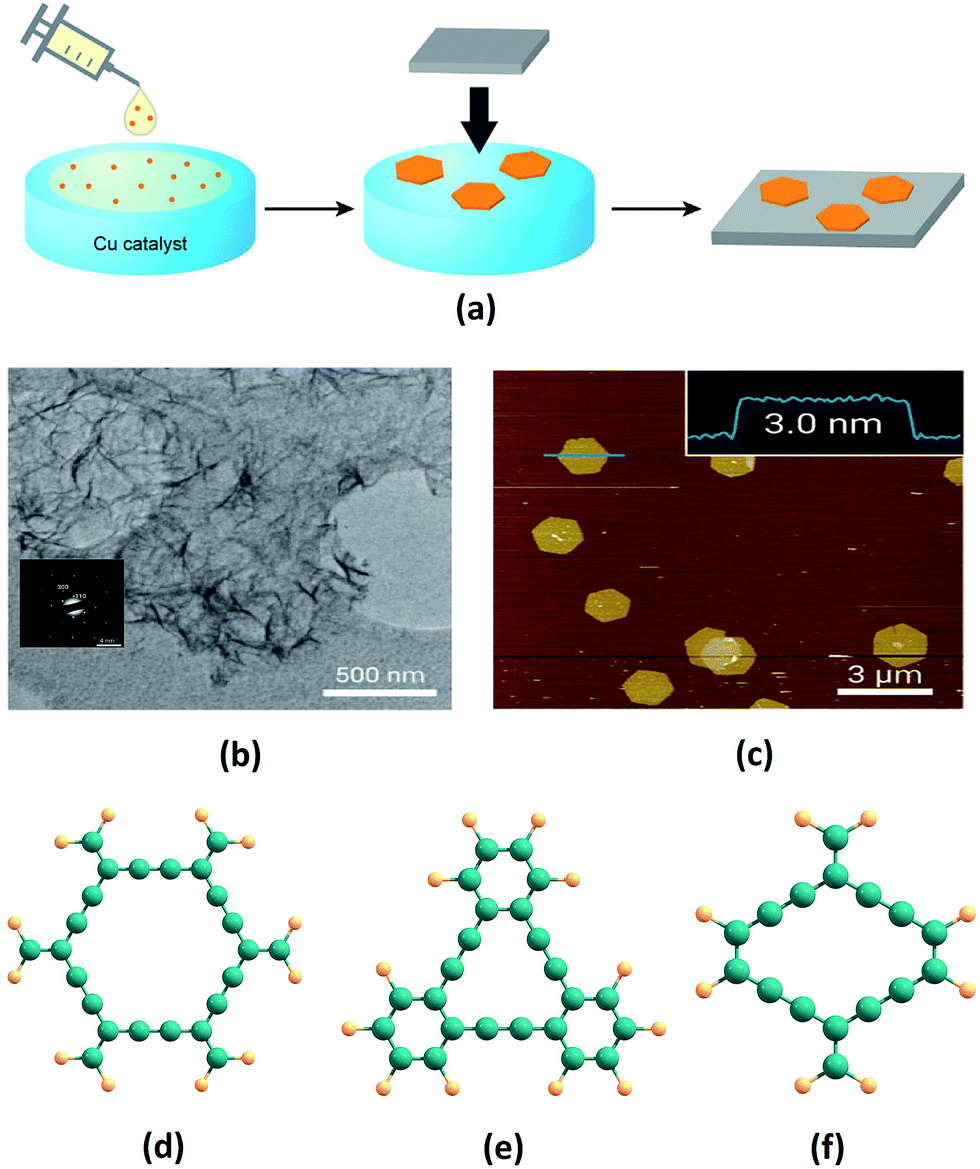 | ||
| Fig. 3 (a) Illustration of the synthetic procedure and transfer of GDY thin films on copper substrates as adopted by Nishihara and co-workers. (b) Transmission electron microscopy image and selected-area electron diffraction pattern (inset) at the liquid–liquid interface, and (c) atomic force microscopy topographical image at the gas–liquid interface of GDY nanosheets synthesised by a bottom-up approach. Molecular sub-units of (d) α-GY, (e) γ-GY and (f) rhombic-GY. Parts (a–c) are adapted from ref. 60 with permissions from the American Chemical Society. | ||
Another example of bottom-up approach for the synthesis of γ-GYs is oligotrimerization and cyclotrimerization of graphyne subunits. In recent advances, the hexakis-benzenes are proven to be the best graphyne subunits. While they seem to be the easy route, they have certain drawbacks such as additional cyclo-oligomeric and/or polymeric products often resulting in a low yield of the desired materials.63 Intramolecular cyclization is yet another powerful method for the construction of more complex structures of the GY networks. However, Haley showed that it produces a meagre yield. To overcome this problem, considering the high efficiency of alkyne metathesis, Haley combined it with pre-organized propynyl groups obtaining a higher yield of 20%.63 Wu et al. also developed a similar alkyne metathesis, synthesising graphyne-like porous networks whose thickness can be tuned by adjusting the reaction time.64 To minimise other side products, various methods such as intramolecular alkylation with suitable molecules like bis(dibenzylideneacetone)palladium(0) (Pd(dba)2) under high dilution conditions were also discovered.
Ding’s group found that small carbyne chains on nano-active materials have a curved polyynic structure on less active metal surfaces (Cu), whereas, they possess a linear configuration on active metals like Ni.65 Hence, a synthesis method of GY sheets by self-assembly of the carbyne chains at low temperature via carbon cluster sputtering was suggested.65 It has been demonstrated that certain functionalization leads to high selectivity in alkyne homocoupling forming polymer strands which exhibit mutual lateral attraction yielding multiple stranded assemblies that are stable at room temperature. Klappenberger et al. constructed high quality, flexible CN-functionalized 3–2 GDY nanowires (with three phenyl and two alkyne(terminal) moieties) via on-surface covalent coupling of terminal alkyne building blocks.66 From these results, the authors also concluded that secondary functional groups drastically increased the coupling efficiency. Jia and co-workers synthesised thin films of rhombic-GY-2 on copper substrates using a solvent-phase reaction.36 Thin films were synthesised using tetraethynylethene molecules in pyridine solvent under N2 protected atmosphere. Apart from these successful syntheses, many dehydroannulene and radialene molecules are reported to be synthesised in literature, which can be considered as the molecular sub-units of GYs. A few representative molecular model systems of GYs are shown in Fig. 3d–f.
3 Graphynes for water desalination
Graphynes are intrinsically nanoporous one-atom-thick membranes with good mechanical and chemical stability. Unlike graphene, GYs can be used for molecular filtration without any post-synthetic modifications. Well-defined porous structures of GYs prevent defect formation over the membranes. Water desalination, one of the key aspects of molecular filtration is becoming increasingly important with the depletion of natural freshwater resources. Recent molecular simulation studies and experiments have demonstrated that GYs are much better membranes for water desalination compared to the commercially available forward osmosis (FO) and reverse osmosis (RO) membranes. Liu and co-workers have used the γ-GY-2 membranes synthesised over Cu foils to remove lead ions from water.67 Acetylenic linkages in γ-GY-2 were shown to interact strongly with metal ions and their adsorption helps in removal of heavy metal ions.67 However, most of the studies related to water desalination are carried out theoretically and the results are yet to be realized experimentally.In 2014, Bartolomei et al. studied the interactions between γ-GYs and water molecules using second-order Moller–Plesset perturbation theory calculations (MP2C) with extended basis set.68 Calculations were carried out on the smallest precursors of GYs, namely annulenes. Lengths of the sides of the triangular pores of γ-GY-1, γ-GY-2 and γ-GY-3, were estimated to be 1.3, 3.9 and 6.4 Å respectively, while the van der Waals diameter of water is in the range of 3.15–3.28 Å. There was a finite barrier for the penetration of water through γ-GY-1 and γ-GY-2 while γ-GY-3 allowed the barrierless flow of water. However, hydrogen bonding interaction from a second water molecule on the other side of the graphyne pore can reduce the barrier for water penetration, as illustrated in Fig. 4a. A comparative study of MP2C calculations with Lennard-Jones (LJ) and improved Lennard-Jones (ILJ) pair potentials was carried out by Bartolomei and co-workers.68 ILJ force field (FF) optimization curves serve as a good approximation for water–graphyne interactions (Fig. 4b). The ILJ pair potential is given by
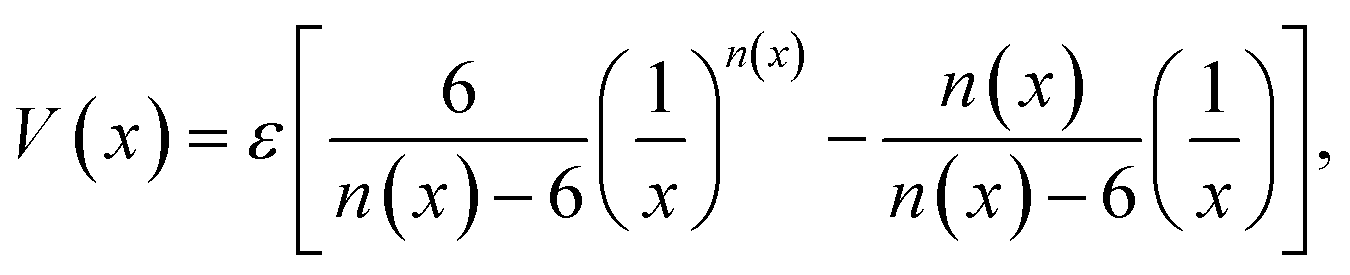 | (1) |
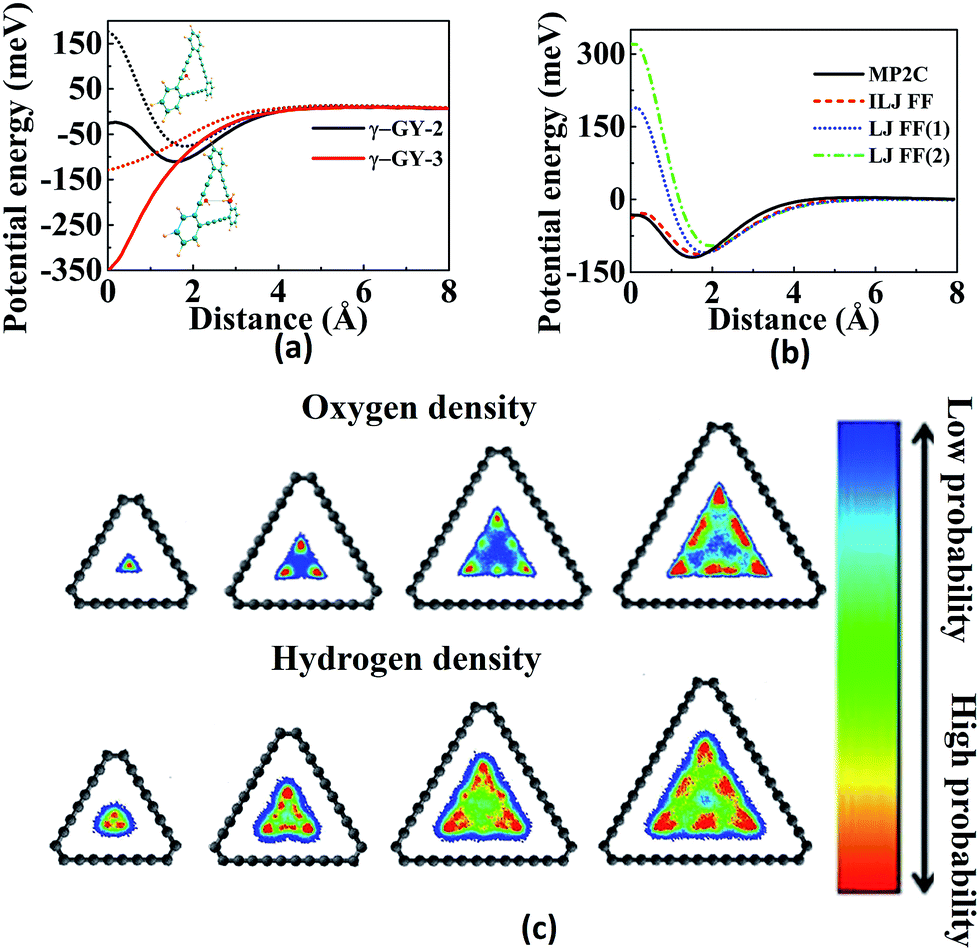 | ||
| Fig. 4 Water permeation through GYs: (a) effect of hydrogen bonding on the energy barrier for water penetration through γ-GYs. (b) Comparison of potential energy scans of water penetration through nanopores of γ-GY-2 calculated using MP2C as well as ILJ and LJ FFs. (c) Oxygen atom and hydrogen atom densities inside the pores of various γ-GYs. Parts (a) and (b) are reproduced from ref. 68 with permissions from the American Chemical Society. Part (c) is reproduced from ref. 69 with permission from the Nature Publishing Group. | ||
Employing molecular dynamics (MD) simulations, Zhang et al. demonstrated that γ-GY membranes could be used as FO membranes to purify water.70 Water fluxes through a series of GY-N membranes (N = 3, 4, 5 and 6) were studied and they were all found to be of the same order of magnitude. In the simulation, no linear correlation between structural properties like pore size and water flux during FO was observed. Water flux through the membrane was influenced by hydrogen bonding, electrostatic and van der Waals interactions between water molecules, solutes and membranes. An increase in the number of average hydrogen bonds was observed with the increase in pore size of the membrane, which resulted in the slowing of water flow through pores. Similar to RO, the salt rejection efficiency of GY as FO membrane is dependent on the size exclusion effect. γ-GY-3 gives a complete salt rejection while γ-GY-N membranes with N > 3 showed decreased salt rejection. Water flux through GY membranes is found to be around 10 l cm−1 h−1, which is three to four orders of magnitude higher than the commercial RO and FO membranes. As GYs are one-atom-thick membranes, concentration polarization phenomenon, which results in a reduction of water flux through FO membranes is minimal. Water flux through each pore and pore density on the sheet are two important factors that determine water permeability across GY membranes.69 Water flow per unit pore for GY-Ns increases stepwise from N = 3 to N = 6. As the pore size of the GY sheet increases, the pore density starts to decrease. As a result, effective water flow per unit area is the highest for γ-GY-4. Qin et al. have observed that the water molecules near the interface of GY show peculiar periodicity and higher mass density compared to the bulk.71 The higher water viscosity can be attributed to the formation of layers of in-plane hydrogen-bonded networks near the membrane. Passage of water molecules through the pores requires the breaking of this ordered structural arrangement. The water molecule at the centre of the triangular pore of γ-GY forms a hydrogen bond with another water molecule across the membrane helping in permeation. As soon as the water permeates through the membrane, water molecule rotates in such a way that it forms two hydrogen bonds with the water layer and a third hydrogen bond with the next incoming water molecule. Thus, at a time, a single water molecule passes through the membrane resulting in quantized water transport through GYs.69 Hydrogen bond formation between water molecules across the membrane helps in the formation of short single-file water structure, promoting fast water transmission as well as an increase in the concentration of water molecules close to the membrane surface.72 Due to the rotation, there exists no consistency in the orientation of the water dipole while permeating through GY membranes. However, other membrane channels like CNTs retain the dipole orientations during permeation.72
Kou and co-workers carried out MD simulations to study permeation of water through graphyne membranes.72 Water molecules were able to permeate through γ-GY-3 while the smaller pores of γ-GY-2 do not allow water molecules to pass through. Later, Kou et al. demonstrated that the γ-GY-3 membrane shows complete salt rejection for hydrostatic pressure varying from 0 to 350 MPa, while other graphyne analogues with higher pore size show decrease in salt rejection with an increase in pressure.73 It was also observed that an increase in salt concentration decreases water permeability through GYs. With the increase in salt concentration, more water molecules become part of hydration structure and thus prevent random motion of molecules and permeability. Similar to other nanoporous membranes, the passage of ions through GY membranes is dependent on the hydrodynamic radii of the ions. Using MD simulations, Xue et al. have shown that the α-GY-1 and β-GY-1 membranes give 100% salt rejection.74 γ-GY-4 pores allow the permeation of K+ and Na+ ions. Water flux across the membranes for different GYs, α-GY-1, β-GY-1, γ-GY-3 and γ-GY-4 was estimated to be 5.5, 8.11, 11.7 and 14.31 l cm−2 per day per MPa, respectively. Xue and co-workers mapped the distribution characteristics of oxygen density while water molecules passed through the GY membranes (Fig. 4c). The density distributions for α-GY and β-GY are observed to be round in shape, with the highest probability of water density at the centre of the pore. For γ-GY-3 and γ-GY-4, the density distribution map is triangular. γ-GY-3 has one channel for water passage while γ-GY-4 has three apparent channels for water to pass through.74 Energy profiles for the passage of water molecules and ions are different across α-GY-1. Minor valleys present near the graphyne pore help in the formation of the hydrogen-bonded single-file configuration of water molecules on both sides of the membrane, which in turn helps in higher water flux. GYs are found to be more efficient than conventional RO membranes in both water flux as well as salt rejection.
Graphyne membranes possess acetylenic linkages which can get hydrogenated or hydroxylated in the presence of protons or radicals in water. The evaluation of water desalination performance of functionalized GYs is also important. Raju et al. studied the desalination performance of bare as well as hydrogenated GYs.75 Hydrogenation of GYs results in 20–40% reduction in pore area. Water flux through the GY membranes decreases in the following order: γ-GY-4 > hydrogenated γ-GY-4 (H γ-GY-4) > γ-GY-3 > α-GY-1 > γ-GY-2 > hydrogenated γ-GY-3 (H γ-GY-3) > hydrogenated α-GY-1 (H α-GY-1) > hydrogenated γ-GY-2 (H γ-GY-2). Hydrogenated γ-GY-2 does not allow any water passage up to a pressure of 100 MPa. Except for γ-GY-4, all membranes show high salt rejection efficiency (greater than 75%) for pressures up to 2 GPa. γ-GY-2, γ-GY-3, α-GY-1, H α-GY-1 and H γ-GY-3 show a significant water flux as well as salt rejection efficiency that is required for an efficient RO membrane. Hydrogenation of GYs does not change the shape of probability density map of oxygen passing through membranes, but the area of oxygen density distribution is significantly reduced. Similar to graphene nanopores, pore functionalization can introduce selectivity for the passage of the ions. H γ-GY-4 membrane allows selective permeation of negatively charged ions, while hydroxylation of the γ-GY-4 pore results in the passage of positively charged ions and rejection of negatively charged ions.
4 Graphynes for gas separation
The separation of gases is considered to play a vital role in various processes, from industrial-scale to small-scale applications, such as H2 production from syngas, separation of atmospheric gases for medical and industrial utilization, and isotope separation for usage in nuclear power.76–79 In recent times, people are searching for clean and renewable energy due to the depletion of fossil fuels and increased environmental problems. Owing to natural abundance and environmental friendliness, hydrogen has been identified for this purpose. Therefore, it is very essential to separate hydrogen from other undesirable species in an efficient and cost-effective way. Among various processes of gas separation, membrane technology proves to be one of the well accepted approaches due to its several advantages including facile operation, low energy consumption, and easy maintenance.80 Among diverse membranes, graphene-based membranes are of profound interest and have a great potential for gas separation due to their one-atom-thickness, and consequent high efficiency.81,82 Even small atoms like He cannot permeate through graphene sheet.83,84 It is therefore imperative to create pores using experimental techniques85,86 to achieve gas permeability. However, the creation of pores in graphene often leads to defects in the lattice and is not cost effective. Thus, GYs are proposed as alternative materials for gas separation as they possess large natural pores formed by the replacement of some carbon–carbon bonds in graphene with acetylenic linkages.Jiao et al. suggested GDY as the membrane for achieving the separation of H2 from CH4 and CO using density functional theory (DFT) and transition state theory (TST).87 The uniformly distributed pores, with sizes in between the van der Waals diameter of H2 and CH4/CO make GDY a good option for hydrogen sieving from syngas. The selectivity for the separation of binary mixtures of gases (H2/O2, H2/N2, H2/CO2, H2/CH4) was studied by Zhao and co-workers using MD simulations.88 Permeance of H2 was found to increase as the pressure increases from 47 MPa to 1.5 GPa and decrease with increasing pressure after 1.5 GPa. Cranford et al. investigated temperature and force dependence of the hydrogen diffusion and its separation from syngas through GDY pores using MD simulations.89 The energy barrier for H2 molecule is determined to be 0.11 eV. High temperature is needed for the permeation of CO and CH4, whereas H2 can pass through at room temperature. A driving force can enhance the selectivity as well as permeation of H2 up to a limit of 40 pN. Increasing the force above this limit results in permeation of CO and further increase results in CH4 permeation. The introduction of nitrogen atoms in GDY by replacing three sp2 hybridized carbon atoms enhances the hydrogen selectivity over syngas.90 The energy barrier for the permeance of hydrogen decreases whereas the barrier height increases for other gases. These methods for hydrogen purification involve complexity due to chemical modification of the nanopores. Tan et al. investigated whether the selectivity for hydrogen purification can be enhanced by the introduction of positive charge in GDY.91 GDY displays a good performance for the separation of H2 from mixtures of large molecules like CH4. However, the pore structure appears to be too large to accomplish ideal H2 separation from small molecules such as CO and N2. Zhang et al. explored the use of rhombic-GY-1, whose pore size is in between that of γ-GY-1 and GDY for the advancement of selectivity of hydrogen purification from CO2/N2/CH4.92 The separation is not based on the kinetic diameter but according to the physical and chemical interactions between the pores and the penetrating molecules.43
The purification of H2 is also investigated using α-GY and γ-GY. Sang et al. modified the triangular pores in γ-GY-1 by rupturing one-third of acetylenic bonds and replacing the unsatisfied valences using nitrogen and hydrogen atoms to obtain γ-GYN and γ-GYH, respectively.93 The increased pore size which is in between that of γ-GY-1 and γ-GY-2 results in a decrease of the energy barrier for the selective separation of H2 from other gases using both γ-GYN and γ-GYH. It is found that there is no electron density overlap between H2 and γ-GYX (X = N, H). MD and DFT results show that both modified membranes are acceptable candidates for the sieving of H2 even though γ-GYN is the best among them. As temperature increases, the selectivity was found to decrease as shown in Fig. 5a. Alaghemandi et al. used hydrogenated α-GY with and without defects as membranes for molecular sieving of pairs of H2/N2 and H2/CH4.94 Since CH4 molecule is impermeable, the selectivity of H2 is infinity and is independent of temperature, whereas in case of H2/N2, the selectivity of H2 decreases with increase in temperature due to high kinetic energy and permeation probability of N2. In case of hydrogenated α-GY with a defect (which is obtained by the removal of a carbon atom that is surrounded by three carbon atoms), H2 selectivity increases with increase in temperature due to rearrangements of sp3 free arms at the defect position.
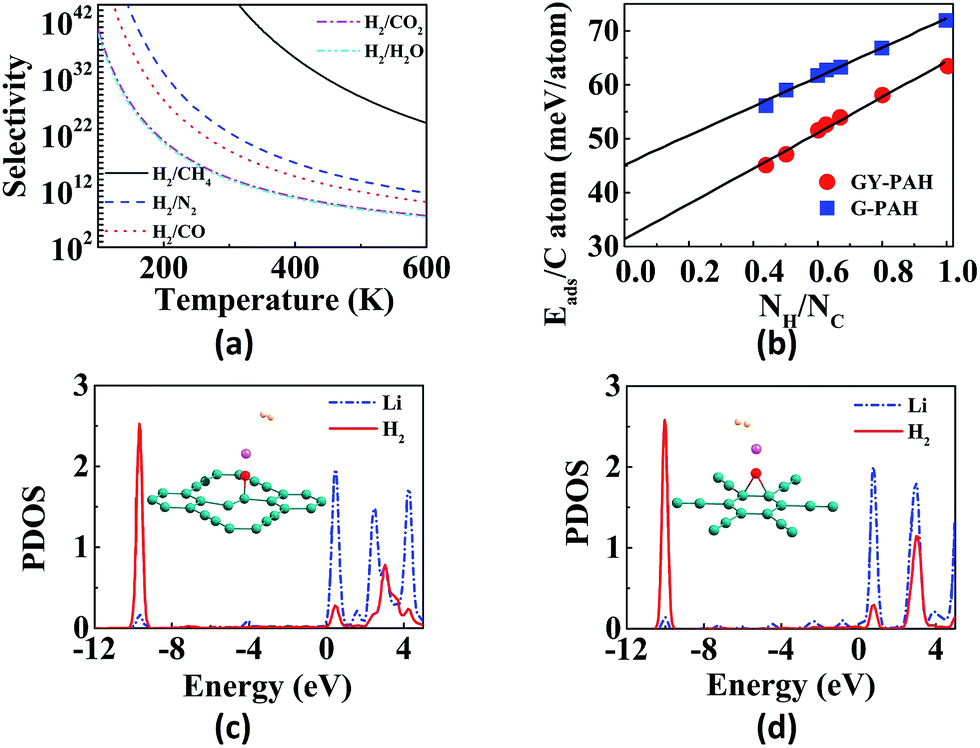 | ||
Fig. 5 Illustrations of separation, adsorption and sensing using GYs: (a) H2 selectivity over H2O, CO2, N2, CO and CH4 as a function of temperature for γ-GYN. (b) Adsorption energy per carbon atom as a function of H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratio of PAH molecules for binding on graphene (G-PAH) and γ-GY-1 (GY-PAH). Partial density of states (PDOS) of Li 2s and H2 1s orbitals suggesting the strong interaction between H2 and Li-decorated oxidised GY at (c) T1 site and (d) B1 site. Part (a) is reproduced from ref. 93 with permission from the Elsevier. Part (b) is reproduced from ref. 110 with permission from the Wiley Periodicals, Inc. Parts (c) and (d) are reproduced from ref. 114 with permission from the AIP Publishing LLC. :C ratio of PAH molecules for binding on graphene (G-PAH) and γ-GY-1 (GY-PAH). Partial density of states (PDOS) of Li 2s and H2 1s orbitals suggesting the strong interaction between H2 and Li-decorated oxidised GY at (c) T1 site and (d) B1 site. Part (a) is reproduced from ref. 93 with permission from the Elsevier. Part (b) is reproduced from ref. 110 with permission from the Wiley Periodicals, Inc. Parts (c) and (d) are reproduced from ref. 114 with permission from the AIP Publishing LLC. | ||
Oxygen purification from toxic gases is essential since it is of great importance in medical field. Meng et al. studied the separation of O2 gas from a mixture of toxic gases using γ-GY-2.95 The oxidation of acetylenic bond does not occur since the process is associated with a large energy barrier. The electron density overlap of atoms of the triangular pore rims of γ-GY-2 and O2 is very less and hence the barrier is easily surmountable. Separation of CO2 and N2 is a challenge as it decreases the energy content of natural gas as well as corrodes the pipelines. Zhao et al. investigated the separation of CO2 and N2 from CH4 using modified GDY obtained by removing one-third of acetylenic linkages and replacing with H, F and O atoms forming GDY-H, GDY-F and GDY-O, respectively.96 GDY-F and GDY-O are the best candidates for the separation of CO2/CH4 and N2/CH4 due to the strong electronic interactions between CO2 and the membranes, which decrease the barrier height for transmission. GDY-O can separate CO2/N2 since the permeance of CO2 is higher by two orders of magnitude. The permeances of CO2 and N2 through GDY-F and GDY-O are higher than in case of nanoporous graphene, making them a good choice for the separation process. Increasing the temperature increases the kinetic energy leading to the permeation of both gas molecules, thereby causing a decrease in selectivity.
Lei et al. explored the adsorption and diffusion of H2S and CH4 through multilayer γ-GY-N (N = 1–3) (MGN-N) at various temperatures and pressures.97 The adsorption of H2S is much higher than that of CH4 since its polarity results in strong interactions between the adsorbent and the adsorbate species. Under low pressure, MGN-1 exhibits the best performance for the adsorption of both the species, whereas at high pressure, adsorption of H2S is higher in case of MGN-3 due to the formation of molecular clusters. At low pressure, H2S selectivity increases but at higher values of pressure, the selectivity decreases. Increasing temperature decreases the selectivity of H2S due to the increased permeance of CH4.
3He is known to be a rare and highly precious gas, obtained as a byproduct during the radioactive decay of tritium. It has high demand in low-temperature research institutes and cryogenic industries.98 It is heavily used in dilution refrigerators. 3He also has applications in large neutron scattering facilities.99 Recently, investigation of the quantum effects during the transmission of 3He and 4He through a single GDY sheet has demonstrated GDY to be a potential He isotope separator.100 A comparison between the zero-point energy and tunneling effects depicts that both effects are highly relevant at low temperatures (20–30 K). The zero-point energy of 3He is higher than that of 4He, allowing the faster transmission of 4He. At low temperature, tunneling effect is found to be slightly higher than the contribution from zero-point energy which favours the selective permeation of the lighter isotope. Bartolomei et al. employed quantum dynamics simulations for probing the efficiency of GDY and 2D-polyphenylene towards achieving He separation from natural gas.101 Since the pore size of GDY is larger than the van der Waals diameter of He, it has better efficiency for the separation of He. Improved Lennard-Jones potential is used to describe the interactions between He atoms and the membrane. As the temperature increases, the selectivity for He/Ne, Ne/CH4 and He/CH4 decreases.
5 Graphynes for molecular adsorption and gas sensing
Gas sensors have a wide range of applications such as monitoring air pollution, medical diagnosis, leakage detection etc.102–104 The efficiency of gas sensors depends on selectivity as well as sensitivity of the electronic properties of the substrates. Two-dimensional nanoporous materials such as graphene and GYs can be used for gas detection (Fig. 5). Due to uniformly distributed pores and high sensitivity, GYs have the potential to be used in gas sensing technology. The adsorption of gas molecules leads to charge transfer, which causes a change in conductance of the system. The sensitivity as well as the adsorption propensity can further be enhanced by metal decoration, defects and doping.Pristine GYs are of great interest in the detection of various gases due to their unique properties. Formaldehyde is a colourless gas which causes environmental pollution and is even considered to be carcinogenic. Majidi et al. investigated whether GYs can be used for the detection of HCHO using DFT.105 The small binding energy and large equilibrium distance imply the physisorption of HCHO on GYs (α, β and γ-GYs). To increase the efficiency of detection of HCHO, Chen et al. used Sc and Ti decorated GDY.106 GDY, which is a metal, transforms into an n-type semiconductor upon the approach of HCHO. The binding energy for adsorption is higher in case of pristine GDY compared to pristine γ-GY-1 due to increased number of π electrons. It is found that decoration with Sc and Ti enhances the binding energy, with Sc resulting in higher binding strength. The change in the electronic property of γ-GY-1 upon adsorption of H2O2 was studied by Majidi and co-workers.107 The preferential orientation of H2O2 is the configuration in which the O–O bond is perpendicular to the sheet and it gets adsorbed at the triangular pore. Increasing the number of H2O2 molecules decreases the band gap and increases the conductivity and hence γ-GY-1 transforms to an n-type semiconductor. Deb et al. studied the adsorption of boron halides (BF3, BCl3, BI3) on γ-GY and monitored the change in electronic properties on adsorption.108 It was found that the order of binding strength is BF3 < BCl3 < BI3 for the adsorption process. The GYs transform to n-type semiconductors on the adsorption of BCl3 and BI3, whereas in case of BF3 adsorption, they transform as p-type semiconductors. Maximum electron transfer occurs in case of BI3, indicating that GY senses BI3 much better than the other two halides. The dipole moment of γ-GY is high in the case of adsorption of BI3 due to the strong electron transfer. Majidi et al. studied the electronic properties and the binding energies for the adsorption of tetracyanoethylene molecules on GYs.109 The small binding energy and the large adsorption distance imply physisorption of molecules on the sheet. α and β-GYs which are semi-metals transform to semiconductors due to the adsorption of tetracyanoethylene that accepts electrons from the sheets.
PAHs (polycyclic aromatic hydrocarbons) are formed during the incomplete ignition of organic molecules and many of them are considered to be toxic. Arriagada et al. investigated the adsorption of PAHs on graphene and GYs.110 The binding energies for the adsorption of PAHs on GYs are low due to the porous structures and decreased π system of GYs compared to graphene, as shown in Fig. 5b. Smaller PAHs prefer stacked orientation due to increased π–π interactions, whereas for larger PAHs, the preferred orientation is bridge or rotated. The electron transfer from PAHs to GYs decreases the band gap and hence increases the conductivity. Ozmaian et al. studied the diffusion and controlling motion of C60 on γ-GY-N (N = 1–5) sheets.111 γ-GY-1 has high binding energy due to high carbon atom density. The diffusion of C60 on the sheets can be controlled by using various GYs. In case of γ-GY-1, mobile assemblies are formed whereas stable assemblies are formed in case of γ-GY-2 and γ-GY-5.
Zhang et al. investigated the adsorption of gas molecules on γ-GY-1.112 It is found that CO, NH3 and SO2 bind weakly, whereas H2S, NO2 and NO bind strongly through charge transfer and change γ-GY-1 from semiconductor to metal. The strong binding of NO and NO2 indicates that γ-GY-1 can be used for their detection. The competitive adsorption of CO2 and H2 on γ-GY-1 was studied by Kwon and co-workers using DFT.113 The gas molecules can be adsorbed on three sites (i) triangular (ii) bridge and (iii) hexagonal. Considering the adsorption properties, both CO2 and H2 preferably adsorb on the triangular pores (hollow sites), however due to the electrostatic field caused by atomic charges on the sheet, the preference of binding is for CO2.
The effect of adsorption of O2 on the electronic properties of GYs is theoretically studied by Kang and co-workers115 and it was observed that the binding energy of O2 with various GYs is in the order: α-GY > β-GY > γ-GY. Adsorption of oxygen atoms over α and β-GYs results in the formation of oxides of GYs opening up the band gap in these materials. Omidvar et al. investigated the enhancement of CO sensing ability of γ-GY upon metal decoration.116 Pristine GY is not suitable for the sensing of CO due to the weak interaction between the sheet and CO, resulting in only slight variation in the electronic properties. However, there is considerable variation in the band gap of the system upon the adsorption of CO in case of metal-decorated GY. Beheshtian et al. explored the change in electronic properties upon the adsorption of HCN on pristine γ-GY-1 and Si-doped γ-GY-1.117 Upon HCN adsorption on pristine GY, the decrease in band gap is less, implying weak adsorption thereby making γ-GY-1 unsuitable for HCN detection. The adsorption of HCN on Si-doped GY decreases the band gap due to the donation of electrons from the nitrogen atom to the LUMO of Si and hence increases the conductivity. The electronic properties of HCN/Si-doped γ-GY show that doped GYs can be used for HCN detection. Phosgene is a pale yellow poisonous gas used as a chemical weapon during World War II. Felegari et al. explored the effect of adsorption of phosgene on boron, nitrogen and Si-doped γ-GYs.118 Phosgene molecule binds to electron deficient Si sites in such a way that the electron-rich oxygen atoms face the GY sheets. The charge transfer from phosgene to doped GY sheet increases its conductivity.
Peyghan et al. investigated the effect of doping γ-GYs with Ni and Si as the dopants on gas sensing.119 The resulting GYs were used to study the variation in electronic properties upon the adsorption of NH3. In the case of pristine γ-GYs, the adsorption was very weak, which indicates small variation in electronic properties. Doped GYs (designated as MGY-N where M = Ni, Si; N = 1, 2) are obtained by replacing carbon atoms from hexagonal and triangular pores of γ-GYs with Ni/Si, respectively. The density of states (DOS) plot indicates that doping enhances the adsorption strength of NH3 with maximum enhancement reported for Si-doped GYs. Lu et al. studied the effect of doping on the gas sensing ability of pristine γ-GY-1 using DFT calculations.120 Except O2, all other gas molecules (CO, CH4, CO2, NH3 and NO) undergo physisorption over γ-GY-1, causing negligible change in band gap. Thus, γ-GY-1 cannot be used as a gas sensor for other gases. The Mn atom can get adsorbed at the triangular pore of γ-GY-1. The decrease in the band gap, due to the introduction of Mn results in enhancement of the gas sensing ability. The selectivity as well as sensitivity are enhanced by the doping of Mn over γ-GY, since the gas molecules are adsorbed via chemisorption.
6 Graphynes for energy storage
6.1 Hydrogen storage
Hydrogen is the cleanest fuel and is considered to be the future replacement for natural fuels. Currently, storing of this potential energy source is a challenge due to its low critical temperature.121 Many research initiatives are directed towards exploring an efficient way for the storage and transportation of hydrogen. The techniques for the storage include liquefaction, compression, chemical hydrides and physisorption.121,122 The liquefaction as well as compression do not guarantee safety since high pressure is required, whereas use of appropriate adsorption materials assure storage at low pressures.123 GYs, which are 2D nanoporous materials with uniform pores and high pore densities can be used for hydrogen storage. Zhang et al. investigated the hydrogen storage capacity of bilayer γ-GY-2 using DFT calculations.129 Bilayer γ-GY-2 has overlapping triangular pores that can trap H2 molecules. The H2 molecules, upon permeation through the first triangular pore get adsorbed in between the two layers in bilayers, resulting in hydrogen storage. The small barrier height for desorption can be easily surmounted, ensuring the unloading of trapped hydrogen molecules. The limited hydrogen storage capacity of pristine GYs can be enhanced through metal decoration. Metal-decorated GYs are widely reported, but the challenge in fabrication is the higher cohesive energies of metals compared to the binding energies of metals with GYs.Li-decorated GYs have been used as storage media for hydrogen in numerous studies. A comparative study of the hydrogen storage capacities of γ-GY-1 and γ-GY-2 was performed using the first-principles calculations by Srinivasu and co-workers.124 The first Li atom adsorbed preferably occupies triangular pores by interacting with the three acetylenic bonds and the binding strength is higher for γ-GY-1 compared to γ-GY-2. As the number of hydrogen molecules adsorbed per Li increases, interaction energy decreases as shown in Fig. 6a. The maximum number of H2 molecules adsorbed per cationic Li site is three. Even though the adsorption energy is higher in case of γ-GY-2 than γ-GY-1, the adsorption capacity is in reverse order since the number of carbon atoms per unit cell is higher for γ-GY-2. First-principles study of the hydrogen storage in Li-decorated γ-GY-1 was also performed by Guo and co-workers.130 The γ-GY-1 system is found to have high binding energy with Li due to the low ionization potential of Li which donates its s electron to the GY sheet. The hydrogen storage capacity is found to be 18.6 wt%, and each Li adsorbs four H2 molecules. The interaction between H2 and sheet is due to the electric field induced by the ionic Li. The adsorption of the fourth H2 molecule is due to the charge transfer through the polarized H2 molecules that are already adsorbed. In a study carried out by Zhang et al., it is found that double-sided Li-decorated GY adsorbs seven molecules per Li atom with a capacity of 15.15 wt%, wherein the adsorption is attributed to the polarization interaction mechanism.131 Lu and co-workers used Li-decorated 6,6,12-GY, which consists of rhombus-like acetylenic (SA) rings apart from triangular acetylenic (TA) rings for the hydrogen storage.132 Li occupies the centre of SA rings since number of sp hybridized carbons is more in SA rings. The adsorption of hydrogen is stronger at the TA site even though the electron donation of Li to 6,6,12-GY is facile at the SA site. It is found that storage capacity increases to 19.3 wt% upon Li decoration, where the enhanced adsorption is due to the electric field produced by the polarized H2 molecules. Kumar et al. used Li-decorated GY networks for hydrogen storage.133 Each GY linker binds two Li atoms forming Li–GY complexes. It was found that each Li atom adsorbs three H2 molecules weakly through the polarisation of Li forming metal-GY framework-Li8-24H2 with a storage capacity of 6.4 wt%. The adsorption energy decreases with increasing number of H2. Since adsorption of H2 is weak, increasing the temperature leads to desorption. Yan et al. used Li-decorated oxidised GY (obtained by oxidation of the acetylenic bond and binding Li with the oxygen atom) as a hydrogen sensor.114 The oxygen atom forms (i) epoxy bond with two C atoms of hexagonal pore (B1 site) or triangular pore (B2 site), (ii) bond with one C atom of triangular pore (T1 site) or hexagonal pore (T2 site). It is found that Li atom binds strongly to T1 site followed by B1 site. Partial density of states (PDOS) indicates the overlap of the 2s orbital of Li with the σ bond of H2 in both sites as shown in Fig. 5c and d. Each Li atom adsorbs four or five H2 molecules with a storage capacity of 12.03 wt%. Lu et al. investigated the storage capacity of metal-decorated boron-doped γ-GY.134 The storage capacity is low for pristine GYs due to weak interaction between H2 and sheet, which can be enhanced by the adsorption of Li on GY sheets. Since the cohesive energy of Li is higher than the interaction energy, clustering occurs resulting in low adsorption. This can be solved by boron doping of the γ-GY sheet which transforms it to a p-type semiconductor. The binding of H2 with the hexagonal pore is favoured since at the triangular pore, strong charge transfer from Li to the GY sheet occurs, resulting in low adsorption energy.
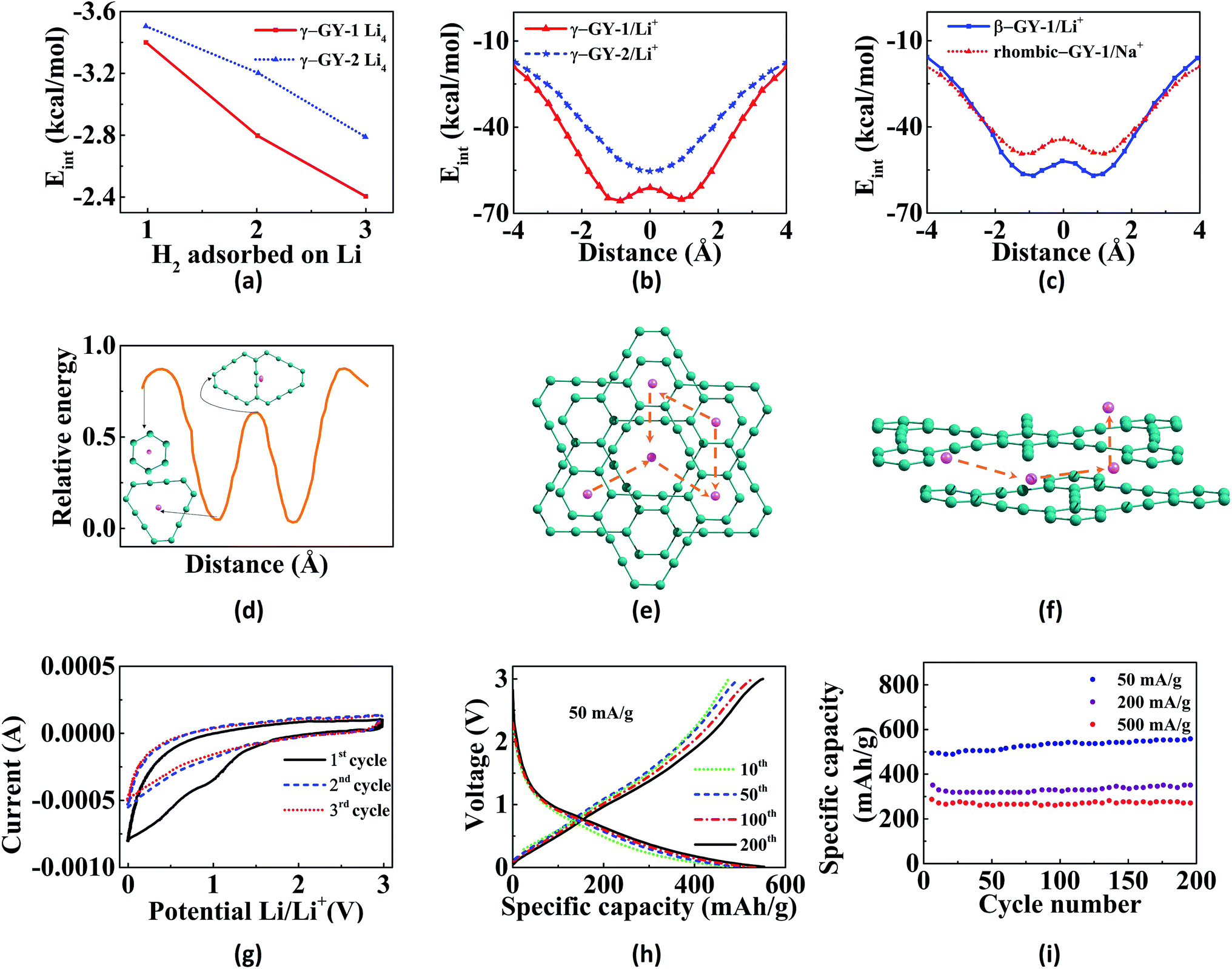 | ||
| Fig. 6 Applications of GYs in energy storage: (a) variation in interaction energy with increase in number of H2 adsorbed per Li. Potential energy scans for the passage of (b) Li+ through γ-GY-1 and γ-GY-2 and (c) Li+ and Na+ through β-GY-1 (triangular pore) and rhombic-GY-1, respectively. (d) Diffusion pathway of Li atom as it moves through different adsorption sites on γ-GY-1. Schematics of the (e) in-plane and (f) out-of-plane diffusion of Li through various sites of bulk γ-GY-1. (g) Cyclic voltammetric curves at initial three cycles with scan rate as 1 mV s−1, (h) galvanostatic charge/discharge profiles with current density as 50 mA g−1 and (i) cycle performance of lithium-ion batteries with GDY as the anode. Part (a) is reproduced from ref. 124, parts (b) and (c) are reproduced from ref. 125 and parts (d–f) are reproduced from ref. 126 with permissions from the American Chemical Society. Part (g) is reproduced from ref. 127 with permission from the Elsevier. Parts (h) and (i) are reproduced from ref. 128 with permission from the Royal Society of Chemistry. | ||
Apart from Li, other alkali metals and alkaline earth metals are also used for hydrogen storage. Liu et al. used Na-decorated single-sided and double-sided γ-GY and BN-yne (GY doped with BN) for the storage of hydrogen by DFT calculations.138 Decorating GY with Na leads to the transfer of s electrons from Na to the sheets. In case of Na-decorated single-sided and double-sided GY, each Na atom adsorbs three hydrogen molecules and each of them has a capacity of 3.49 and 5.98 wt%, respectively. The BN-yne decorated with Na on both sides also adsorbs three hydrogens per Na atom and each of them has a capacity of 5.48 wt%. Hwang et al. studied the storage of H2 molecules in Ca-decorated α, β, γ-GYs.139 It was found that Ca occupies hexagonal pores in α and triangular pores in β and γ-GYs. As the number of H2 increases, adsorption energy decreases in all these cases. They found that transition metals undergo clustering on graphyne sheet, implying that they would be poor choice for metal decoration. Li et al. also found that triangular pores are stable adsorption sites for Ca.140 The hybridization of the σ states of H2 and 3d orbitals of Ca indicates weak adsorption. In case of Ca-decorated single-sided and double sided γ-GY-1, six H2 molecules can be trapped per Ca atom. The storage capacity of Ca-decorated double-sided graphyne was found to be 9.6 wt%. Guo et al. investigated the hydrogen storage capacity of different metal-decorated GYs.141 The metal adsorption is found to be preferable at triangular pores of γ-GYs. The metal donates s electrons to GY sheet and GY back-donates electrons to 3d orbitals of metal. Sc and Ti bind strongly resulting in higher adsorption strength when compared to other metal-decorated GYs. The hydrogen storage capacity is found to be higher for Li and Ca-decorated GYs. GNTs are also used to study the trapping of H2. First-principles calculations were employed to study the storage of H2 in Ca-decorated GNTs.142 Since the binding energy of Ca with GNT is higher than the cohesive energy of Ca and the Ca–Ca distance is large, clustering of Ca on GNTs is ruled out. The hydrogen molecules bind to Ca due to the polarization and hybridization between the σ state of H2 and 3d orbitals of Ca. The hydrogen storage capacity was found to be 7.47 wt%. Due to curvature effect, the metal atoms bind strongly with GNTs preventing clustering.143
The application of external field prevents metal aggregation and enhances the hydrogen storage capacity. Employing DFT calculations, Liu et al. explored the enhancement of hydrogen storage of Mg-decorated γ-GY in presence of applied electric field.144 The binding energy exceeds cohesive energy on applying an external electric field, which prevents the clustering of Mg. The polarization and hybridization between H2 and Mg lead to strong adsorption. Zhang et al. studied the hydrogen storage in Ti-decorated GY in presence of external electric field of varying strengths by first-principles calculations.145 The Ti atom donates electron to the γ-GY sheet, which in turn back-donates electrons to the 3d orbital of Ti. The adsorption of H2 is due to electrostatic interactions between the sheet and H2 molecules, which is the result of polarization effect caused by the applied electric field.
6.2 Lithium-ion batteries
Graphite rods and graphene sheets are extensively explored materials as anodes for lithium-ion batteries. Lithium ions can get intercalated in between the sheets in graphite. Graphyne sheets containing both sp2 and sp hybridized carbons can potentially adsorb more metal atoms. The porous nature of GYs provides additional tools to finetune their properties thereby making them better anodic materials in batteries. γ-GY has two types of adsorption sites for metal atoms. Site with the smaller pore (site A) is made up of six-membered phenyl ring and the site with the larger pore (site B) is made up of twelve-membered ring composed of both sp2 and sp hybridized carbons. Compared to site A, lithium is adsorbed strongly over site B. Binding of the first two lithium atoms on a unit cell of γ-GY-1 occurs in-plane to the carbon surface and they occupy the two B sites.124 Further addition of metal atoms results in occupation of positions above and below the C6 rings. Binding energies for first lithium atom are estimated to be 64.5 kcal mol−1 and 61.9 kcal mol−1 for γ-GY-1 and γ-GY-2, respectively, for binding at site B.124 γ-GY-1 nanopores can only adsorb one atom in site B, while γ-GY-2 pores can adsorb up to three lithium atoms.146In graphite and multilayer graphenes, diffusion of Li is confined to the interlayer spacing.147 However, both in-plane and out-of-plane diffusion of ions are allowed in intrinsically nanoporous graphyne membranes (Fig. 6).126,146 γ-GY-1 and γ-GY-2 provide high in-plane mobility and the barriers for hopping between adsorption sites are less than 20 kcal mol−1 as shown in Fig. 6d and e. The barrier for the out-of-plane diffusion of Li ion through the C6 ring is estimated to be 190 kcal mol−1. Thus, out-of-plane diffusion through C6 ring is not possible under normal conditions. The larger triangular pore in γ-GY-1 provides a barrier of approximately 4 kcal mol−1, allowing fast diffusion (Fig. 6). Higher mobility of metal ions helps in achieving higher current density as well as complete reversibility during multiple charging–discharging cycles. Chandra Shekar et al. studied the rattling motion or out-of-plane mobility of different alkali and alkaline-earth metal ions through various GY sheets.125,148 The binding energies, adsorption heights and barrier heights for passage through the pores of various GYs for lithium and sodium are provided in Table 2. Potential energy curves for the out-of-plane rattling motion of metal ions through various GYs are shown in Fig. 6b and c. Lower diffusion barriers for metal ions through GYs help in fast charging of battery. However, in the case of discharging, the process becomes difficult energetically and requires application of a voltage across anode and cathode.126 Adsorption of lithium atoms on GY is followed by charge transfer from the metal to the sheet. These electrons occupy the conduction band and result in electrostatic interaction between metal adatoms and GY sheet. Zhang et al. attribute the site preference to the charge transfer from Li to the larger triangular pore since the conduction band lies along this C12 ring.126
| Graphynes | Metal ions | Binding energy (kcal mol−1) | Adsorption height (Å) | Barrier height (kcal mol−1) |
|---|---|---|---|---|
| γ-GY-1 | Li+ | 64.22 | 1.10 | 3.54 |
| Na+ | 46.36 | 1.70 | 35.60 | |
| γ-GY-2 | Li+ | 58.01 | 0.0 | 0.0 |
| Na+ | 50.14 | 0.0 | 0.0 | |
| α-GY-1 | Li+ | 49.42 | 0.0 | 0.0 |
| Na+ | 40.10 | 0.0 | 0.0 | |
| β-GY-1 (hexagonal pore) | Li+ | 53.62 | 0.0 | 0.0 |
| Na+ | 45.34 | 0.0 | 0.0 | |
| Rhombic-GY-1 | Li+ | 62.57 | 0.0 | 0.0 |
| Na+ | 45.71 | 1.01 | 1.96 |
Adsorption sites of lithium on α-GY are in-plane and slightly off-centre towards the edge of hexagon.135 Lithium adsorption over α-GY results in in-plane distortion of carbon atoms in the sheet. It is predicted that lithium can be dispersed on α-GY-2 to give a maximum composition of up to C6Li11,18 and corresponding specific capacity is 4259 mA h g−1. One of the important factors affecting the applicability of GYs as anode materials is the structural stability of sheets during intercalation and de-intercalation. Calculated cohesive energies for α-GY and γ-GY are 189.1 and 196 kcal mol−1, respectively, while the cohesive energy for graphite is 209.9 kcal mol−1.136 Detailed study of structural stability and phase transformation (from GY to graphite) is required to address this issue.136 Theoretical predictions for boron-doped γ-GY were made by Lu et al. for the storage of lithium and dihydrogen.134 Calculations were carried out using Grimme DFT-D2 dispersion correction method. Up to three boron atoms were doped at the phenyl ring of γ-GY (1BG, 2BG, 3BG respectively) and the adsorption energy of lithium adatom increased with the number of borons. The lithiation potentials (Li/Li+) were 3.50, 2.66, 4.00 and 2.97 V in 1Li@1BG (one lithium adatom adsorbed on 1BG), 6Li@1BG, 1Li@3BG and 6Li@3BG, respectively, while lithiation potentials in graphite and 4Li–GY are 0.5 and 1.7 V, respectively. Specific capacity of lithium battery is predicted to be 1125 V for 6Li@1BG and increases nominally on further doping. The theoretically predicted maximum energy storage capacities and lithiation potentials for different types of graphynes as anodic material have been summarised in Table 3.
| System | Composition | Specific capacity (mA h g−1) | Volumetric capacity (mA h cm−3) | Lithiation potential (V) |
|---|---|---|---|---|
| a Values are taken from ref. 124.b Values are taken from ref. 135.c Values are taken from ref. 136.d Values are taken from ref. 137.e Values are taken from ref. 134. | ||||
| Graphite | C6Li | 372a,b | 818b | ∼0.5c |
| γ-GY-1 | C3Li | 623.4a (1117c) | 1589b | 2.8–1.7a |
| γ-GY-2 | C2,25Li | 788.4a | — | 2.7–1.9a |
| α-GY-1 | C6Li3 | 1117c | 1364c | — |
| α-GY-2 | C6Li7,31 | 2719c | 2032c | 0.85–0.35c |
| Rhombic-GY-2 | C5Li6 | 2680d | — | 0.75–0.23d |
| Boron-doped γ-GY-1 (3B) | — | 1144e | — | 4.0–2.97e |
| Boron-doped γ-GY-1 (1B) | — | 1125e | — | 3.5–2.66e |
Xu et al. have proposed the usage of GYs as anode materials in sodium-ion battery.149 Storage capacity of γ-GY-1 and γ-GY-2 was predicted to be NaC4 and NaC3, respectively. One sodium can bind with the hexagonal C12 ring in γ-GY-1 sheets, while three atoms can bind in the C18 ring of γ-GY-2. However, the binding of three atoms results in blockage of out-of-plane diffusion through the pore. Sodium atom can also bind to the γ-GY sheets by positioning above the C6 ring. However, combined experimental and theoretical study carried out by Zhang et al. has shown that sodium ions bind only to the C18 ring in well-stacked γ-GY-2 sheets except at defective sites in stacking.150 Observed capacity of GDY-based sodium-ion battery is 261 mA h g−1 at 50 mA g−1 current density.
Experimental studies on the lithium-ion batteries have proved the superior quality of γ-GY-2 as an anode material.151,152 Wang et al. have synthesised both lithium-ion batteries and capacitors with excellent cyclic stability and good capacitance or energy storage performance.127 They observed reversible capacities of 908 mA h g−1 and 526 mA h g−1 at 0.05 A g−1 and 1 A g−1 current densities, respectively. Specific capacitance observed was 208 F A g−1 at 1 A g−1 current density. Cyclic voltammetric curves (Fig. 6g) do not show any distinguishable reduction and oxidation peaks, except in the first cycle. Low initial coulombic efficiency and irreversibility in the first cathodic scan as shown in cyclic voltammogram trace is due to formation of a stable solid electrolyte interface film at the electrode.127,128,152 Due to the unique porous nature of GYs, the charging and discharging cycles are almost reversible after few initial cycles.128 As depicted in Fig. 6h, the charge and discharge capacities in Li-ion batteries with γ-GY-2 anodes remain almost constant after 10th cycle. Similarly, a high specific capacity is retained over many cycles reversibly (Fig. 6i). γ-GY-2 based battery achieved an energy density of around 100 W h kg−1 at both low and high power densities.152 The battery also showed 94.7% retention of energy density after 1000 cycles. Hydrogen-substituted γ-GY-2 is fabricated using coupling of triethynylbenzene. The flexible electrode based on hydrogen-substituted GDY is reported to have a specific capacity of 650 mA h g−1 after 100 cycles. Nitrogen-doped γ-GY-2 (N-GDY) based lithium-ion batteries were synthesised by Yang et al. and Zhang and co-workers.153,154 Nitrogen doping was carried out by Zhang et al. using the prolonged exposure of ammonia at 873 K. While, Yang et al. fabricated N-GDY using derivatized pyrimidine and pyridine monomers. Anodes based on N-GDY showed a better electrochemical performance compared to the pristine ones. Yang and co-workers observed the specific capacities of batteries to be 1168 and 1165 mA h g−1, respectively for anodes with pyridine and pyrimidine-based GDYs at a current density of 100 mA g−1.
7 Graphynes for catalysis
We now review some of the applications of GYs in the area of catalysis. First-principles calculations have been performed on various pristine and doped GYs to design optimal catalysts for reactions such as oxygen reduction reaction, oxygen evolution reaction, CO oxidation etc. Experiments along these lines are only beginning to emerge. The rate of an oxygen reduction reaction (ORR) in fuel cells is usually improved by using Pt-based catalysts. However, there are several limitations for Pt catalysts and hence other alternatives are widely being researched.155,156 Out of these, carbon-based catalysts are of greater interest because of their ease of availability. Doped graphene and CNTs have shown good ORR electrocatalytic activity.157–160 The improved catalytic activity is due to the positive charges on the doped systems. Since pristine graphene has no such positive charges, it does not show any catalytic activity. In contrast to graphene, GYs have uneven charge distribution, making them an excellent choice for the catalysis of ORR.161 GYs are expected to show better catalytic activity than doped graphene and CNTs because they have a large number of positively charged sites for catalysis. Using DFT calculations, Wu et al. reported the electrocatalytic activity of α-GY-1 for ORRs in acidic fuel cells.161 In an acidic fuel cell, there are two possible pathways for the first electron transfer in the ORR process. One is the adsorption of OOH+ on α-GY-1 and the other is the direct adsorption of O2 on α-GY-1, which then reacts with an H+ to form OOH+. Adsorption of OOH+ is a more favourable process than that of O2. The next H+ may move to the O atom bound to the C atom (path I) or the oxygen bound to the H atom (path II). Fig. 7a shows the adsorption energy differences between the reactants and the products for each step and as we can see from the figure, path I is more favourable compared to path II. Thus, α-GY-1 proves to be an excellent catalyst for the ORR process that happens via a 4-electron pathway, which includes an O–O bond breakage and formation of a H2O molecule.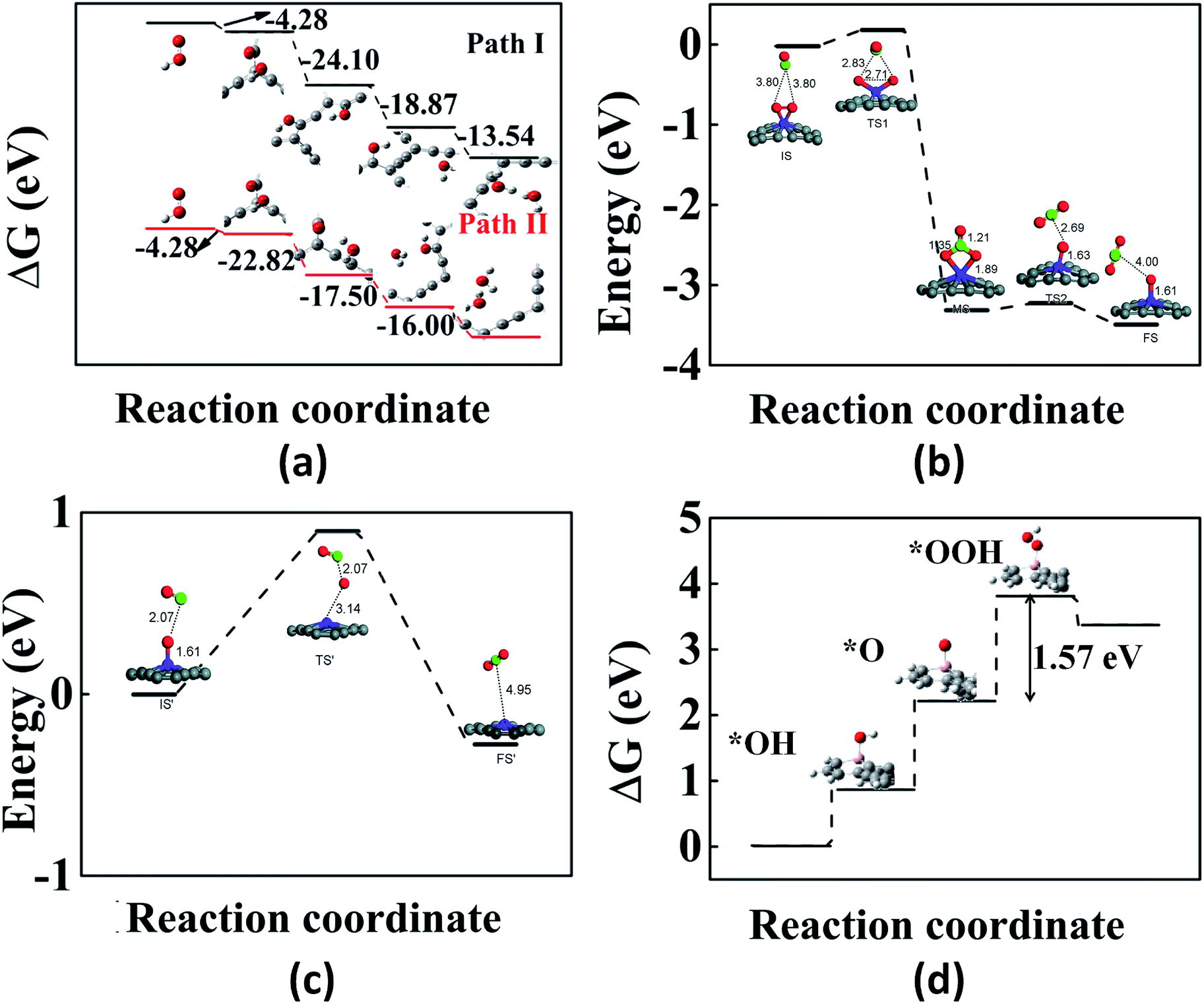 | ||
| Fig. 7 Illustrations of GY-catalyzed reactions: (a) energy differences between reaction intermediates (via path I and path II) of the α-GY-catalyzed ORR. (b and c) The first and second steps of CO oxidation catalyzed by γ-GY-supported Fe atom. (d) Free energy plot for the OER on B-doped γ-GY. Part (a) is reproduced from ref. 161 with permission from the American Chemical Society. Parts (b) and (c) are reproduced from ref. 162 with permissions from the Royal Society of Chemistry. Part (d) is reproduced from ref. 163 with permission from the Elsevier. | ||
Incorporation of transition metals (TM) on GYs changes the electronic structure and magnetic properties in such a way that the ORR catalytic activity is improved. Srinivasu et al. studied Fe, Co and Ni decorated GYs as catalysts for ORR.164 DFT studies showed that the most favourable position for metal binding is above the triangular pores of γ-GY-1. γ-GY–TM systems have higher binding energies than the corresponding graphene–TM systems due to the presence of sp hybridized carbon atoms. The most preferred site for O2 adsorption is just above the TM. O2 adsorbs via the side-on mode in γ-GY–Fe case and via the end-on mode for Ni and Co systems. The order of reactivity of the metals is given by Fe > Co > Ni, indicating that γ-GY–Fe is the best ORR catalyst among these systems. In acid medium, γ-GY–TM systems proceed through a 4-electron process and the γ-GY–Co system shows a higher over-potential than γ-GY–Fe, making the γ-GY–Fe system a better catalyst. In alkaline medium, γ-GY–Co proceeds through a 2-electron mechanism, whereas the γ-GY–Fe system proceeds via a 4-electron mechanism. Similar to acid medium, γ-GY–Fe is a better catalyst than γ-GY–Co in alkaline medium too. From the computed free energy profiles, it was concluded that γ-GY–Fe shows enhanced catalytic efficiency in the alkaline medium compared to the acid medium.
Metal-free ORR catalysts with good stability and tolerance to CO and methanol are of growing interest and the heteroatom-doped GYs (excluding transition metal atoms as the heteroatoms) seem to exhibit these desired properties due to the electronegativity differences between C atoms and the heteroatoms. Kong et al. investigated the effect of B doping on γ-GY-1 for its use as a catalyst.165 B doping changes the electronic structure of γ-GY-1 and introduces positive charge density on the sheet which enhances the catalytic ability. Using first-principles study, Chen et al. studied the mechanism of ORR catalysis by B, N single-doped α-GY-1, γ-GY-1 and co-doped α-GY-1.166 B and N single-doped α-GY-1 showed low catalytic efficiency due to high over-potential in the reduction steps, whereas the single-doped γ-GY-1 showed much better catalytic efficiency. Thus, the configuration of the doped GY also plays an important role in deciding the catalytic ability. Initially, the authors studied three co-doped systems – α-B1N2G, α-B1N3G, α-B1N4G and except for α-B1N2G, all steps for all the systems had positive reversible potential. These systems did not show much improvement in catalytic activity and therefore α-B1(N3)4G with increased N doping was studied. It showed a higher efficiency than α-B1N4G due to improved onset potential. N-doped β-GY-1 could also efficiently catalyze ORR via a 4-electron process.167 The β-GY-1 with an sp C replaced by N showed higher efficiency than the GY with an sp2 C replaced by N because of a higher positive charge created in the former case. Das and co-workers compared the influence of B doping on α-GY, β-GY, γ-GY, δ-GY, 6,6,12-GY, and rhombic-GY using first-principles calculations.168 All systems show a 4-electron pathway for ORR, but only the Rsp2-rhombic system (Rsp2 represents replacing sp2 C at the corner of the rectangular or square ring of rhombic-GY with the dopant) showed a monotonically exothermic ORR pathway. All other systems have an endothermic step in between. Thus, it is the B-doped rhombic-GY that is of greater interest for future ORR applications. N-doped GDY shows a catalytic efficiency comparable to the Pt/C system with improved stability and tolerance to methanol.169 The imine N enhanced the positive effect on the sheet to a larger extent than the pyridinic N atom. Recently, Lv et al. treated the N-doped GDY to high temperature to get porous N-doped GDY with superior catalytic activity.170 N-GDY was further calcined in an ammonia atmosphere to increase the nitrogen content. The N,N′-GDY thus prepared showed an even higher catalytic activity. The effect of co-doping GDY with multi-elements for improving the ORR rate was investigated by Zhang and co-workers.171 The GDY was co-doped with N and S, N and B, N and F successfully and out of these, N and F co-doped GDY showed the best catalytic performance with good stability and improved CO and methanol tolerance.
Another interesting and emerging class of carbon-based catalysts for ORR is GY nanotubes. Recently, Chen studied the effect of the doped γ-GNT on ORR catalytic efficiency.172 The GNTs were doped with N, B, P and S atoms and their catalytic activities were studied using DFT. S-GNT is a poor ORR catalyst and the efficiencies of the other three catalysts follow the order: N-GNT > B-GNT > P-GNT. The O2 molecule is adsorbed in the end-on mode for N and B-GNTs, whereas ORR is initiated by the bridge adsorption of O2 in P-GNT.
GDYs also act as metal-free catalysts for CO oxidation.173 It is again the positively charged C atoms that facilitate the CO oxidation via the Eley–Rideal (ER) mechanism. The adsorption energy of O2 is higher than that of CO and even in a mixture of CO and O2, the GDY system will be majorly covered by O2 because of its higher binding energy. Initially, the O2 adsorbs in a parallel fashion to the GDY (side-on mode). Next, there is an O–O bond cleavage with a carbonate-like intermediate formation which is finally followed by a CO2 formation. CO oxidation is also catalyzed by GY-supported single Fe atom.162 The non-uniform charge distribution favours the adsorption of single Fe atom on γ-GY-1 and results in a better distribution of the metal catalyst unlike in the case of pristine graphene, where the metal atoms move to the edges. The metal atom shows preferential adsorption over the hollow site above the centre of the acetylenic ring. As in the previous case, O2 adsorption is preferred over CO and the CO oxidation proceeds via the ER mechanism as shown in Fig. 7b and c.
In a similar manner, GDY acts as an excellent support for Co nanoparticles to catalyze oxygen evolution reaction (OER).174 A support material like GY helps in preventing the decrease in catalytic efficiency caused by the aggregation of the nanoparticles. Li et al. successfully synthesised a 3D Cu@GDY/Co electrode that shows good OER catalytic activity. The bare Cu foam or the Cu foam with GDY does not show any catalytic activity on its own. It is the combination of Cu foam, GDY and Co nanoparticle that shows high catalytic activity. The GDY layer supported on the Cu foam improves the conductivity and prevents the aggregation of the Co catalyst. Using DFT calculations, it was shown that 2D B-doped γ-GY-1 is a promising metal-free catalyst for OER (Fig. 7d).163 The effect of N and B doping at different sites of γ-GY-1 on the catalytic activity was investigated and it was found that B doping of γ-GY-1 resulted in better catalytic performance than N doping and within B doping, replacing sp2 carbons by B atoms gives lowest over-potential and hence results in better OER catalysts.
GYs were recently investigated as potential candidates to support Pt for the oxidation of ethanol.175 The binding energies of Pt on γ-GY, β-GY, GDY and graphene were compared and they follow the order: graphene < GDY < γ-GY < β-GY. Binding on GYs leads to higher binding energies compared to pristine graphene due to more reactive sp carbon atoms present in these systems. Adsorption of a Pt cluster (Pt4) was also studied and in this case the binding energy followed a different order: graphene < β-GY < γ-GY < GDY. However, Pt-supported GYs did not show any remarkable enhancement of ethanol oxidation.
GYs also show interesting catalytic properties for the dehydrogenation of light metal complex hydrides.176 The decrease in the energy required for the removal of H atoms from three systems, namely LiAlH4, LiBH4 and NaAlH4 was studied using DFT calculations. All the three systems showed positive binding energies with GDY and the greatest stabilization on hydrogen removal was achieved by LiBHx (x = 2, 3 and 4). The catalytic enhancement observed on using GDY can be ascribed to three major reasons: (i) the sideways orientation of the alkali metal and hydride results in interaction between B/Al and C atoms, (ii) the smaller distance between the metal and GDY helps in the removal of H atom from the complex and (iii) the interaction between alkali metal and C increases the electron density on the C atom on removal of the H atom, which causes a decrease in the energy required to remove H from hydride.
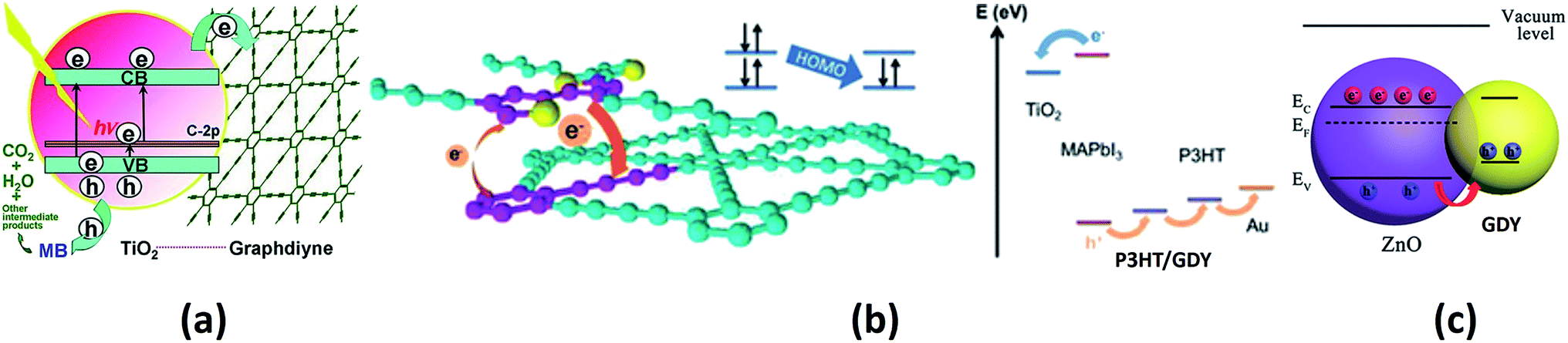
The use of GDY in the field of photocatalysis is another emerging aspect that has many applications in the field of environmental remediation. Wang and co-workers studied the activity of titania–GDY nanocomposites (P25–GDY) of different compositions as photocatalysts for the degradation of methylene blue.177 P25-GDY showed improved activity than bare P25 due to the conjugated nature of GDY. The excited electron moves from titania to GDY by a percolation mechanism, thereby suppressing charge recombination. A schematic representation of this process is shown in Fig. 8a. Further, experimental studies were carried out to understand the photocatalytic properties of GDY-modified TiO2.178 Yang et al. compared the catalytic ability of TiO2–GDY and TiO2–graphene composites on the photodegradation of methylene blue and it was seen that the TiO2–GDY composite showed higher catalytic performance. The enhanced charge separation, higher oxidation ability and the presence of larger impurity levels in TiO2–GDY composite are the major reasons attributed to its better performance. Another GDY hybrid, Ag/AgBr/GO/GDY has been successfully implemented as a photocatalyst for the degradation of methyl orange.179 The GO/GDY part of the hybrid suppresses the charge recombination and the hybrid shows 100% methyl orange degradation. A novel GDY–ZnO hybrid was investigated as a catalyst for the photodegradation of azo dyes.180 GDY–ZnO nanohybrid showed improved photocatalytic activity towards the degradation of methylene blue and rhodamine B. It also showed a superior catalytic efficiency towards the photodegradation of phenol compared to bare ZnO nanoparticles. Thus, we see that, a majority of the suggested applications of various forms of GYs in catalysis have emerged out of first-principles calculations and are yet to be realized in experiments. The authors believe that recent experiments demonstrating the role of GDY in photocatalysis would provide an impetus to researchers towards probing the other forms of GYs for related applications.
 | ||
| Fig. 8 Schematics of various photo-processes assisted by GYs: (a) photocatalytic degradation of methylene blue over P25–GDY. (b) Illustration of energy levels in P3HT/GDY-based perovskite solar cell. (c) Illustration of hole transfer from ZnO NP to GDY NP that enhances UV photodetection. Parts (a), (b) and (c) are reproduced from ref. 177, 182 and 186, respectively, with permissions from the John Wiley and Sons. | ||
8 Graphynes for optoelectronics
Recent research has indicated that GDY can be employed to achieve improved performance of optoelectronic devices such as solar cells and photodetectors. In this section, we describe some of the applications of GDYs in optoelectronics. The effect of GDY doping on the efficiency of polymer solar cells was investigated by Du and co-workers.181 Different ratios of GDY was doped into poly(3-hexathiophene) (P3HT):[6,6]-phenyl-C61-butyric acid methyl ester (PCBM) active layer and the short-circuit current, Jsc and power conversion efficiency (PCE) were calculated. The doped cells showed superior PCE and higher Jsc due to better electron transport via the GDY percolation paths. Perovskite-based solar cells may or may not contain a hole transporting material (HTM) layer. However, the HTM layer is usually necessary to improve the efficiency as it suppresses charge recombination and helps in hole transportation/separation. Inspired by the improved efficiency of GDY-doped P3HT in organic solar cells, Xiao et al. employed GDY doping on P3HT hole transporting material of perovskite solar cell and observed a higher PCE.182 The strong π–π stacking between GDY and P3HT enhances the hole transporting capacity (Fig. 8b). The GDY aggregates also enhance the light absorption of the solar cell. The doping of GDY in the electron transport layer (ETL) of perovskite has also been investigated. A work by Kuang et al. showed a remarkable PCE enhancement when the ETL component of PCBM was doped with GDY.183 GDY improved electron mobility, conductivity, charge extraction as well as the ETL coverage on the cell. The enhancement of Jsc is attributed to the high conductivity of GDY. Very recently, improved electron transport was achieved in CH3NH3PbI3 perovskite solar cell by doping both PCBM and ZnO film of the cell with GDY.184 The dual-doped solar cell showed a remarkable enhancement of PCE and stability. The dual doping with GDY decreased the surface defects of PCBM and ZnO, thereby suppressing the charge recombination and current leakage. GDY has also been used to enhance the catalytic activity of dye-sensitized solar cells (DSSC).185 GDY nanosheets with Pt nanoparticles were shown to improve electron transfer and catalytic activity when used as the counter electrode of a DSSC. The GDY-based substrates showed better performance than bare Pt and Pt nanoparticle-reduced graphene oxide counter electrodes.A photoelectrochemical cell (PEC) generates hydrogen by conversion of the solar energy into a photoelectrode. The higher surface area and light harvesting abilities make quantum dots (QDs) an excellent material for photoelectrodes. They are mainly used as photoanodes because electron transfer through the QDs can take place easily.187,188 However, the transport of holes through QDs is difficult and hence QD-based photocathodes are limited. Therefore, researchers are trying to develop materials that can facilitate the hole transportation through photocathodes. Li et al. investigated the use of GDY as the HTM of the QD-sensitized photocathode for hydrogen production in a PEC water splitting cell and it was seen that the high hole mobility through GDY and the strong π–π interaction between GDY and mercaptopyridine surface-functionalized CdSe QDs enhance the performance of the PEC.189 Another study by Gao et al. utilizes the high carrier mobility through GDY to enhance the PEC behaviour of BiVO4 by synthesising a GDY/BiVO4 composite photoanode for the PEC.190 The faster extraction of the carriers from BiVO4 by GDY helps in reducing unwanted carrier recombinations and prevents the oxidation of the BiVO4 layer by the holes, thereby increasing the stability of the electrode.
GDY is also being used to improve the performance of photodetectors (PDs). ZnO is one of the most common materials used to fabricate ultraviolet (UV) PDs. However, there are still efforts to improve the performance of UV PDs. Recently, GD:ZnO nanocomposites have been synthesised and they exhibit enhanced photoresponse.186 The GDY layer, as explained before, reduces charge recombination by trapping the holes that are generated by UV irradiation. Fig. 8c is an illustration of the hole transfer from ZnO NP to GDY NP. The PN junction depletion region width quickly decreases (increases) upon light illumination on (off) and this results in improved photoresponse. Semiconducting single-walled carbon nanotubes (s-SWNTs) are generally used for IR detection, but the poor separation of the excitons by s-SWNTs and exciton quenching by small quantities of metallic nanotubes present in the s-SWNTs raise the demand for discovering new materials that can combine with s-SWNTs to overcome these problems. Zheng et al. found that the introduction of GDY layers can improve the photoelectric conversion of the s-SWNTs.191 The GDY layer helps in faster dissociation of the excitons and increases carrier density, which in turn improves the photoresponse.
9 Conclusions and outlook
Overall, “graphynes” is the latest buzzword in carbon flatland. The family of graphynes is structurally diverse, offering researchers a variety of nanoporous architectures to play around with. The real challenge in this area of course lies in achieving successful large-scale synthesis of α-, β-, γ-, δ-, 6,6,12-GYs and rhombic-GYs. Bulk syntheses of γ-GY-2 and rhombic-GY-2 are only reported to date. Attempts are being made by several research groups to synthesise the other forms and the authors believe that they would soon be realised in experiments. Despite limited success in the synthetic protocols, theorists have taken strides towards understanding the fundamental properties and proposing newer applications of GYs. Some of the interesting properties of GYs include Dirac cone-like electronic structure, high carrier mobility, anisotropic electrical conductivity, stress or strain induced variation in band gap, mechanical robustness etc., most of which are summarized in a recent review by Li and co-workers.24 Structural diversity of the GY family has been showcased earlier by Ivanovskii.25 The porous architectures of GNTs can result in potential applications such as drug delivery, molecular encapsulation etc. Their low thermal conductivity and superplasticity in contrast to CNTs are attributed to the acetylenic linkages. In this review article, we focused on describing the theoretical as well as experimental aspects of the applications of GYs, emerging out of their interesting porous architectures. The applications considered range from water desalination, gas sensing and separation, molecular adsorption, energy storage, photovoltaics to catalysis. Theoretical studies have proposed that GYs could be excellent membranes for water desalination. The scope of GYs in forward osmosis is another exciting aspect that is being investigated in great detail. Gas separation using GYs is theoretically found to be a better option when compared to the conventional separation techniques. The variation in electrical conductivity of GYs on molecular adsorption contributes to their sensing ability. The uniform porous structures of GYs imply that they are a viable option for hydrogen storage. Doping of GY sheets and metal decoration have emerged as strategies for fine-tuning the properties like gas sensing and gas storage. Research has indicated improvement in storage capacity of Li-ion batteries on doping, fuelling interest in controlling the efficiencies of Na and Mg ion batteries as well. The unique electronic structure of GYs makes them promising materials for photovoltaics, eventually aiming at the fabrication of artificial photosynthetic devices. GYs, in pristine as well as doped form, are suggested as alternatives for metals in electrochemical actuators, molecular switches and drug delivery. Most of the efforts thus far have been in the direction of probing if GYs could emerge as viable alternatives to graphene for various purposes. It is interesting to note that, despite numerous theoretical and experimental studies on investigating the interactions between graphene and biopolymers such as DNA and proteins, a parallel effort involving GYs has hardly been undertaken.192,193 We believe that studies probing the interfaces of GYs with biological systems will soon emerge. Therefore, it is clear that researchers have only scratched the surface of the numerous possibilities that the family of GYs has to offer and there are many more opportunities for theorists as well as experimentalists towards realizing the full potential of GYs.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
RSS acknowledges the Science and Engineering Research Board (SERB), Government of India for financial support through the SERB Women Excellence Award Scheme. AJ, CJ, CHO, and SC thank IISER-TVM for financial support. SM thanks the Department of Science and Technology (DST), Government of India for financial support through the INSPIRE fellowship.References
- A. Coustenis and T. Encrenaz, Life beyond Earth: the search for habitable worlds in the Universe, Cambridge University Press, Cambridge, 2013, pp. 1–287 Search PubMed.
- H. B. Liu, J. L. Xu, Y. J. Li and Y. L. Li, Acc. Chem. Res., 2010, 43, 1496–1508 CrossRef PubMed.
- H. Y. Zheng, Y. J. Li, H. B. Liu, X. D. Yin and Y. L. Li, Chem. Soc. Rev., 2011, 40, 4506–4524 RSC.
- H. B. Liu, Y. L. Li, L. Jiang, H. Y. Luo, S. Q. Xiao, H. J. Fang, H. M. Li, D. B. Zhu, D. P. Yu, J. Xu and B. Xiang, J. Am. Chem. Soc., 2002, 124, 13370–13371 CrossRef PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef PubMed.
- S. Bae, H. Kim, Y. Lee, X. Xu, J. Park, Y. Zheng, J. Balakrishnan, L. Tian, H. Ri Kim, Y. I. Song, Y. Kim, K. Kim, B. Ozyilmaz, J. Ahn, B. Hee Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef PubMed.
- J. Wu, H. A. Becerril, Z. Bao, Z. Liu, Y. Chen and P. Peumans, Appl. Phys. Lett., 2008, 92, 263302 CrossRef.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef PubMed.
- J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, ACS Nano, 2009, 3, 301–306 CrossRef PubMed.
- C. Berger, Z. M. Song, T. B. Li, X. B. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912–19916 CrossRef.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, E. H. Marchenkov, A. N. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef PubMed.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef PubMed.
- H. Wang, L. F. Cui, Y. Yang, C. H. Sanchez, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef PubMed.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2008, 9, 72–75 CrossRef PubMed.
- L. Pan, X.-D. Zhu, K.-N. Sun, Y.-T. Liu, X.-M. Xie and X.-Y. Ye, Nano Energy, 2016, 30, 347–354 CrossRef.
- P. Long, Z. Xiao-Dong, X. Xu-Ming and L. Yi-Tao, Adv. Funct. Mater., 2015, 25, 3341–3350 CrossRef.
- Y.-T. Liu, X.-D. Zhu, Z.-Q. Duan and X.-M. Xie, Chem. Commun., 2013, 49, 10305–10307 RSC.
- P. R. Wallace, Phys. Rev., 1947, 71, 622–634 CrossRef.
- H. P. Boehm, A. Clauss, G. O. Fischer and U. Hofmann, Z. Anorg. Allg. Chem., 1962, 316, 119–127 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef PubMed.
- Y. J. Li, L. Xu, H. B. Liu and Y. L. Li, Chem. Soc. Rev., 2014, 43, 2572–2586 RSC.
- A. L. Ivanovskii, Prog. Solid State Chem., 2013, 41, 1–19 CrossRef.
- J. O. Sofo, A. S. Chaudhari and G. D. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153401 CrossRef.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610–613 CrossRef PubMed.
- J. Zhou, Q. Wang, Q. Sun, X. S. Chen, Y. Kawazoe and P. Jena, Nano Lett., 2009, 9, 3867–3870 CrossRef PubMed.
- W. Zhao, J. Gebhardt, F. Späth, K. Gotterbarm, C. Gleichweit, H.-P. Steinrück, A. Görling and C. Papp, Chem.–Eur. J., 2015, 21, 3347–3358 CrossRef PubMed.
- R. H. Baughman, H. Eckhardt and M. Kertesz, J. Chem. Phys., 1987, 87, 6687–6699 CrossRef.
- B. G. Kim and H. J. Choi, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 115435 CrossRef.
- F. Diederich, Nature, 1994, 369, 199–207 CrossRef.
- U. H. F. Bunz, Y. Rubin and Y. Tobe, Chem. Soc. Rev., 1999, 28, 107–119 RSC.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- Y.-Q. Zhang, N. Kepčija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V. Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed.
- Z. Jia, Z. Zuo, Y. Yi, H. Liu, D. Li, Y. Li and Y. Li, Nano Energy, 2017, 33, 343–349 CrossRef.
- Z. Jia, Y. Li, Z. Zuo, H. Liu, C. Huang and Y. Li, Acc. Chem. Res., 2017, 50, 2470–2478 CrossRef PubMed.
- J. Zhong, J. Wang, J.-G. Zhou, B.-H. Mao, C.-H. Liu, H.-B. Liu, Y.-L. Li, T.-K. Sham, X.-H. Sun and S.-D. Wang, J. Phys. Chem. C, 2013, 117, 5931–5936 CrossRef.
- Y. Yang and X. Xu, Comput. Mater. Sci., 2012, 61, 83–88 CrossRef.
- Y. Pei, Physica B, 2012, 407, 4436–4439 CrossRef.
- S. Lin and M. J. Buehler, Nanoscale, 2013, 5, 11801–11807 RSC.
- A. L. Ivanovskii, Prog. Solid State Chem., 2013, 41, 1–14 CrossRef.
- C. Owais, A. James, C. John, R. Dhali and R. S. Swathi, J. Phys. Chem. B, 2018, 122, 5127–5146 CrossRef PubMed.
- S. W. Cranford and M. J. Buehler, Carbon, 2011, 49, 4111–4121 CrossRef.
- A. N. Enyashin and A. L. Ivanovskii, Phys. Status Solidi B, 2011, 248, 1879–1883 CrossRef.
- S. W. Cranford, D. B. Brommer and M. J. Buehler, Nanoscale, 2012, 4, 7797–7809 RSC.
- D. Malko, C. Neiss, F. Viñes and A. Görling, Phys. Rev. Lett., 2012, 108, 086804 CrossRef PubMed.
- Y. Y. Zhang, Q. X. Pei and C. M. Wang, Appl. Phys. Lett., 2012, 101, 081909 CrossRef.
- Q. Peng, W. Ji and S. De, Phys. Chem. Chem. Phys., 2012, 14, 13385–13391 RSC.
- V. R. Coluci, S. F. Braga, S. B. Legoas, D. S. Galvão and R. H. Baughman, Nanotechnology, 2004, 15, S142–S149 CrossRef.
- X.-M. Wang and S.-S. Lu, J. Phys. Chem. C, 2013, 117, 19740–19745 CrossRef.
- G. Li, Y. Li, X. Qian, H. Liu, H. Lin, N. Chen and Y. Li, J. Phys. Chem. C, 2011, 115, 2611–2615 CrossRef.
- S. Alaei, S. Jalili and S. Erkoc, Fullerenes, Nanotub. Carbon Nanostruct., 2014, 23, 494–499 CrossRef.
- J. Deb, B. Bhattacharya and U. Sarkar, AIP Conf. Proc., 2016, 1731, 050081 CrossRef.
- M. Hu, Y. Jing and X. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 155408 CrossRef.
- J. M. de Sousa, G. Brunetto, V. R. Coluci and D. S. Galvão, Carbon, 2016, 96, 14–19 CrossRef.
- X. Qian, Z. Ning, Y. Li, H. Liu, C. Ouyang, Q. Chen and Y. Li, Dalton Trans., 2012, 41, 730–733 RSC.
- D. Solis, C. F. Woellner, D. D. Borges and D. S. Galvao, MRS Adv., 2017, 2, 129–134 CrossRef.
- V. R. Coluci, S. F. Braga, S. B. Legoas, D. S. Galvão and R. H. Baughman, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 035430 CrossRef.
- R. Matsuoka, R. Sakamoto, K. Hoshiko, S. Sasaki, H. Masunaga, K. Nagashio and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 3145–3152 CrossRef PubMed.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- X. Qian, H. Liu, C. Huang, S. Chen, L. Zhang, Y. Li, J. Wang and Y. Li, Sci. Rep., 2015, 5, 7756 CrossRef PubMed.
- M. M. Haley, Pure Appl. Chem., 2008, 80, 519–532 Search PubMed.
- B. Wu, M. Li, S. Xiao, Y. Qu, X. Qiu, T. Liu, F. Tian, H. Li and S. Xiao, Nanoscale, 2017, 9, 11939–11943 RSC.
- Q. Yuan and F. Ding, Nanoscale, 2014, 6, 12727–12731 RSC.
- F. Klappenberger, R. Hellwig, P. Du, T. Paintner, M. Uphoff, L. Zhang, T. Lin, B. A. Moghanaki, M. Paszkiewicz, I. Vobornik, J. Fujii, O. Fuhr, Y. Q. Zhang, F. Allegretti, M. Ruben and J. V. Barth, Small, 2018, 14, 1704321 CrossRef PubMed.
- R. Liu, J. Y. Zhou, X. Gao, J. Q. Li, Z. Q. Xie, Z. Z. Li, S. Q. Zhang, L. M. Tong, J. Zhang and Z. F. Liu, Adv. Electron. Mater., 2017, 3, 1700122 CrossRef.
- M. Bartolomei, E. Carmona-Novillo, M. I. Hernandez, J. Campos-Martinez, F. Pirani, G. Giorgi and K. Yamashita, J. Phys. Chem. Lett., 2014, 5, 751–755 CrossRef PubMed.
- C. Zhu, H. Li, X. C. Zeng, E. G. Wang and S. Meng, Sci. Rep., 2013, 3, 3163 CrossRef PubMed.
- X. Zhang and J. G. Gai, RSC Adv., 2015, 5, 68109–68116 RSC.
- Z. Qin and M. J. Buehler, Nano Lett., 2015, 15, 3939–3944 CrossRef PubMed.
- J. L. Kou, X. Y. Zhou, Y. Y. Chen, H. J. Lu, F. M. Wu and J. T. Fan, J. Chem. Phys., 2013, 139, 064705 CrossRef PubMed.
- J. L. Kou, X. Y. Zhou, H. J. Lu, F. M. Wu and J. T. Fan, Nanoscale, 2014, 6, 1865–1870 RSC.
- M. M. Xue, H. Qiu and W. L. Guo, Nanotechnology, 2013, 24, 505720 CrossRef PubMed.
- M. Raju, P. B. Govindaraju, A. C. T. van Duin and M. Ihme, Nanoscale, 2018, 10, 3969–3980 RSC.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef PubMed.
- G. Arora and S. I. Sandler, Fluid Phase Equilib., 2007, 259, 3–8 CrossRef.
- M. Hankel, H. Zhang, T. X. Nguyen, S. K. Bhatia, S. K. Gray and S. C. Smith, Phys. Chem. Chem. Phys., 2011, 13, 7834–7844 RSC.
- T. M. Nenoff, R. J. Spontak and C. M. Aberg, MRS Bull., 2006, 31, 735–741 CrossRef.
- P. Bernardo, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef.
- Y. Li, Z. Zhou, P. Shen and Z. Chen, Chem. Commun., 2010, 46, 3672–3674 RSC.
- S. Blankenburg, M. Bieri, R. Fasel, K. Müllen, C. A. Pignedoli and D. Passerone, Small, 2010, 6, 2266–2271 CrossRef PubMed.
- J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano Lett., 2010, 8, 2458–2462 CrossRef PubMed.
- O. Leenaerts, B. Partoens and F. M. Peeters, Appl. Phys. Lett., 2010, 93, 193107 CrossRef.
- M. D. Fischbein and M. Drndć, Appl. Phys. Lett., 2008, 93, 113107 CrossRef.
- G. Compagnini, F. Giannazzo, S. Sonde, V. Raineri and E. Rimini, Carbon, 2008, 47, 3201–3207 CrossRef.
- Y. Jiao, A. Du, M. Hankel, Z. Zhu, V. Rudolph and S. C. Smith, Chem. Commun., 2011, 47, 11843–11845 RSC.
- W.-h. Zhao, L.-f. Yuan and J.-l. Yang, Chin. J. Chem. Phys., 2012, 25, 434–440 CrossRef.
- S. W. Cranford and M. J. Buehler, Nanoscale, 2012, 4, 4587–4593 RSC.
- Y. Jiao, A. Du, S. C. Smith, Z. Zhu and S. Z. Qiao, J. Mater. Chem. A, 2015, 3, 6767–6771 RSC.
- X. Tan, L. Kou, H. A. Tahini and S. C. Smith, Mol. Simul., 2015, 42, 573–579 CrossRef.
- H. Zhang, X. He, M. Zhao, M. Zhang, L. Zhao, X. Feng and Y. Luo, J. Phys. Chem. C, 2012, 116, 16634–16638 CrossRef.
- P. Sang, L. Zhao, J. Xu, Z. Shi, S. Guo, Y. Yu, H. Zhu, Z. Yan and W. Guo, Int. J. Hydrogen Energy, 2017, 42, 5168–5176 CrossRef.
- M. Alaghemandi, Chem. Phys. Lett., 2015, 629, 65–69 CrossRef.
- Z. Meng, X. Zhang, Y. Zhang, H. Gao, Y. Wang, Q. Shi, D. Rao, Y. Liu, K. Deng and R. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 28166–28170 CrossRef PubMed.
- L. Zhao, P. Sang, S. Guo, X. Liu, J. Li, H. Zhu and W. Guo, Appl. Surf. Sci., 2017, 405, 455–464 CrossRef.
- G. Lei, C. Liu, Q. Li and X. Xu, Fuel, 2016, 182, 210–219 CrossRef.
- A. Cho, Science, 2009, 326, 778–779 CrossRef PubMed.
- W. P. Halperin, Nat. Phys., 2014, 10, 467–470 Search PubMed.
- M. I. Hernández, M. Bartolomei and J. Campos-Martínez, J. Phys. Chem. A, 2015, 119, 10743–10749 CrossRef PubMed.
- M. Bartolomei, E. Carmona-Novillo, M. I. Hernández, J. Campos-Martínez, F. Pirani and G. Giorgi, J. Phys. Chem. C, 2014, 118, 29966–29972 CrossRef.
- X. Bévenot, A. Trouillet, C. Veillas, H. Gagnaire and M. Clément, Sens. Actuators, B, 2000, 67, 57–67 CrossRef.
- S. Ryabtsev, A. Shaposhnick, A. Lukin and E. Domashevskaya, Sens. Actuators, B, 1999, 59, 26–29 CrossRef.
- W. Tsujita, A. Yoshino, H. Ishida and T. Moriizumi, Sens. Actuators,B, 2005, 110, 304–311 CrossRef.
- R. Majidi and A. R. Karami, Phys. E, 2014, 59, 169–173 CrossRef.
- X. Chen, P. Gao, L. Guo, Y. Wen, D. Fang, B. Gong, Y. Zhang and S. Zhang, Phys. Lett. A, 2017, 381, 879–885 CrossRef.
- R. Majidi and A. R. Karami, Phys. E, 2013, 54, 177–180 CrossRef.
- J. Deb, B. Bhattacharya, N. B. Singh and U. Sarkar, Struct. Chem., 2016, 27, 1221–1227 CrossRef.
- R. Majidi and A. Karami, Stud. Univ. Babes-Bolyai, Chem., 2016, 61, 177–184 Search PubMed.
- D. Cortés-Arriagada, Int. J. Quantum Chem., 2017, 117, 1–7 CrossRef.
- M. Ozmaian, A. Fathizadeh, M. Jalalvand, M. R. Ejtehadi and S. M. Allaei, Sci. Rep., 2016, 6, 21910 CrossRef PubMed.
- P. Zhang, Q. Song, J. Zhuang and X.-J. Ning, Chem. Phys. Lett., 2017, 689, 185–189 CrossRef.
- H. J. Kwon, Y. Kwon, T. Kim, Y. Jung, S. Lee, M. Cho and S. Kwon, AIP Adv., 2017, 7, 125013 CrossRef.
- Z. Yan, L. Wang, J. Cheng, L. Huang, C. Zhu, C. Chen, L. Miao and J. Jiang, J. Appl. Phys., 2014, 116, 174304 CrossRef.
- B. Kang, H. Liu and J. Y. Lee, Phys. Chem. Chem. Phys., 2014, 16, 974–980 RSC.
- A. Omidvar and A. Mohajeri, Mol. Phys., 2015, 113, 3900–3908 CrossRef.
- J. Beheshtian, A. A. Peyghan, Z. Bagheri and M. B. Tabar, J. Struct. Chem., 2013, 25, 1–7 Search PubMed.
- Z. Felegari and S. Hamedani, Results Phys., 2017, 7, 2626–2631 CrossRef.
- A. A. Peyghan, S. F. Rastegar and N. L. Hadipour, Phys. Lett. A, 2014, 378, 2184–2190 CrossRef.
- Z. Lu, P. Lv, D. Ma, X. Yang, S. Li and Z. Yang, J. Phys. D: Appl. Phys., 2018, 51, 065109 CrossRef.
- A. Zuttel, Naturwissenschaften, 2004, 91, 157–172 CrossRef PubMed.
- D. J. Durbin and C. Malardier-Jugroot, Int. J. Hydrogen Energy, 2013, 38, 14595–14617 CrossRef.
- A. Delahaye, A. Aoufi, A. Gicquel and I. Pentchev, AIChE J., 2002, 48, 2061–2073 CrossRef.
- K. Srinivasu and S. K. Ghosh, J. Phys. Chem. C, 2012, 116, 5951–5956 CrossRef.
- S. C. Shekar and R. S. Swathi, J. Phys. Chem. C, 2015, 119, 8912–8923 CrossRef.
- H. Y. Zhang, M. W. Zhao, X. J. He, Z. H. Wang, X. J. Zhang and X. D. Liu, J. Phys. Chem. C, 2011, 115, 8845–8850 CrossRef.
- K. Wang, N. Wang, J. J. He, Z. Yang, X. Y. Shen and C. S. Huang, Electrochim. Acta, 2017, 253, 506–516 CrossRef.
- S. L. Zhang, H. B. Liu, C. S. Huang, G. L. Cui and Y. L. Li, Chem. Commun., 2015, 51, 1834–1837 RSC.
- H. Zhang, X. Zhao, M. Zhang, Y. Luo, G. Li and M. Zhao, J. Phys. D: Appl. Phys., 2013, 46, 495307 CrossRef.
- Y. Guo, K. Jiang, B. Xu, Y. Xia, J. Yin and Z. Liu, J. Phys. Chem. C, 2012, 116, 13837–13841 CrossRef.
- H. Zhang, M. Zhao, H. Bu, X. He, M. Zhang, L. Zhao and Y. Luo, J. Appl. Phys., 2012, 112, 084305 CrossRef.
- J. Lu, Y. Guo, Y. Zhang and J. Cao, Int. J. Hydrogen Energy, 2014, 39, 17112–17117 CrossRef.
- S. Kumar and T. J. Dhilip Kumar, ACS Appl. Mater. Interfaces, 2017, 9, 28659–28666 CrossRef PubMed.
- R. Lu, D. Rao, Z. Meng, X. Zhang, G. Xu, Y. Liu, E. Kan, C. Xiao and K. Deng, Phys. Chem. Chem. Phys., 2013, 15, 16120–16126 RSC.
- B. Jang, J. Koo, M. Park, H. Lee, J. Nam, Y. Kwon and H. Lee, Appl. Phys. Lett., 2013, 103, 263904 CrossRef.
- H. J. Hwang, J. Koo, M. Park, N. Park, Y. Kwon and H. Lee, J. Phys. Chem. C, 2013, 117, 6919–6923 CrossRef.
- M. Makaremi, B. Mortazavi and C. V. Singh, Appl. Mater. Today, 2018, 10, 115–121 CrossRef.
- Y. Liu, W. Liu, R. Wang, L. Hao and W. Jiao, Int. J. Hydrogen Energy, 2014, 39, 12757–12764 CrossRef.
- H. J. Hwang, Y. Kwon and H. Lee, J. Phys. Chem. C, 2012, 116, 20220–20224 CrossRef.
- C. Li, J. Li, F. Wu, S.-S. Li, J.-B. Xia and L.-W. Wang, J. Phys. Chem. C, 2011, 115, 23221–23225 CrossRef.
- Y. Guo, X. Lan, J. Cao, B. Xu, Y. Xia, J. Yin and Z. Liu, Int. J. Hydrogen Energy, 2013, 38, 3987–3993 CrossRef.
- Y. S. Wang, P. Fei Yuan, M. Li, W. Fen Jiang, Q. Sun and Y. Jia, J. Solid State Chem., 2013, 197, 323–328 CrossRef.
- J. Lu, Y. Guo, Y. Zhang, Y. Tang and J. Cao, J. Solid State Chem., 2015, 231, 53–57 CrossRef.
- P.-P. Liu, H. Zhang, X.-L. Cheng and Y.-J. Tang, Appl. Surf. Sci., 2016, 371, 44–49 CrossRef.
- L. Zhang, S. Zhang, P. Wang, C. Liu, S. Huang and H. Tian, Comput. Theor. Chem., 2014, 1035, 68–75 CrossRef.
- C. H. Sun and D. J. Searles, J. Phys. Chem. C, 2012, 116, 26222–26226 CrossRef.
- K. Persson, Y. Hinuma, Y. S. Meng, A. Van der Ven and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125416 CrossRef.
- S. C. Shekar and R. S. Swathi, J. Phys. Chem. A, 2013, 117, 8632–8641 CrossRef PubMed.
- Z. M. Xu, X. Lv, J. Li, J. G. Chen and Q. S. Liu, RSC Adv., 2016, 6, 25594–25600 RSC.
- S. L. Zhang, J. J. He, J. Zheng, C. S. Huang, Q. Lv, K. Wang, N. Wang and Z. G. Lan, J. Mater. Chem. A, 2017, 5, 2045–2051 RSC.
- C. S. Huang, S. L. Zhang, H. B. Liu, Y. J. Li, G. T. Cui and Y. L. Li, Nano Energy, 2015, 11, 481–489 CrossRef.
- H. P. Du, H. Yang, C. S. Huang, J. J. He, H. B. Liu and Y. L. Li, Nano Energy, 2016, 22, 615–622 CrossRef.
- Z. Yang, X. Shen, N. Wang, J. He, X. Li, X. Wang, Z. Hou, K. Wang, J. Gao, T. Jiu and C. Huang, ACS Appl. Mater. Interfaces, 2018 DOI:10.1021/acsami.8b01823.
- S. Zhang, H. Du, J. He, C. Huang, H. Liu, G. Cui and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 8467–8473 CrossRef PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef PubMed.
- Z. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542–7543 CrossRef PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef PubMed.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef PubMed.
- P. Wu, P. Du, H. Zhang and C. Cai, J. Phys. Chem. C, 2012, 116, 20472–20479 CrossRef.
- P. Wu, P. Du, H. Zhang and C. Cai, Phys. Chem. Chem. Phys., 2015, 17, 1441–1449 RSC.
- X. Kong, Y. Huang and Q. Liu, Carbon, 2017, 123, 558–564 CrossRef.
- K. Srinivasu and S. K. Ghosh, J. Phys. Chem. C, 2013, 117, 26021–26028 CrossRef.
- X.-k. Kong, Q.-w. Chen and Z. Sun, RSC Adv., 2013, 3, 4074–4080 RSC.
- X. Chen, Q. Qiao, L. An and D. Xia, J. Phys. Chem. C, 2015, 119, 11493–11498 CrossRef.
- B. Kang, H. Shi, S. Wu, W. Zhao, H. Ai and J. Y. Lee, Carbon, 2017, 123, 415–420 CrossRef.
- B. K. Das, D. Sen and K. K. Chattopadhyay, Phys. Chem. Chem. Phys., 2016, 18, 2949–2958 RSC.
- R. Liu, H. Liu, Y. Li, Y. Yi, X. Shang, S. Zhang, X. Yu, S. Zhang, H. Cao and G. Zhang, Nanoscale, 2014, 6, 11336–11343 RSC.
- Q. Lv, W. Si, Z. Yang, N. Wang, Z. Tu, Y. Yi, C. Huang, L. Jiang, M. Zhang, J. He and Y. Long, ACS Appl. Mater. Interfaces, 2017, 9, 29744–29752 CrossRef PubMed.
- S. Zhang, Y. Cai, H. He, Y. Zhang, R. Liu, H. Cao, M. Wang, J. Liu, G. Zhang, Y. Li, H. Liu and B. Li, J. Mater. Chem. A, 2016, 4, 4738–4744 RSC.
- X. Chen, Phys. Chem. Chem. Phys., 2015, 17, 29340–29343 RSC.
- P. Wu, P. Du, H. Zhang and C. Cai, Phys. Chem. Chem. Phys., 2014, 16, 5640–5648 RSC.
- J. Li, X. Gao, X. Jiang, X.-B. Li, Z. Liu, J. Zhang, C.-H. Tung and L.-Z. Wu, ACS Catal., 2017, 7, 5209–5213 CrossRef.
- G. Tian, Z. Qi, W. Ma and Y. Wang, ChemistrySelect, 2017, 2, 2311–2321 CrossRef PubMed.
- H. Yu, A. Du, Y. Song and D. J. Searles, J. Phys. Chem. C, 2013, 117, 21643–21650 CrossRef.
- S. Wang, L. Yi, J. E. Halpert, X. Lai, Y. Liu, H. Cao, R. Yu, D. Wang and Y. Li, Small, 2012, 8, 265–271 CrossRef PubMed.
- N. Yang, Y. Liu, H. Wen, Z. Tang, H. Zhao, Y. Li and D. Wang, ACS Nano, 2013, 7, 1504–1512 CrossRef PubMed.
- X. Zhang, M. Zhu, P. Chen, Y. Li, H. Liu, Y. Li and M. Liu, Phys. Chem. Chem. Phys., 2015, 17, 1217–1225 RSC.
- S. Thangavel, K. Krishnamoorthy, V. Krishnaswamy, N. Raju, S. J. Kim and G. Venugopal, J. Phys. Chem. C, 2015, 119, 22057–22065 CrossRef.
- H. Du, Z. Deng, Z. Lü, Y. Yin, L. Yu, H. Wu, Z. Chen, Y. Zou, Y. Wang, H. Liu and Y. Li, Synth. Met., 2011, 161, 2055–2057 CrossRef.
- J. Xiao, J. Shi, H. Liu, Y. Xu, S. Lv, Y. Luo, D. Li, Q. Meng and Y. Li, Adv. Energy Mater., 2015, 5, 1401943 CrossRef.
- C. Kuang, G. Tang, T. Jiu, H. Yang, H. Liu, B. Li, W. Luo, X. Li, W. Zhang, F. Lu, J. Fang and Y. Li, Nano Lett., 2015, 15, 2756–2762 CrossRef PubMed.
- J. Li, T. Jiu, C. Duan, Y. Wang, H. Zhang, H. Jian, Y. Zhao, N. Wang, C. Huang and Y. Li, Nano Energy, 2018, 46, 331–337 CrossRef.
- H. Ren, H. Shao, L. Zhang, D. Guo, Q. Jin, R. Yu, L. Wang, Y. Li, Y. Wang, H. Zhao and D. Wang, Adv. Energy Mater., 2015, 5, 1500296 CrossRef.
- Z. Jin, Q. Zhou, Y. Chen, P. Mao, H. Li, H. Liu, J. Wang and Y. Li, Adv. Mater., 2016, 28, 3697–3702 CrossRef PubMed.
- K. Tvrdy, P. A. Frantsuzov and P. V. Kamat, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 29–34 CrossRef PubMed.
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316–4324 CrossRef PubMed.
- J. Li, X. Gao, B. Liu, Q. Feng, X. B. Li, M. Y. Huang, Z. Liu, J. Zhang, C. H. Tung and L. Z. Wu, J. Am. Chem. Soc., 2016, 138, 3954–3957 CrossRef PubMed.
- X. Gao, J. Li, R. Du, J. Zhou, M. Y. Huang, R. Liu, J. Li, Z. Xie, L. Z. Wu, Z. Liu and J. Zhang, Adv. Mater., 2017, 29, 1605308 CrossRef PubMed.
- Z. Zheng, H. Fang, D. Liu, Z. Tan, X. Gao, W. Hu, H. Peng, L. Tong, W. Hu and J. Zhang, Adv. Sci., 2017, 4, 1700472 CrossRef PubMed.
- S. Chandra Shekar and R. S. Swathi, J. Phys. Chem. C, 2014, 118, 4516–4528 CrossRef.
- N. Parvin, Q. Jin, Y. Wei, R. Yu, B. Zheng, L. Huang, Y. Zhang, L. Wang, H. Zhang, M. Gao, H. Zhao, W. Hu, Y. Li and D. Wang, Adv. Mater., 2017, 29, 1606755 CrossRef PubMed.
Footnotes |
| † Present address: Institute for Computational Physics, University of Stuttgart, Allmandring 3, 70569 Stuttgart, Germany. |
| ‡ Present address: Presidency University, Bangalore, India-560064. |
| This journal is © The Royal Society of Chemistry 2018 |