
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid inoculation of single bacteria into parallel picoliter fermentation chambers†
Christopher
Probst‡
,
Alexander
Grünberger‡
,
Nadja
Braun
,
Stefan
Helfrich
,
Katharina
Nöh
,
Wolfgang
Wiechert
and
Dietrich
Kohlheyer
*
Institute of Bio- and Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: d.kohlheyer@fz-juelich.de; Fax: +49 2461 61 3870; Tel: +49 2461 61 2875
First published on 15th October 2014
Abstract
Microfluidic single-cell cultivation devices have been successfully utilized in a variety of biological research fields. One major obstacle to the successful implementation of high throughput single-cell cultivation technology is the requirement for a simple, fast and reliable cell inoculation procedure. In the present report, an air-bubble-based cell loading methodology is described and validated for inoculating single bacteria into multiple picoliter sized growth chambers arranged in a highly parallel manner. It is shown that the application of the injected air bubble can serve as a reproducible mechanism to modify laminar flow conditions. In this way, convective flow was temporarily induced in more than 1000 cultivation chambers simultaneously, which under normal conditions operate exclusively under diffusive mass transport. Within an inoculation time of 100 s, Corynebacterium glutamicum cells were inoculated by convection at minimal stress level and single bacteria remain successfully trapped by cell-wall interactions. The procedure is easy, fast, gentle and requires only minimal fluidic control and equipment. The technique is well suited for microbial cell loading into commonly used microfluidic growth sites arranged in parallel intended for high throughput single-cell analysis.
Introduction
Single-cell analysis based on microfluidic cultivation systems has been successfully developed to investigate a variety of living organisms in vivo.1–6 In contrast to all other procedures, this technology facilitates exceptional environmental control as well as full spatial and temporal resolution at the single-cell level and is thus well suited to investigate cellular dynamics for microbiology and biotechnology.7,8 The functional microfluidic cultivation geometries utilized so far can be classified into simple cultivation wells,9 shallow growth sites restricting cellular growth to a monolayer,10,11 growth tracks harboring single cells arranged in a straight queue12,13 and single-cell traps holding exclusively one cell.14,15Typically these micrometer and sub micrometer sized cultivation sites are arranged in a highly parallel manner thereby incorporating up to several thousand regions of interest in a single device. During operation, preferably a single cell is inoculated into each cultivation site facilitating spatiotemporal investigations on isogenic microcolonies, for example by automated time-lapse microscopy. One major obstacle to the successful implementation of high throughput microfluidic single-cell cultivation technology is the prerequisite for simple and fast but reliable cell inoculation from the pre-culture suspension.
Inoculation procedures differ and depend to a large extent on the microfluidic cultivation geometry applied. However, the vast majority of cell seeding routines are based on random cell trapping while the cell suspension is infused into the microfluidic device. As illustrated in Fig. 1a, this is straightforward if simple trapping obstacles and barriers are directly infused with the cell suspension during loading.15–19 These barrier structures facilitate excellent medium exchange and constant cultivation conditions by continuous convective flow. However, cells may be washed out and become lost as soon as the drag force exceeds the force of the barrier structure's trapping mechanisms and convection may be undesirable for many applications.
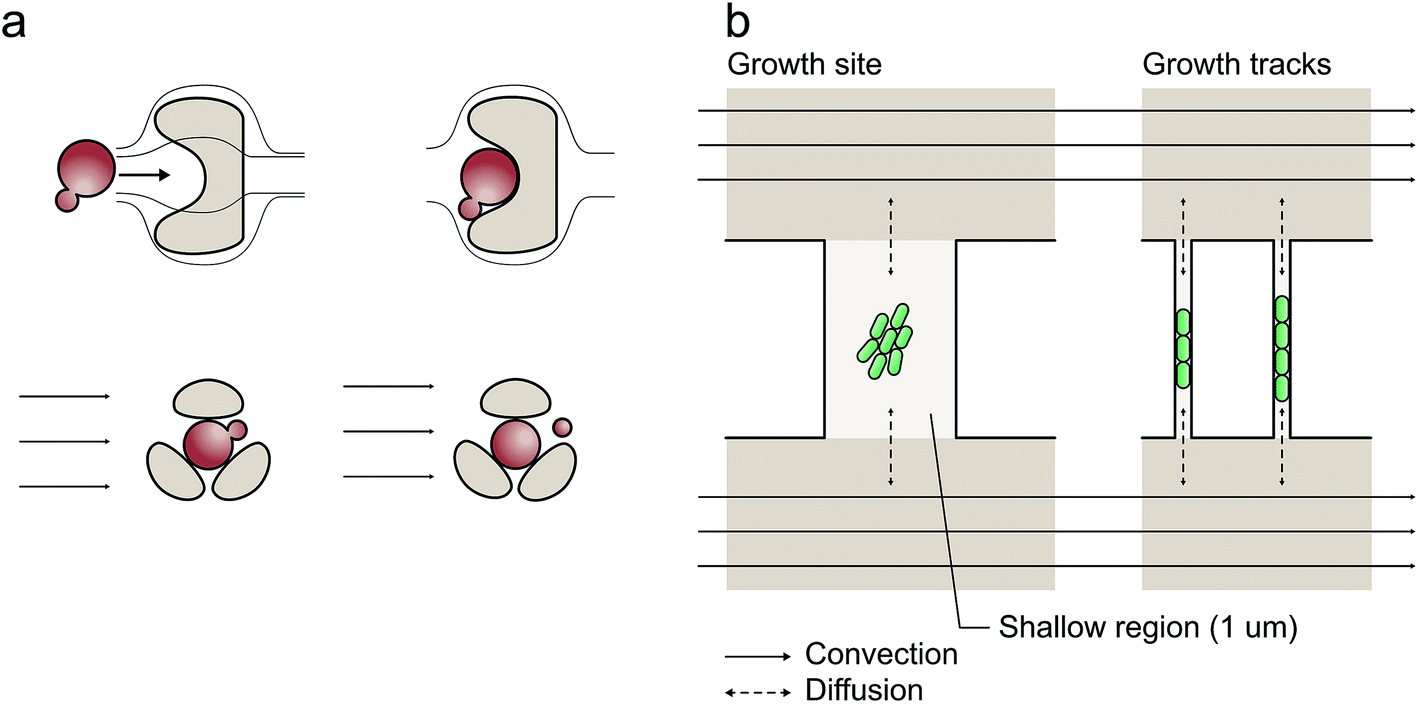 | ||
| Fig. 1 Examples of microfluidic single-cell cultivation geometries: (a) microfluidic barrier structures for single-cell cultivation with continuous laminar flow; (b) cultivation chambers and growth tracks facilitating exclusively diffusion-based mass transport if the two parallel media volume flows are equal. | ||
As depicted in Fig. 1b, cultivation chambers and channels have therefore been placed perpendicular to the flow and interconnected between parallel media supply channels avoiding convection and shear stress.10,12,20 This latter configuration facilitates exclusively diffusion-based mass transport if the two media-supplying volume flows are equal. The cultivation sites can have different geometries,10,12,21,22 which have been applied for bacteria,10,22 yeast11 and eukaryotic cells.23
At short micrometer length scales, diffusive mass transport is typically very efficient thus guaranteeing stable cultivation conditions. However, cell inoculation remains a bottleneck since cells are not directly flushed into the cultivation sites (Fig. 1b). To overcome this limitation, several workarounds have been reported to load single cells into the chambers, for example, “manually applied pressure pulses to the channels to induce a momentary flow change”,24 whole-device centrifugation for inoculation by centrifugal forces,13 incorporation of additional seeding channels elaborately operated by flexible valves,25 controlled but low throughput cell seeding by optical tweezers26 and interconnected under-pressure channels to actively draw cells into the cultivation sites.27,28 Clearly, there is an urgent requirement for improvement, particularly in view of the challenge of implementing a reliable, user-friendly, fast and perhaps commercially applicable procedure.
In the present report, a simple, fast, gentle and reproducible method is described for microbial cell inoculation into microfluidic cultivation sites, in which during cultivation exclusively diffusive mass transport is present whereas convective flow occurs only inside the adjacent supply channels. Our inoculation procedure utilizes an entrapped nanoliter sized air bubble to temporarily modify the flow conditions. The bubble induces a momentary convective flow also through the cultivation chambers, thus enabling cell transport into the chambers. In contrast to previously described procedures, the present chip device remains inside the temperature incubator the whole time and the connected tubing is not removed during operation, which is ideal for a fast and gentle cell transferal from the pre-culture to the main microfluidic chip culture. The principle is limited to gas permeable device material since the bubble removal is based on gas diffusion through the interfacing wall material. The proposed gas-bubble inoculation requires neither a complex fabrication procedure nor elaborate control equipment. Furthermore, by applying multiple bubbles it is highly parallelizable because the gas–liquid interface adapts to any channel geometry. The procedure was successfully validated with the organism Corynebacterium glutamicum, which subsequently exhibited normal growth behavior.
Material & methods
Chip fabrication
Single-use microfluidic cultivation devices were fabricated by standard polydimethylsiloxane (PDMS) (Sylgard 184 Silicone Elastomer, Dow Corning Corporation, Midland, USA) molding, casting a 100 mm silicon wafer carrying appropriately designed SU-8 (MicroChem Corp, USA) structures processed by cleanroom photolithography. To this end, SU-8 layers 1 μm and 10 μm in height were spin-coated and processed separately, resulting in a layered dual resist configuration. The molded PDMS chips had supply channels of 10 μm height and cultivation sites 1 μm in height. Prior to the experiments, each chip was thoroughly cleaned and permanently bonded onto 170 μm thick glass plates (SCHOTT Malaysia) after oxygen plasma treatment and finally connected to tubing (Saint Gobain; VWR International GmbH, Germany). A detailed fabrication procedure has been published previously.29Device configuration
The present PDMS-glass microfluidic device incorporates four individual perfusion channels with a single inlet and outlet respectively (Fig. 2a and b). Each inlet channel (100 μm width) branches into two separate cultivation arrays having a partly beveled air bubble entrapment zone (430 μm width) in front (Fig. 2c). By this configuration one single chip combines 1184 growth chambers in total. Growth chambers are arranged in between and perpendicular to the flow. Therefore, two opposing chamber openings are connected to the supply channels (30 μm width, 10 μm height) for continuous media supply. In the present design two different growth chamber sizes were implemented (W × L × H), namely: 40 μm × 60 μm × 1 μm and 50 μm × 60 μm × 1 μm, as depicted in Fig. 2d. An exemplary SEM image is shown in Fig. 2e.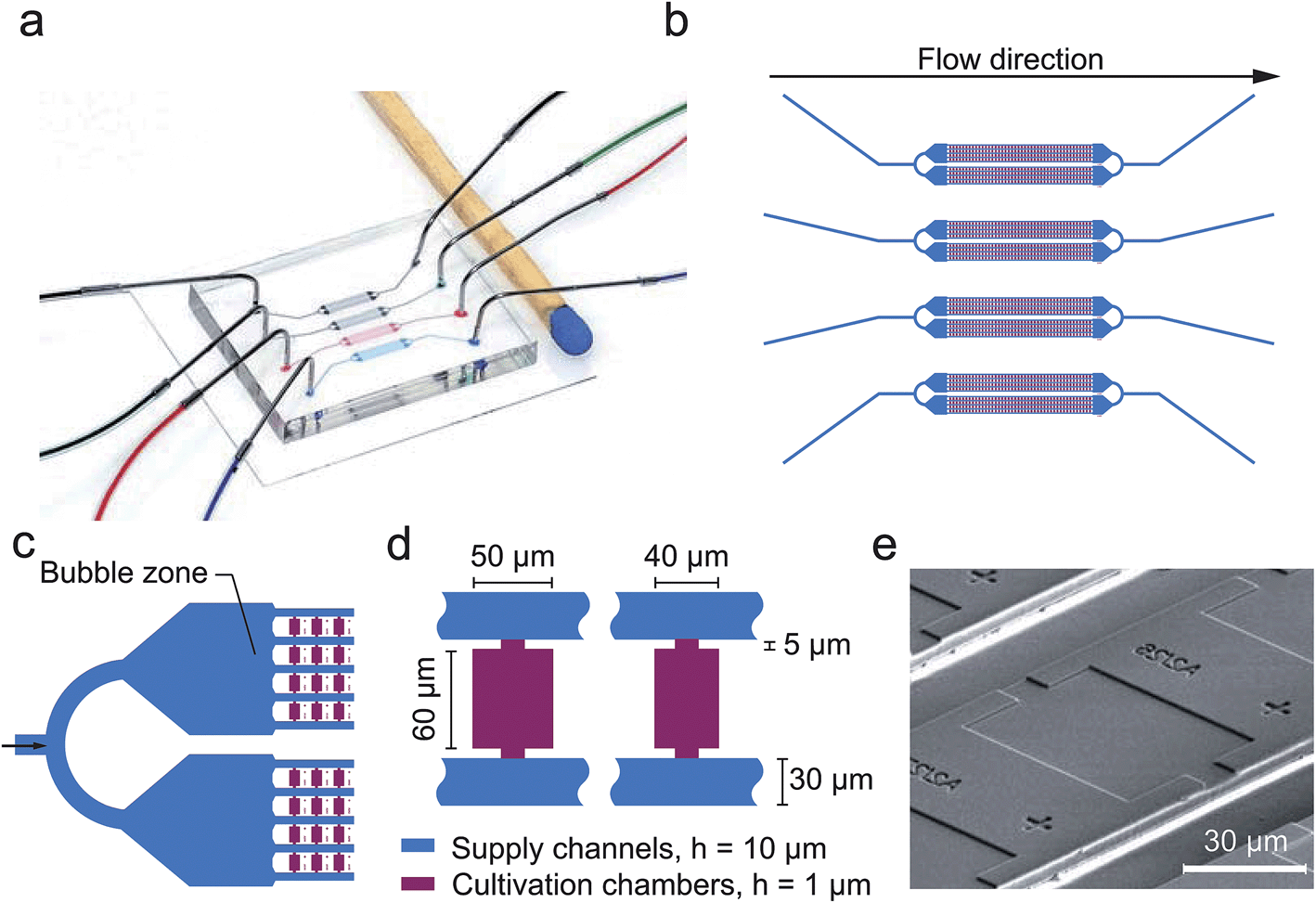 | ||
| Fig. 2 Microfluidic single cell cultivation device developed for air bubble based cell inoculation. (a) Microfluidic PDMS chip (h = 3 mm) bonded to a glass plate (h = 170 μm) and channels filled with differently colored dye; (b) each chip incorporates 4 separate channels for multiple analysis in parallel; (c) each channel branches into two cultivation arrays having an air bubble zone arranged in front; (d) two different cultivation chambers are arranged perpendicular to the flow interconnected between parallel supply channels; (e) SEM image of a single cultivation chamber. | ||
Experimental setup
Experiments were carried out using an inverted time-lapse microscope (Nikon TI-Eclipse, Nikon, Japan), equipped with an temperature incubator (PeCon GmbH, Germany). Images were recorded with a 10× objective during air bubble removal and a 100× objective during cultivation. Prior to cell inoculation desired air bubbles were injected using a pressure-driven pumping system (MCFS, Fluigent, France) as well as 2 external electromagnetic 3/2 valves (Cetoni GmbH, Germany), as illustrated in Fig. 3a. Afterwards and during cultivation, a syringe pump (NeMESYS, Cetoni GmbH, Germany) was used instead to continuously supply the cells with fresh medium at a flow rate of 200 nl min−1.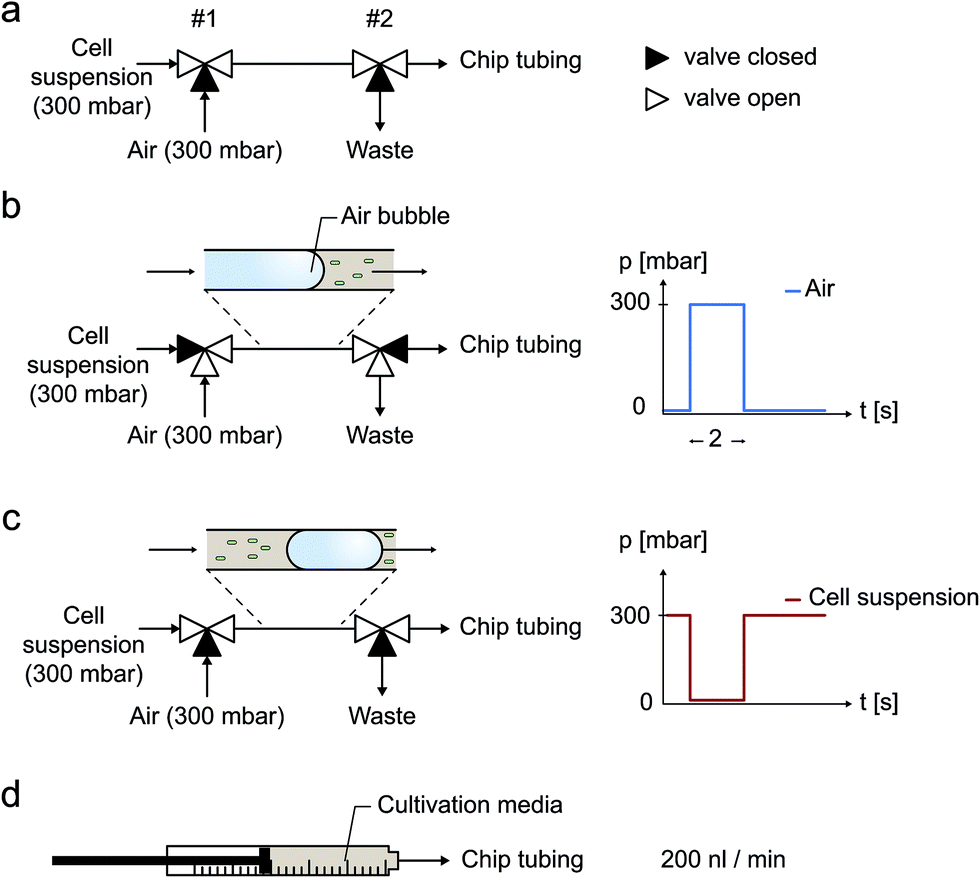 | ||
| Fig. 3 Air bubble injection into the microfluidic cultivation device using externally set-up electromagnetic valves. (a) Cell suspension flow, (b) air bubble injection, (c) cell suspension flow, (d) continuous cultivation media supply at 200 nl min−1. | ||
Bubble injection and cell inoculation procedure
Two electromagnetic 3/2 way valves connected in series were used to quickly and reproducible switch between cell suspension, pressurized air and a waste outlet. This external configuration was connected to the microfluidic chip inlets. Desired air bubbles for cell inoculation were induced by an automated valve actuation sequence to realize following procedure (Fig. 3a):(a) Cell suspension supply from a 300 mbar pressurized container (Fig. 3a).
(b) Within a 2 second pulse, air is injected at 300 mbar into the tubing (Fig. 3b).
(c) Cell suspension supply at 300 mbar is restored and the resulting entrapped air bubble is pushed through the tubing into the microfluidic channels (Fig. 3c).
(d) After the successful inoculation procedure, cultivation medium is supplied continuously (Fig. 3d).
Once in its operational position located in front of the fivefold channel junctions, air bubbles disappeared by continuous gas diffusion through the interfacing PDMS walls under constant pressure conditions (Fig. 4 and 6). Several pressure levels (200, 300, 400 and 500 mbar) were experimentally characterized. Phase contrast images of shrinking air bubbles were captured every second to derive geometrical parameter information by image analysis. After cell inoculation, 1184 growth chambers were optically inspected and trapped cells were counted to derive the trapping efficiency.
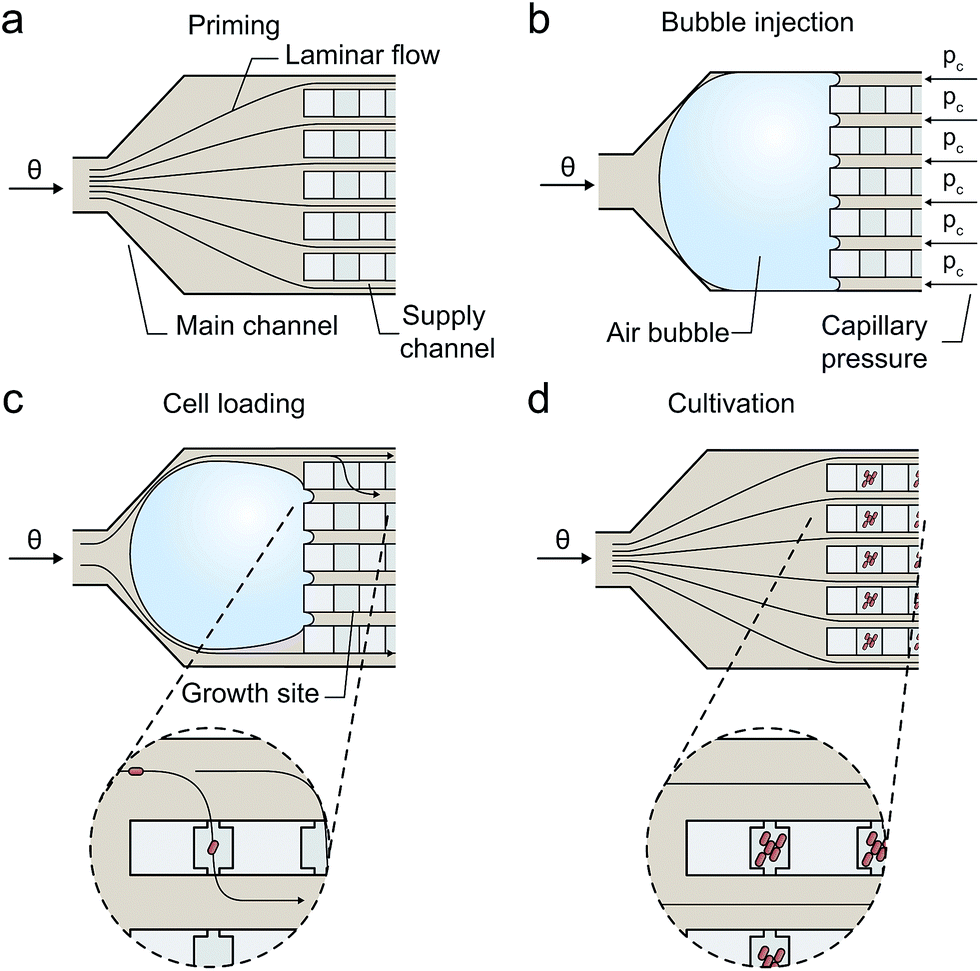 | ||
| Fig. 4 Air bubble based inoculation procedure for microbial single-cells: (a) priming: flow inside all supply channels is homogeneous and solely diffusion based mass transport occurs inside the growth sites. (b) Injection: air bubble is injected and blocks the multifold channel junction. The sum of capillary pressures pc counteracts the applied externally pressure keeping the air bubble at its operating position. (c) Cell inoculation: the air bubble temporary distorts the flow profile resulting in an inhomogeneous flow through the parallel supply channels and convection through the growth sites. Single cells get inoculated. (d) Cultivation: growth media is supplied continuously and mass transport inside the growth sites is based on diffusion only. | ||
Fluorescent flow tracer analysis
Flow characterization was performed by infusing fluorescently labeled latex beads of 1 μm diameter (blue fluorescent 350/440) and 200 nm diameter (yellow-green fluorescent 505/515), respectively. FluoSpheres® carboxylate-modified microspheres (2% solids) were purchased from Molecular Probes (Invitrogen, USA). 1 μm beads were applied to visualize the laminar flow profile during air-bubble-assisted cell loading thereby mimicking cell trapping. 200 nm beads were applied to track the diffusive particle behavior during subsequent normal cultivation conditions. Prior to flow visualization, the channels were primed with 0.1% BSA solution for 60 minutes to minimize unspecific bead adhesion. All bead suspensions were diluted (5 μl in 1000 μl of 0.1% BSA). An exposure time of 500 ms was used to record the trajectories of the 1 μm beads, whereas an exposure time of 10 s was used to track the 200 nm beads. Fluorescently labeled beads were excited using a fluorescent light source (Intensilight, Nikon, Japan) at the maximum intensity and using appropriate optical filters.Cultivation of C. glutamicum
C. glutamicum wild type was pre-cultured in 20 ml of fresh CGXII medium29 in 100 ml shake flasks and shaken at 30 °C at 150 rpm overnight. Prior to inoculation into the chip, 500 μl from the overnight culture was transferred to 20 ml of fresh CGXII and incubated until an optical density of 1 was reached. After successful inoculation, fresh CGXII medium was infused.Images of growth sites were recorded every 10 minutes to derive the growth rate. Growth of single-cell microcolonies was analyzed by using a customized semi-automated image analysis toolkit. Advanced image analysis methods were adapted for time-lapse image stacks and implemented as a plugin for the ImageJ platform.30
Results and discussion
Single-cell inoculation procedure
The present device operates with a simple microfluidic geometry, thereby minimizing technological efforts and improving applicability. The configuration facilitates exclusively diffusion based mass transport inside the cultivation chambers if the two parallel media supplying volume flows are equal and no pressure gradient is present through the cultivation site. This is achieved by configuring the single inlet channel so that it branches into multiple parallel supply channels of identical hydrodynamic resistance (Fig. 2c). Successfully inoculated cells grow under minimal shear stress and with efficient media exchange by diffusion based mass transport.By utilizing a nanoliter sized air bubble entrapped in front the supply channels, a simple and reproducible inoculation procedure was realized as illustrated in Fig. 4. The procedure follows following sequence:
(a) Device priming (homogenous flow conditions inside the channels and solely diffusion based mass transport inside the cultivation chambers);
(b) air bubble injection; air bubble blocks the five-fold channel junction.
(c) cell suspension infusion and cell inoculation (inhomogeneous flow conditions inside the supply channels supporting convection across the chambers until bubble fully disappears);
(d) continuous medium supply and cell cultivation (homogenous flow conditions along the channels and solely diffusion across the chambers).
To initiate single cell inoculation, air was injected during a 2 s pulse from a pressurized air supply into the external tubing by actuating two electromagnetic valves in automated sequence (Fig. 3). The resulting air bubble was then pushed with the cell suspension flow downstream the tubing into the connected microfluidic chip where it blocked the multifold junction to the parallel supply channels due to the counteracting capillary force as illustrated in Fig. 4b (ESI section S1†).
Once in its operating position at the channel junction, the bubble volume continuously decreases due to the high gas permeability of the interfacing PDMS wall material. However, the bubble must not be pushed through the entire device at higher pressure levels, since cells may be detached from the cultivation chambers by the propagating air–liquid interface. The transient blockage of several media supply channels creates an flow imbalance in the parallel supply channels, thereby inducing convection through the cultivation chambers supporting the inoculation of single microbes. Infused cells simply remain trapped inside the shallow cultivation chambers by cell-wall interactions. During this stochastic filling process cells are distributed inside the chambers. Preferably, single cells become trapped inside sufficient number of chambers. Typically these stochastic filling processes follow a well-known Poisson distribution. However, in the present configuration the inoculation rate is not constant over time and the individual chambers, thus Poisson is not applicable. The trapping efficiency depends on various parameters, for example, the inoculation period, cell suspension density, flow velocities, cell size, and was optimized experimentally. As soon as the air bubble completely disappears, the inoculation procedure is finished and the cultivation medium is continuously pumped through the channels instead.
Flow tracer characterization
Fluorescently labeled beads and long exposure times during fluorescence microscopy were used to visualize the particle trajectories during normal cultivation conditions and inoculation conditions as shown in Fig. 5a and c. Under normal cultivation conditions the volume flows were equal in all supply channels as visualized by identical tracer patterns (Fig. 5a and b). Fluorescent beads entering the growth chambers exclusively revealed diffusive and random motion (Fig. 5c).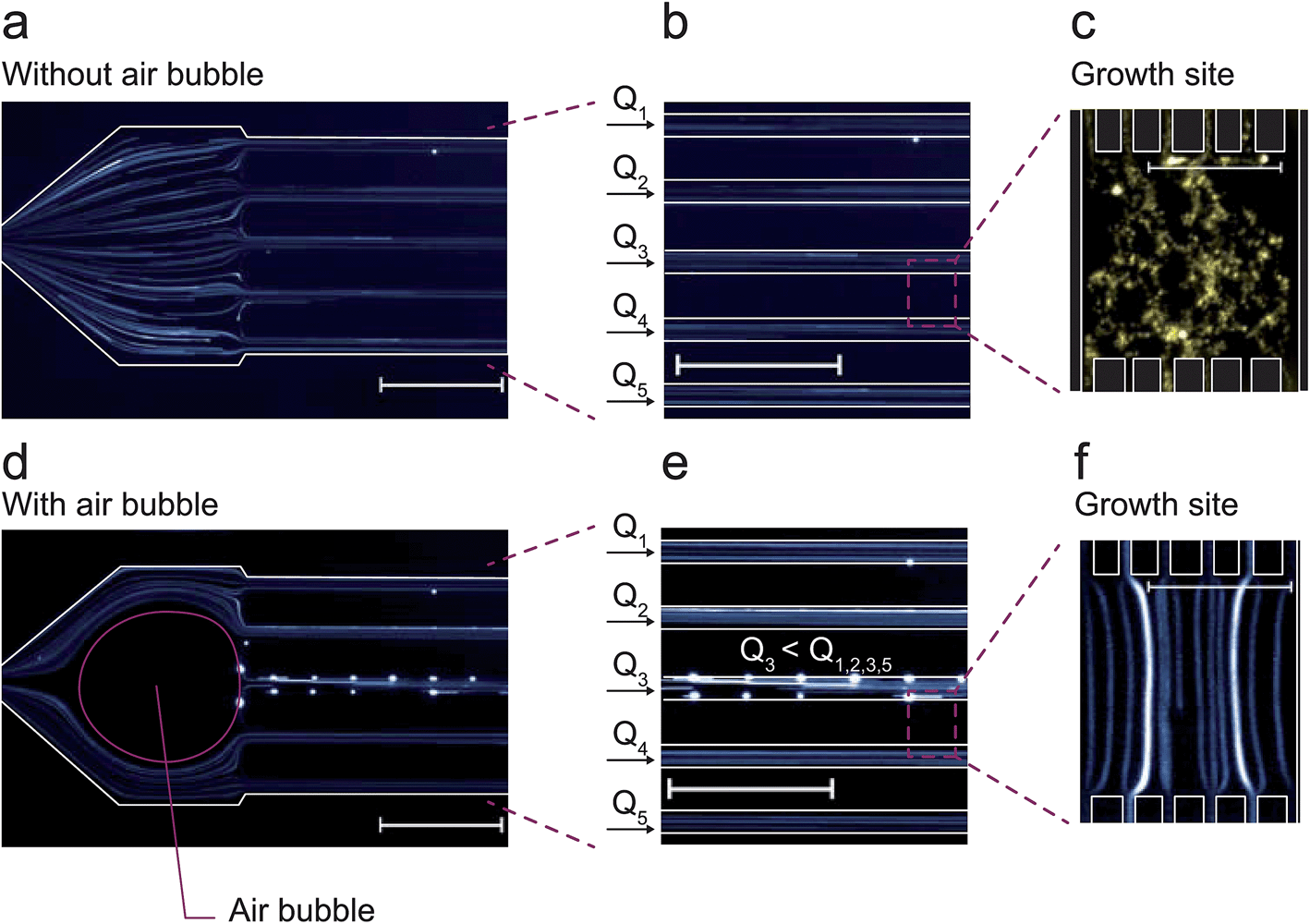 | ||
| Fig. 5 Fluorescent flow tracer analysis: (a–c) normal cultivation conditions with equal flow rates inside all supply channels; (c) diffusive mass transport conditions inside the cultivation chambers; (d) air bubble is deployed at the fivefold channel junction resulting in (e) a temporary inhomogeneous laminar flow profile and different volume flows Φ1, Φ2, Φ3, Φ4 and Φ5 and thus (f) laminar flow through the cultivation chambers (scale bar 25 μm). | ||
As soon as the air bubble was injected (Fig. 5d and e), tracer analysis revealed unbalanced flow rates and resulting fluid convection through the cultivation chambers. The fluorescent beads then followed straight laminar flow lines through the chamber (Fig. 5f). Several beads attached to the channel walls resulting in noticeably bright sports in the image (Fig. 5d and e) not further influencing the analysis. The flow situation was completely reversed once the air bubble disappeared by diffusion in less than two minutes.
Air bubble injection
During bubble injection the applied flow pressure should not exceed the sum of the counteracting capillary pressures to avoid loss of the functional bubble. The maximum pressure during the procedure results from the sum of the counteracting capillary pressures at the liquid air interface (Fig. 4b). For a rectangular channel with constant cross-section and width (w) ≫ height (h), the capillary pressure can be estimated from eqn (1) (more details see ESI S1†).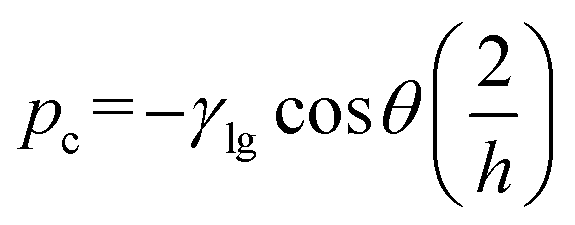 | (1) |
The capillary pressure (pc) of one single supply channel with a height of h = 10 μm, a surface tension γ = 0.07 N m−1 and a contact angle θ = 111° results in approximately pc = 120 mbar. Thus in the present geometry, the resulting fivefold counteracting pressure is about 600 mbar. This was in good agreement with our experimental findings, as the bubble was ineffectively pushed through the junction at an inlet pressure around 600 mbar. By adjusting the device layout, this restriction can be easily increased to specific requirements, but flow rates during inoculation were satisfactory in our experiments. At a later phase during cell inoculation and a smaller bubble size, it still remains trapped at its position mainly due the surface tension and the flow diverging around the bubble.
Air bubble diffusion
The available time window for inoculation is greatly dependent on the gas permeation rate through the surrounding wall material. The procedure is limited to PDMS or other materials or device configurations supporting gas diffusion.Fig. 6a shows a time-lapse image series of an injected air bubble, which then continuously shrank due to diffusion at the applied pressure p = 300 mbar. It can be seen from Fig. 6b that a minimal pressure of 300 mbar was needed to remove the air bubble effectively. Each inoculation sequence was repeated 3 times and the measured cross-sectional bubble area plotted over time. A simple first-order decay expression was used to fit the bubble removal rate31 (details in ESI S2†).
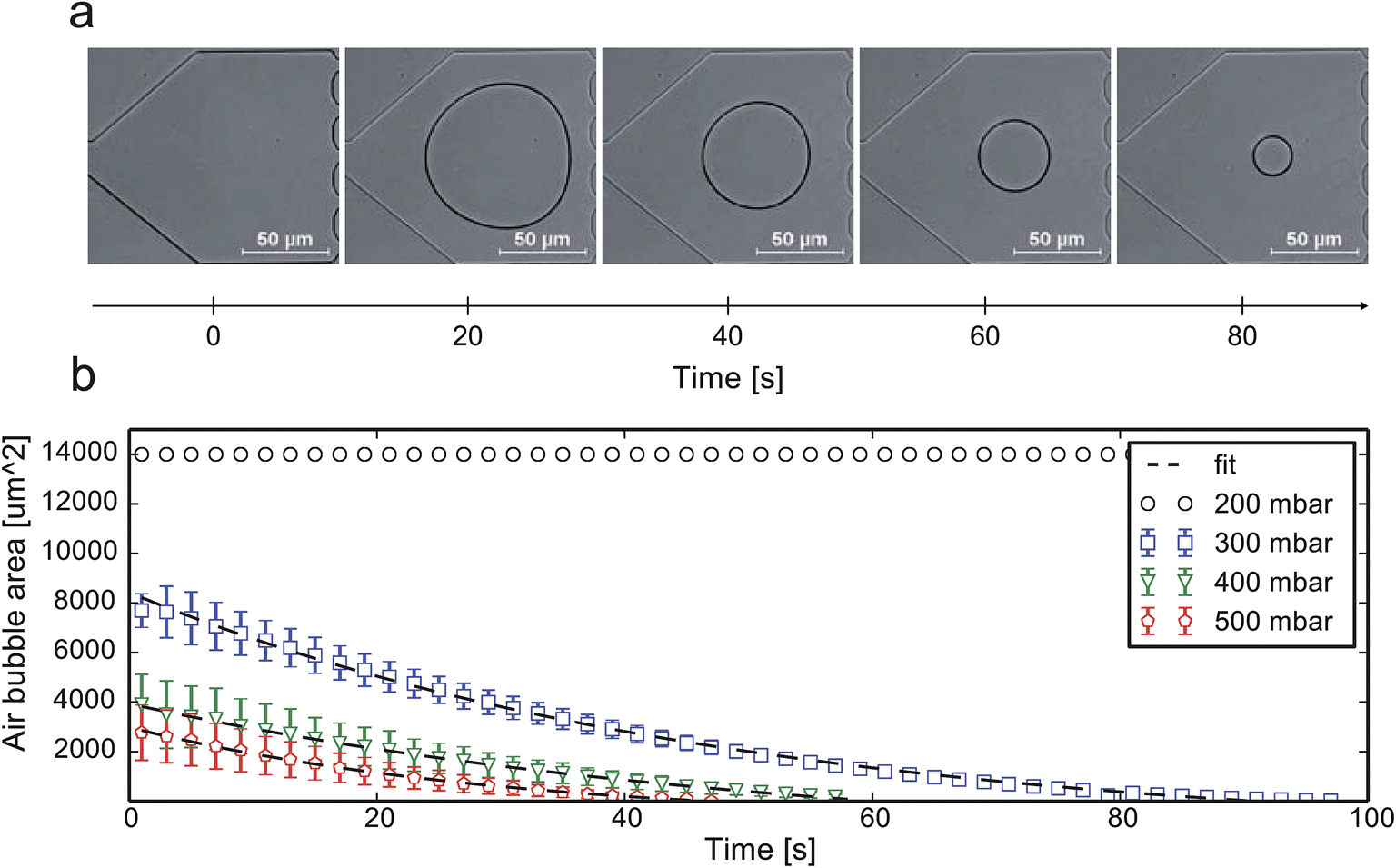 | ||
| Fig. 6 Experimental air bubble removal validation: (a) time-lapse images after the successful injection of the air bubble and continuous decay at 300 mbar; (b) air bubble surface area decay vs. time for various pressure settings and respective fit. | ||
Cell loading performance
The presented method was validated using the microorganism C. glutamicum. In order to guarantee isogenic starting conditions of each microcolony, only one cell per growth side needs to be trapped during inoculation. During cell division of C. glutamicum two related cells often remain attached to each other.32 These cell pairs were also considered as one cell since they obviously arise from the same lineage.The inoculation was performed at 300 mbar inoculation pressure and a cell suspension with an optical cell density of OD 0.1 and OD 1. Each cultivation chamber was inspected by microscopy afterwards and the entrapped cell number was counted. Fig. 7a and c shows the distribution of trapped cells per growth chamber (50 μm × 60 μm) for both cell suspension densities. In addition, the position distributions of all trapped cells inside the growth chamber is depicted in Fig. 7b and d. It is noted that a central cell positioning is most desirable to allow homogenous and radial colony growth with maximum diameter. Inoculation was performed most efficiently at a low cell density (OD 0.1). On average, 52 chambers out of 592 were successfully inoculated with one individual cell (technical replicate: N = 3). In the same device 57 out of 592 chambers (40 μm × 60 μm) were satisfactory inoculated with a single cell as shown in Fig. 7e–h. The small size difference did not significantly impact cell loading efficiency. At first sight, these numbers seem low, but typically time lapse microscopy can hardly handle more regions of interest if appropriate time resolution in the order of a few minutes is anticipated. Inoculation at 300 mbar within 100 s resulted in a trapping efficiency of around 10%. Even higher cell density led to an increased number of chambers with more than one cell and more cells being trapped at the inlet of the growth sites rather than in the center of the chamber.
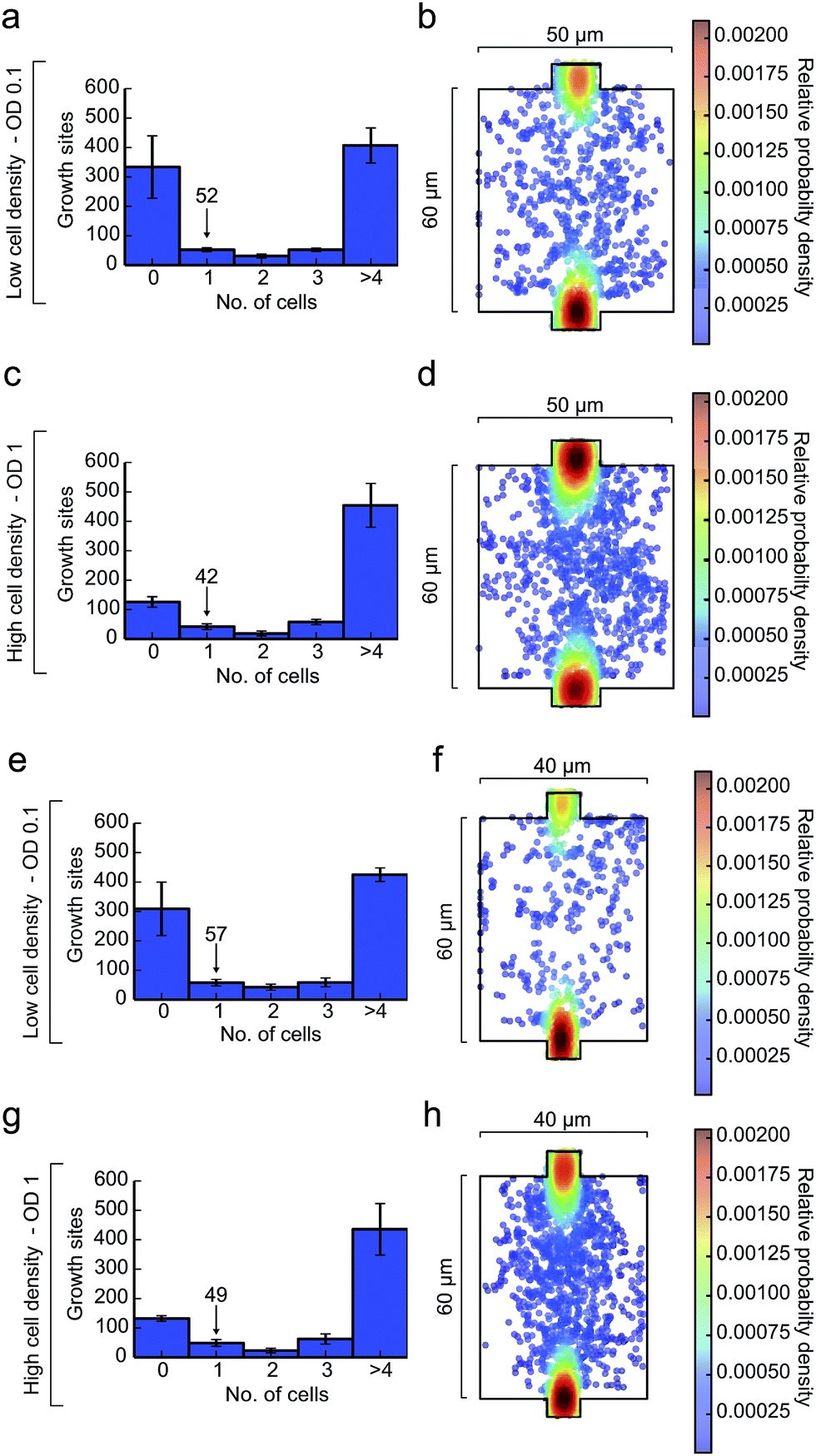 | ||
| Fig. 7 Cell trapping analysis (N = 3) of C. glutamicum at 300 mbar and different cell suspension densities for growth sites of 50 μm × 60 μm (N = 592) and 40 μm × 60 μm (N = 592). (a and b, e and f) OD 0.1: inoculation efficiency distribution and single-cell location plot; (c and d, g and h) OD 1: inoculation efficiency distribution and single-cell location plot. | ||
Viability and cell growth
Finally, the growth of C. glutamicum after our inoculation procedure was validated. Each colony exhibited excellent viability and showed no impact on cellular growth or morphology. Fig. 8a shows microcolony growth for 9 hours in one of the observed growth sites. As an example, 23 microcolonies were analyzed in more detail and single-cell growth rates μmax were derived as shown in Fig. 8b and c. We found the maximum average growth rate μmax_mean to be 0.6 ± 0.03 h−1, which corresponded very well to previously published results obtained with C. glutamicum in microfluidic devices.33 The shifts in time of the different growth curves can be explained by different cell cycle states of the individual mother cells.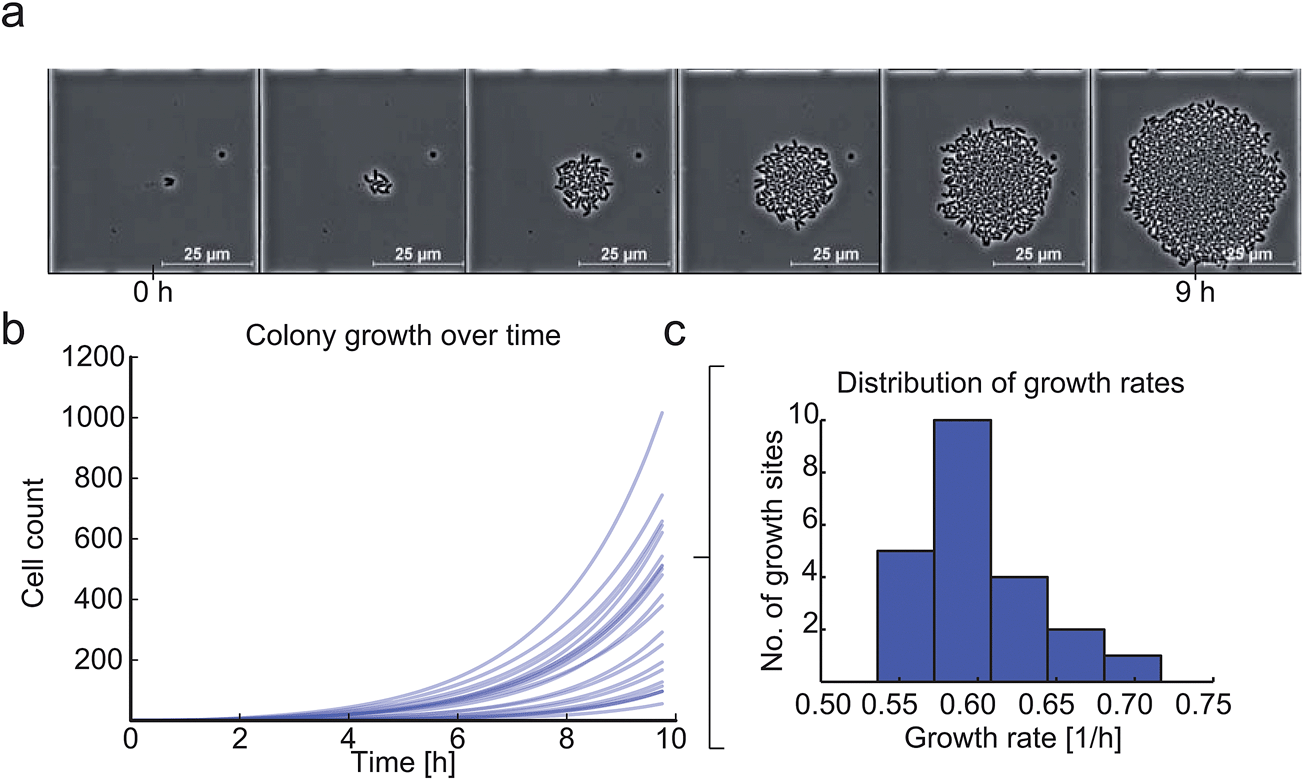 | ||
| Fig. 8 Viability analysis of C. glutamicum after the inoculation procedure: (a) time-lapse image series of C. glutamicum colony growing in one growth site over 9 hours. (b and c) microcolony growth curves of multiple microcolonies (N = 23) revealing an average maximum growth rate μmax_mean = 0.6 ± 0.04 h−1. | ||
Conclusion
A fast, gentle and simple microbial cell inoculation procedure for microfluidic single-cell analysis is described. Air bubbles are often considered as a negative side effect in microfluidics.34 However, we have shown that the application of an injected air bubble can serve as a reproducible mechanism to modify laminar flow conditions. In this way, convective flow is temporarily induced in more than 1000 cultivation chambers in parallel, which under normal conditions operate exclusively under diffusive mass transport. Within an inoculation time of 100 s, C. glutamicum cells were inoculated by convection and single bacteria remain successfully trapped by cell-wall interactions. During cell trapping, the air bubble continuously reduced in size due to gas permeation through the surrounding. Once the bubble had been removed, normal flow conditions were fully restored. The inoculation efficiency of 10% was sufficient for our experiments but can certainly be further enhanced. The method is not restricted to the geometries presented here and can be adapted to other microfluidic single-cell analysis devices. Inoculated C. glutamicum cells survived the procedure well as shown by their vital growth and morphology.Acknowledgements
This work was performed in part at the Helmholtz Nanoelectronic Facility (HNF) of Forschungszentrum Jülich GmbH. The authors would like to thank the HNF for help and support. The German Federal Ministry of Education and Research (031A095A) and the Helmholtz Association (VH-NG-1029) are gratefully acknowledged for funding. The DFG is acknowledged for funding within the priority programme SPP1617. Dr Jan Eijkel (Twente University, NL) is acknowledged for his support.References
- F. S. O. Fritzsch, C. Dusny, O. Frick and A. Schmid, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 129–155 CrossRef CAS PubMed.
- K. Klepárník and F. Foret, Anal. Chim. Acta, 2013, 800, 12–21 CrossRef PubMed.
- V. Lecault, A. K. White, A. Singhal and C. L. Hansen, Curr. Opin. Chem. Biol., 2012, 16, 381–390 CrossRef CAS PubMed.
- K. R. Love, S. Bagh, J. Choi and J. C. Love, Trends Biotechnol., 2013, 31, 16–22 CrossRef PubMed.
- B. Okumus, S. Yildiz and E. Toprak, Curr. Opin. Biotechnol., 2014, 25, 30–38 CrossRef CAS PubMed.
- H. B. Yin and D. Marshall, Curr. Opin. Biotechnol., 2012, 23, 110–119 CrossRef CAS PubMed.
- A. Schmid, H. Kortmann, P. S. Dittrich and L. M. Blank, Curr. Opin. Biotechnol., 2010, 21, 12–20 CrossRef CAS PubMed.
- A. Grünberger, W. Wiechert and D. Kohlheyer, Curr. Opin. Biotechnol., 2014, 29, 15–23 CrossRef PubMed.
- V. Lecault, M. VanInsberghe, S. Sekulovic, D. J. H. F. Knapp, S. Wohrer, W. Bowden, F. Viel, T. McLaughlin, A. Jarandehei, M. Miller, D. Falconnet, A. K. White, D. G. Kent, M. R. Copley, F. Taghipour, C. J. Eaves, R. K. Humphries, J. M. Piret and C. L. Hansen, Nat. Methods, 2011, 8, 581–593 CrossRef CAS PubMed.
- W. Mather, O. Mondragon-Palomino, T. Danino, J. Hasty and L. S. Tsimring, Phys. Rev. Lett., 2010, 104, 208101 CrossRef.
- J. Uhlendorf, A. Miermont, T. Delaveau, G. Charvin, F. Fages, S. Bottani, G. Batt and P. Hersen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14271–14276 CrossRef CAS PubMed.
- Z. Long, E. Nugent, A. Javer, P. Cicuta, B. Sclavi, M. C. Lagomarsino and K. D. Dorfman, Lab Chip, 2013, 13, 947–954 RSC.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- D. D. Carlo, L. Y. Wu and L. P. Lee, Lab Chip, 2006, 6, 1445–1449 RSC.
- C. Probst, A. Grünberger, W. Wiechert and D. Kohlheyer, Micromachines, 2013, 4, 357–369 CrossRef PubMed.
- A. Grünberger, N. Paczia, C. Probst, G. Schendzielorz, L. Eggeling, S. Noack, W. Wiechert and D. Kohlheyer, Lab Chip, 2012, 12, 2060–2068 RSC.
- H. Kortmann, P. Chasanis, L. M. Blank, J. Franzke, E. Y. Kenig and A. Schmid, Lab Chip, 2009, 9, 576–585 RSC.
- S. S. Lee, I. A. Vizcarra, D. H. E. W. Huberts, L. P. Lee and M. Heinemann, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4916–4920 CrossRef CAS PubMed.
- J. Ryley and O. M. Pereira-Smith, Yeast, 2006, 23, 1065–1073 CrossRef CAS PubMed.
- G. Schendzielorz, M. Dippong, A. Grünberger, D. Kohlheyer, A. Yoshida, S. Binder, C. Nishiyama, M. Nishiyama, M. Bott and L. Eggeling, ACS Synth. Biol., 2013, 3, 21–29 CrossRef PubMed.
- A. Groisman, C. Lobo, H. J. Cho, J. K. Campbell, Y. S. Dufour, A. M. Stevens and A. Levchenko, Nat. Methods, 2005, 2, 685–689 CrossRef CAS PubMed.
- G. Ullman, M. Wallden, E. G. Marklund, A. Mahmutovic, I. Razinkov and J. Elf, Philos. Trans. R. Soc., B, 2013, 368, 20120025 CrossRef CAS PubMed.
- X. He, H. Kimura and T. Fujii, J. Robot. Mechatron., 2013, 25, 623–630 Search PubMed.
- A. Prindle, P. Samayoa, I. Razinkov, T. Danino, L. S. Tsimring and J. Hasty, Nature, 2012, 481, 39–44 CrossRef CAS PubMed.
- D. Falconnet, A. Niemisto, R. J. Taylor, M. Ricicova, T. Galitski, I. Shmulevich and C. L. Hansen, Lab Chip, 2011, 11, 466–473 RSC.
- C. Probst, A. Grünberger, W. Wiechert and D. Kohlheyer, J. Microbiol. Methods, 2013, 95, 470–476 CrossRef PubMed.
- M. Kolnik, L. S. Tsimring and J. Hasty, Lab Chip, 2012, 12, 4732–4737 RSC.
- L. Wang, X. F. Ni, C. X. Luo, Z. L. Zhang, D. W. Pang and Y. Chen, Biomed. Microdevices, 2009, 11, 679–684 CrossRef CAS PubMed.
- A. Gruenberger, C. Probst, A. Heyer, W. Wiechert, J. Frunzke and D. Kohlheyer, J. Visualized Exp., 2013, e50560 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- J. H. Kang, Y. C. Kim and J.-K. Park, Lab Chip, 2008, 8, 176–178 RSC.
- M. Letek, M. Fiuza, E. Ordóñez, A. Villadangos, A. Ramos, L. Mateos and J. Gil, Antonie van Leeuwenhoek, 2008, 94, 99–109 CrossRef CAS PubMed.
- A. Grünberger, J. van Ooyen, N. Paczia, P. Rohe, G. Schiendzielorz, L. Eggeling, W. Wiechert, D. Kohlheyer and S. Noack, Biotechnol. Bioeng., 2013, 110, 220–228 CrossRef PubMed.
- L. Kim, Y.-C. Toh, J. Voldman and H. Yu, Lab Chip, 2007, 7, 681–694 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ay02257b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |