Highly enantioselective hydrogenation of α-aryl-β-substituted acrylic acids catalyzed by Ir-SpinPHOX†‡
Yi
Zhang
ab,
Zhaobin
Han
b,
Fuying
Li
a,
Kuiling
Ding
*b and
Ao
Zhang
*a
aSynthetic Organic & Medicinal Chemistry Laboratory, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, P.R. China. E-mail: aozhang@mail.shcnc.ac.cn; Fax: (+86) 21 50806035
bState Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, P.R. China. E-mail: kding@mail.sioc.ac.cn; Fax: (+86) 21 64166128
First published on 24th November 2009
Abstract
The enantioselective hydrogenation of a series of challenging substrates, α-aryl-β-substituted acrylic acids, was realized with high efficiency and enantioselectivity (up to 96%) under the catalysis of Ir(I) complex of Spiro-based P,N ligand, SpinPHOX.
Enantiopure α-arylpropanic acid and their derivatives are key intermediates for the synthesis of various biologically important compounds.1 The development of a straightforward approach to their direct access represents one of the most challenging issues in terms of synthetic chemistry, in which asymmetric hydrogenation of the corresponding prochiral α-arylacrylic acid derivatives has proven to be the most promising.2 In the past decades, transition metal catalyzed asymmetric hydrogenation of α,β-unsaturated carbonyl compounds has been very successful.3–5 However, asymmetric hydrogenation of α-aryl-β-substituted acrylic acids with high efficiency remains unsatisfactory. On the other hand, the asymmetric hydrogenation of unsaturated carboxylic acids using chiral Ir catalysts has recently received significant attention. For example, the Ir–PHOX or Ir–SIPHOX complexes (Scheme 1) have been employed in the asymmetric hydrogenation of a variety of α,β-unsaturated acids including 2-phenylacrylic acid6 or α-alkyl-β-aryl/alkyl acrylic acids,7 respectively, affording the corresponding optically active acids in good yields with moderate to excellent ees. Despite of the facts mentioned above, the catalytic asymmetric hydrogenation of α-aryl-β-substituted acrylic acids 1, the most direct approach to a key chiral intermediate for the synthesis of potential antidiabetic drug PSN-GK1 (in phase I clinic),8 still remains a challenging theme (Scheme 1). In the present communication, we report our preliminary results on the use of Ir(I) complexes of a novel class of spiro[4,4]-1,6-nonadiene-based phosphine–oxazoline ligands (SpinPHOX, 2)9 in the catalytic asymmetric hydrogenation of a variety of α-aryl-β-substitued acrylic acids (1), leading to the production of a series of biologically interesting carboxylic acids with excellent yields and up to 96% enantioselectivity.
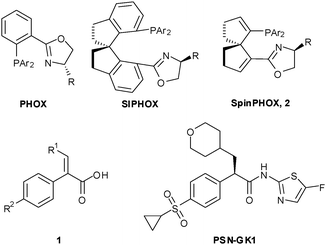 | ||
| Scheme 1 Representative P,N chiral ligands for Ir(I)-catalyzed hydrogenation of α,β-unsaturated carboxylic acids. | ||
The trisubstituted α,β-unsaturated carboxylic acid, (E)-2-phenyl-3-(tetrahydro-2H-pyran-4-yl) acrylic acid (1a), was initially taken as the model substrate in order to optimize the hydrogenation reaction conditions. The iridium complexes of (R,S)-2a and (S,S)-2a were employed as the catalyst (1 mol%).10 After screening of various reaction conditions (see Table S1 in electronic supplementary information, ESI‡) including the effects of solvent, temperature, and hydrogen pressure, the hydrogenation of 1a in methanol at 50 °C under 30 atm of H2 turned out to be optimal. Considering the poor solubility of the substrate in the reaction medium and the potential impact of a carboxylate group on the catalysis, we then examined the effect of base additives on the reaction rate and enantioselectivity of the catalysis. Notably, the addition of 1 equiv. of NEt37,11 (see Table S2 in ESI‡) gave the best result in terms of both conversion of the substrate and enantiomeric excess of the product. In the presence of 1 mol% of Ir(I)/(R,S)-2a, the hydrogenation of 1a proceeded smoothly with 92% conversion in 20 h, affording (–)-2-phenyl-3-(tetrahydro-2H-pyran-4-yl) propanoic acid (3a) with 93% ee (Table 1, entry 1). In order for comparison, Pfaltz’s Ir/PHOX (Ar = Ph, R = iPr) catalyst was also employed in the hydrogenation of 1a, 31% conv. of substrate and 32% ee of the product were obtained. It should be noted in the hydrogenation of the (Z)-isomer of 1a in the presence of (R,S)-2h under identical reaction conditions, the conversion of substrate is negligible.
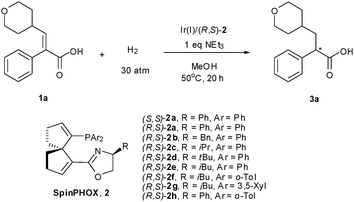
| Entry | Ligand | Conv. (%)b | Ee (%)c |
|---|---|---|---|
| a Reaction conditions: 0.1 mmol scale of 1a with [1a] = 0.1 mol L−1 in MeOH, PH2 = 30 atm, s/c = 100, T = 50 °C. b Determined by 1H NMR. c Determined by HPLC analysis on a Chiralcel AD-H column after esterification with diazomethane. | |||
| 1 | (R,S)-2a | 92 | 93 (–) |
| 2 | (S,S)-2a | > 99 | 69 (+) |
| 3 | (R,S)-2b | 56 | 84 (–) |
| 4 | (R,S)-2c | 59 | 58 (–) |
| 5 | (R,S)-2d | 85 | 92 (–) |
| 6 | (R,S)-2e | 75 | 92 (–) |
| 7 | (R,S)-2f | > 99 | 94 (–) |
| 8 | (R,S)-2g | 76 | 83 (–) |
| 9 | (R,S)-2h | > 99 | 96 (–) |
The chirality at the spiro backbone of ligand 2 was found to have a significant impact on the asymmetric induction of the reaction. The combination of a R configuration in the spiro backbone and an S configuration in the oxazoline component was disclosed to be a matched case as in Ir(I)/(R,S)-2a, whereas the Ir(I) complex of the corresponding diastereomeric ligand (S,S)-2a gave lower enantioselectivity with opposite asymmetric induction, although a complete conversion of 1a was observed in the latter (entry 2 vs. 1 in Table 1). Therefore, the sign of asymmetric induction in the hydrogenation is predominately determined by the chirality of the spiro backbone. The examination of the substituent effect of the oxazoline moiety in ligand (R,S)-2a–e revealed that (R,S)-2a bearing a phenyl group was the best in terms of both reactivity and enantioselectivity (entry 1 vs. entries 3–6 in Table 1). To further improve the catalytic performance of the reaction, three new ligands (R,S)-2f–h bearing different aryl groups at the P atom were synthesized and employed in the hydrogenation of 1a (entries 7–9). Gratifyingly, the catalyst composed of ligand (R,S)-2h with two o-tolyl groups at P-atom showed remarkable reactivity (>99% conv.) and enhanced enantioselectivity (96% ee) (Table 1, entry 9).
With the optimized catalyst Ir(I)/(R,S)-2h in hand, we then investigated its applicability in the catalysis of trisubstituted α,β-unsaturated carboxylic acids 1a–l. The substrates were well-chosen considering the possibility for the use of the hydrogenation products as the key intermediates for the synthesis of analogous compounds of PSN-GK1.8 Accordingly, a variety of α-aryl acrylic acids with various β-substituents (variation at R1) (1a–e) were prepared and subsequently hydrogenated in methanol using Ir(I)/(R,S)-2h (1 mol%) as the catalyst under 30 atm of H2 at 50 °C. The corresponding hydrogenated carboxylic acids were obtained quantitatively with good to excellent enantioselectivities (88–96% ee; Table 2, entries 1–8). It is of note that the reaction of β-alkyl or phenyl substituted acrylic acids (1a, 1c–e) generally affords excellent enantioselectivity (94–96%) while the parent α-phenyl acrylic acid (1b) provides a somewhat inferior result (88% ee), implying that the present catalyst system is particularly suitable for the sterically more demanding substrate at the β-position of acrylic acids. On the other hand, the electronic properties of substituent (R2) at para-position of α-phenyl ring has less impact on the enantioselectivity of the reaction (entry 1 vs. entries 6–8, Table 2).
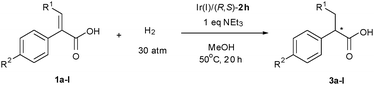
| Entry | R1 | R2 | Ee (%)b |
|---|---|---|---|
| a For the reaction conditions and analysis of reaction system, see footnotes a,b in Table 1. The complete conversion of substrates was observed for all entries. b Determined on a Chiralcel column after esterification with diazomethane. The absolute configuration was determined by comparison of the optical rotation with that reported in the literature.12 c Determined on a Chiralcel AD-H column after being transformed to dehalogenated methyl ester. d s/c = 50. e Determined on a Chiralcel OD-H column after being transformed to their corresponding sulfone derivatives. | |||
| 1 | Tetrahydro-2H-pyran-4-yl (1a) | H | 96 (–) |
| 2 | H (1b) | H | 88 (R) |
| 3 | Me (1c) | H | 94 (R) |
| 4 | Ph (1d) | H | 94 (R) |
| 5 | Cyclopentyl (1e) | H | 95 (–) |
| 6 | Tetrahydro-2H-pyran-4-yl (1f) | MeO | 95 (–) |
| 7 | Tetrahydro-2H-pyran-4-yl (1g) | F | 95 (–) |
| 8 | Tetrahydro-2H-pyran-4-yl (1h) | I | 91c (–) |
| 9 | Tetrahydro-2H-pyran-4-yl (1i) | MeSO2 | 91 (R) |
| 10 | Tetrahydro-2H-pyran-4-yl (1j) | c-PrSO2 | 89 (R) |
| 11 | Tetrahydro-2H-pyran-4-yl (1k) | MeS | 94d,e (R) |
| 12 | Tetrahydro-2H-pyran-4-yl (1l) | c-PrS | 95e (R) |
Both the β-tetrahydro-2H-pyran-4-yl moiety and the sulfonyl unit at the para-position of the α-aryl group of the carboxylic acid have proven to be critically important in the chiral antidiabetic drug PSN-GK1 and analogous molecules. Therefore, the development of the catalyst system for the direct access of these key intermediates is highly desirable. Accordingly, the ideal catalyst must tolerate the presence of both sulfonyl and tetrahydro-2H-pyran-4-yl moieties in the acid substrates or related precursors. As described above, our catalyst system is particularly effective for the hydrogenation of substrate 1a containing β-tetrahydro-2H-pyran-4-yl moiety. The further introduction of an alkylthio- or alkylsulfonyl group to the para-position of the α-phenyl of acrylic acid 1a afforded substrates 1i–l, which were subsequently hydrogenated under the optimized conditions in the presence of Ir(I)/(R,S)-2h. It was good to find that the hydrogenation of all these four substrates gave the corresponding carboxylic acids 3i–l in almost quantitative yields with 89–95% ee (entries 9–12, Table 2). These facts indicate that the present catalyst system not only has the advantage of high enantioselective control in the reaction but also possesses excellent substrate functional group tolerance if one considers the strong coordination tendency of sulfide or sulfone with a transition metal catalyst. Fortunately, the hydrogenation of 1l afforded the corresponding carboxylic acid 3l with 95% ee which can be directly oxidized by mCPBA to give 3j in >99% yield without any racemization. Simple condensation of 3j with 5-fluoro-2-aminothiazole gave the antidiabetic drug PSN-GK1 conveniently in good yield with the same enantiomeric excess. In order to demonstrate the practicality of the catalysis, a hydrogenation with 4 mmol (1.2 g) scale of substrate 1l was carried out in the presence of 1 mol% of (R,S)-2h under 30 atm of H2 at 50 °C, complete conversion of 1l and 95% ee of 3l were obtained. It should be noted that the chirality of the stereocenter in the PSN-GK1 molecule is critically important for its bioactivity, in which the R-enantiomer has the desired clinical actions in vivo while the S-enantiomer is completely inactive, although both R- and S-congeners act equi-potently in vitro.13 Therefore, the present work has not only furnished a facile method for rapid synthesis of the related chiral drug in a large scale for clinical study, but has also provided an excellent opportunity for generating a small library of both enantiomers of analogous compounds with the variation at the α- or β-position of the carboxylic moiety for further structure–activity-relationship (SAR) study to identify more potent leading compounds.
This work is partially supported by NSFC (nos. 20632060, 20821002, 20620140429, 30772625), the Chinese Academy of Sciences, the Major Basic Research Development Program of China (grant no. 2010CB833300), the Science and Technology Commission of Shanghai Municipality, and Merck Research Laboratories.
Notes and references
- (a) For selected reviews, see: A. L. Ong, A. H. Kamaruddin and S. Bhatia, Process Biochem., 2005, 40, 3526 Search PubMed; (b) C. Ghelardini, N. Galeotti, M. N. Romanelli, F. Gualtieri and A. Bartolini, CNS Drug Rev., 2000, 6, 63 CAS; (c) J.-P. Rieu, A. Boucherle, H. Cousse and G. Mouzin, Tetrahedron, 1986, 42, 4095 CrossRef CAS; (d) T. Y. Shen, Angew. Chem., Int. Ed. Engl., 1972, 11, 460 CrossRef CAS ; For selected examples, see; (e) Y. Lu, T. M.-D. Nguyen, G. Weltrowska, I. Berezowska, C. Lemieux, N. N. Chung and P. W. Schiller, J. Med. Chem., 2001, 44, 3048 CrossRef CAS; (f) M. Allegretti, R. Bertini, M. C. Cesta, C. Bizzarri, R. Di Bitondo, V. Di Cioccio, E. Galliera, V. Berdini, A. Topai, G. Zampella, V. Russo, N. Di Bello, G. Nano, L. Nicolini, M. Locati, P. Fantucci, S. Florio and F. Colotta, J. Med. Chem., 2005, 48, 4312 CrossRef CAS; (g) O. Piccolo, F. Spreafico and G. Visentin, J. Org. Chem., 1987, 52, 10 CrossRef CAS.
- (a) J.-P. Genet, in Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH, Weinheim, 2008, pp. 18–22 Search PubMed; (b) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley-Interscience, New York, 1994, pp. 30–33 Search PubMed; (c) L. A. Oro and D. Carmona, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, pp. 14–19 Search PubMed; (d) T. Sugimura, T. Uchida, J. Watanabe, T. Kubota, Y. Okamoto, T. Misaki and T. Okuyama, J. Catal., 2009, 262, 57 CrossRef CAS.
- (a) H.-U. Blaser, Adv. Synth. Catal., 2002, 344, 17 CrossRef CAS; (b) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef; (c) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS.
- For selected examples of Rh catalysis, see: (a) P. H. Briner, M. C. T. Fyfe, J. P. Madeley, P. J. Murray, M. J. Procter and F. Spindler, Int. Pat., WO 2006/016178, 2006 Search PubMed; (b) W. Chen, P. J. McCormack, K. Mohammed, W. Mbafor, S. M. Roberts and J. Whittall, Angew. Chem., Int. Ed., 2007, 46, 4141 CrossRef CAS; (c) T. T. Co, S. C. Shim, C. S. Cho, T. J. Kim, S. O. Kang, W.-S. Han, J. Ko and C.-K. Kim, Organometallics, 2005, 24, 4824 CrossRef CAS; (d) I. N. Houpis, L. E. Patterson, C. A. Alt, J. R. Rizzo, T. Y. Zhang and M. Haurez, Org. Lett., 2005, 7, 1947 CrossRef CAS; (e) R. Hoen, J. A. F. Boogers, H. Bernsmann, A. J. Minnaard, A. Meetsma, T. D. Tiemersma-Wegman, A. H. M. de Vries, J. G. de Vries and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 4209 CrossRef CAS; (f) T. Hayashi, N. Kawamura and Y. Ito, J. Am. Chem. Soc., 1987, 109, 7876 CrossRef CAS.
- For selected examples of Ru catalysis, see: (a) X. Cheng, J.-H. Xie, S. Li and Q.-L. Zhou, Adv. Synth. Catal., 2006, 348, 1271 CrossRef CAS; (b) L. Qiu, F. Y. Kwong, J. Wu, W. H. Lam, S. Chan, W.-Y. Yu, Y.-M. Li, R. Guo, Z. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2006, 128, 5955 CrossRef CAS; (c) X. Cheng, Q. Zhang, J.-H. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2005, 44, 1118 CrossRef CAS; (d) P. E. Maligres, S. W. Krska and G. R. Humphrey, Org. Lett., 2004, 6, 3147 CrossRef CAS; (e) C.-C. Pai, C.-W. Lin, C.-C. Lin, C.-C. Chen and A. S. C. Chan, J. Am. Chem. Soc., 2000, 122, 11513 CrossRef CAS; (f) Q.-H. Fan, C.-Y. Ren, C.-H. Yeung, W.-H. Hu and A. S. C. Chan, J. Am. Chem. Soc., 1999, 121, 7407 CrossRef CAS; (g) T. Uemura, X. Zhang, K. Matsumura, N. Sayo, H. Kumobayashi, T. Ohta, K. Nozaki and H. Takaya, J. Org. Chem., 1996, 61, 5510 CrossRef CAS; (h) T. Manimaran, T.-C. Wu, W. D. Klobucar, C. H. Kolich and G. P. Stahly, Organometallics, 1993, 12, 1467 CrossRef CAS; (i) T. Ohta, H. Takaya, M. Kitamura, K. Nagai and R. Noyori, J. Org. Chem., 1987, 52, 3174 CrossRef CAS.
- A. Scrivanti, S. Bovo, A. Ciappa and U. Matteoli, Tetrahedron Lett., 2006, 47, 9261 CrossRef CAS.
- S. Li, S.-F. Zhu, C.-M. Zhang, S. Song and Q.-L. Zhou, J. Am. Chem. Soc., 2008, 130, 8584 CrossRef CAS.
- (a) L. S. Bertram, D. Black, P. H. Briner, R. Chatfield, A. Cooke, M. C. T. Fyfe, P. J. Murray, F. Naud, M. Nawano, M. J. Procter, G. Rakipovski, C. M. Rasamison, C. Reynet, K. L. Schofield, V. K. Shah, F. Spindler, A. Taylor, R. Turton, G. M. Williams, P. Wong-Kai-In and K. Yasuda, J. Med. Chem., 2008, 51, 4340 CrossRef CAS; (b) M. C. T. Fyfe, J. R. White, A. Taylor, R. Chatfield, E. Wargent, R. L. Printz, T. Sulpice, J. G. McCormack, M. J. Procter, C. Reynet, P. S. Widdowson and P. Wong-Kai-In, Diabetologia, 2007, 50, 1277 CrossRef CAS; (c) K. R. Guertin and J. Grimsby, Curr. Med. Chem., 2006, 13, 1839 CrossRef CAS; (d) R. Sarabu, R. Taub and J. Grimsby, Drug Discovery Today: Therapeutic Strategies, 2007, 4, 111 CrossRef; (e) M. Coghlan and B. Leighton, Expert Opin. Invest. Drugs, 2008, 17, 145 Search PubMed; (f) M. Pal, Drug Discovery Today, 2009, 14, 784 CrossRef CAS; (g) J. Grimsby, S. J. Berthel and R. Sarabu, Curr. Top. Med. Chem., 2008, 8, 1524 CrossRef CAS.
- Z. Han, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2009, 48, 5345 CrossRef CAS.
- Cationic iridium(I) complexes of the SpinPHOX (2) were readily prepared with a standard procedure reported in ref. 9.
- For examples of addition of bases, see: (a) K. Yamamoto, K. Ikeda and L. K. Yin, J. Organomet. Chem., 1989, 370, 319 CrossRef CAS; (b) X. Sun, L. Zhou, C.-J. Wang and X. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2623 CrossRef CAS; (c) M. E. Fox, M. Jackson, I. C. Lennon, J. Klosin and K. A. Abboud, J. Org. Chem., 2008, 73, 775 CrossRef CAS.
- (a) G. Argouarch, O. Samuel and H. B. Kagan, Eur. J. Org. Chem., 2000, 2885 CrossRef CAS; (b) G. Sarakinos and E. J. Corey, Org. Lett., 1999, 1, 1741 CrossRef CAS; (c) see ref. 7; (d) M. C. T. Fyfe, L. S. Gardner, M. Nawano, M. J. Procter, C. M. Rasamison, K. L. Schofield, V. K. Shah and K. Yasuda, Int. Pat., WO 2004/072031, 2004 Search PubMed; (e) G. R. Bebernitz and L. Kirman, Int. Pat., WO 2007/041366, 2007 Search PubMed; (f) S. Chen, P. T. W. Cheng and R. A. Smirk, Int. Pat., WO 2008/005914, 2008 Search PubMed.
- For the impact of molecular chirality on the GK activity, see: J. Grimsby, R. Sarabu, W. L. Corbett, N.-E. Haynes, F. T. Bizzarro, J. W. Coffey, K. R. Guertin, D. W. Hilliard, R. F. Kester, P. E. Mahaney, L. Marcus, L. Qi, C. L. Spence, J. Tengi, M. A. Magnuson, C. A. Chu, M. T. Dvorozniak, F. M. Matschinsky and J. F. Grippo, Science, 2003, 301, 370 Search PubMed.
Footnotes |
| † This article is part of a ChemComm ‘Catalysis in Organic Synthesis’ web-theme issue showcasing high quality research in organic chemistry. Please see our website (http://www.rsc.org/chemcomm/organicwebtheme2009) to access the other papers in this issue. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and characterization data for all new compounds. See DOI: 10.1039/b919902k |
| This journal is © The Royal Society of Chemistry 2010 |