Total synthesis of largamide H†
Shuo
Liang
a,
Zhengshuang
Xu
*ab and
Tao
Ye
*ab
aLaboratory of Chemical Genomics, Peking University Shenzhen Graduate School, University Town of Shenzhen, Xili, Nanshan District, Shenzhen, China 518055. E-mail: yet@szpku.edu.cn; xuzs@szpku.edu.cn; Tel: +86 755 26032697
bDepartment of Applied Biology & Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, China. E-mail: bctaoye@inet.polyu.edu.hk; Fax: +852 22641912; Tel: +852 34008722
First published on 16th November 2009
Abstract
Total synthesis of largamide H has been completed, utilising the oxidative elimination reaction of enantiomerically pure 2-amino-3-(phenylselenyl)butanoic acid residues to stereospecifically install both (Z)- and (E)-2,3-dehydro-2-aminobutanoic moieties.
Secondary metabolites produced by cyanobacteria are a vital source of potential target compounds for the pharmaceutical industry. One particular species, Oscillatoria, is responsible for the production of a considerable number of bioactive molecules.1 The compounds produced by these microorganisms possess a wide array of structures, including cyclopeptides and cyclodepsipeptides that embody both polypeptide and polyketide domains, displaying conformational flexibility. In addition, the possibility of extracting these compounds from their natural sources in significant quantities is very low. This, together with their novel and complex structures, makes these compounds a challenge to synthetic chemists. Indeed, many of these compounds have already been produced synthetically.1 We have been interested for some time in marine cyclopeptides and cyclodepsipeptides,2 and view their syntheses as a key route to structural confirmation, structural modification and subsequent activity control. Here we report the first total synthesis of largamide H.
Largamide H was isolated from the marine cyanobacterium Oscillatoria sp. with the proposed stereochemistry as shown in Fig. 1. Structurally, largamide H comprises ten stereogenic centers, a novel 2,5-dihydroxylated β-amino acid moiety, 2-amino-5-(4′-methoxyphenyl)pentanoic acid, and two residues of the nonstandard 2,3-dehydro-2-aminobutanoic acid (Dab) inscribed in a 31-membered macrocycle. Its structure and absolute configurations were determined by NMR, Mass and Chiral HPLC techniques.3
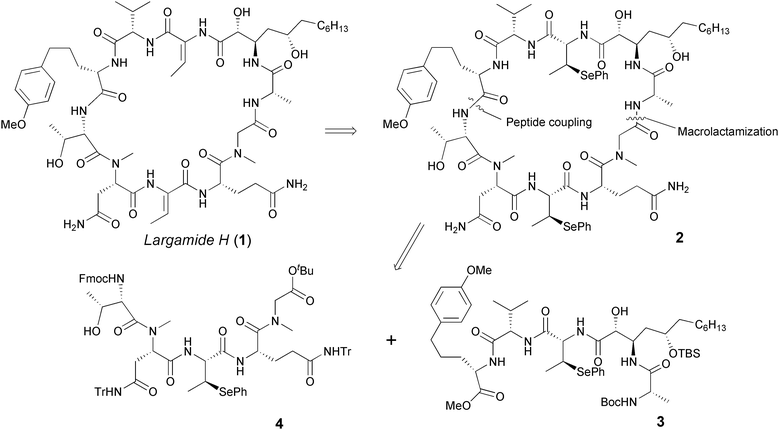 | ||
| Fig. 1 Structure and retrosynthetic analysis of Largamide H. | ||
There are three principal challenges associated with the synthesis of largamide H: (1) the asymmetric construction of α,δ-dihydroxylated β-amino acid, (2) the formation of a 31-membered macrolactam, and (3) the formation and incorporation of both (E)- and (Z)-Dab to the target molecule. Bearing these challenges in mind, we devised a retrosynthetic strategy toward largamide H (1) as illustrated in Fig. 1. In general, dehydroamino acids result in low peptide-coupling yields because they are less reactive electrophiles toward amide-bond formation and are also known as fairly reactive Michael acceptors that react readily with soft nucleophiles.4 Therefore, we decided to employ 3-methyl-Se-phenylselenocysteines to serve as the masked 2,3-dehydro-2-aminobutanoic acid residues for incorporating in the macrocycle and to be converted into the dehydroamino acids at a late stage, preferably after global deprotection. The rationale behind this decision is supported by the well-known syn stereochemistry of selenoxide elimination.5 We also chose to construct the macrolactam ring via an intramolecular coupling between L-alanine and glycine. Coupling reactions employing activated esters of glycine residues are generally very efficient. In addition, since glycine lacks an α-substituent, any possibility of epimerization during the course of the activation/coupling sequence would be avoided. Thus our target fragments for the assembly of the macrocycle were peptides 3 and 4, incorporating threo- and erythro-selenocysteine derivatives, respectively.
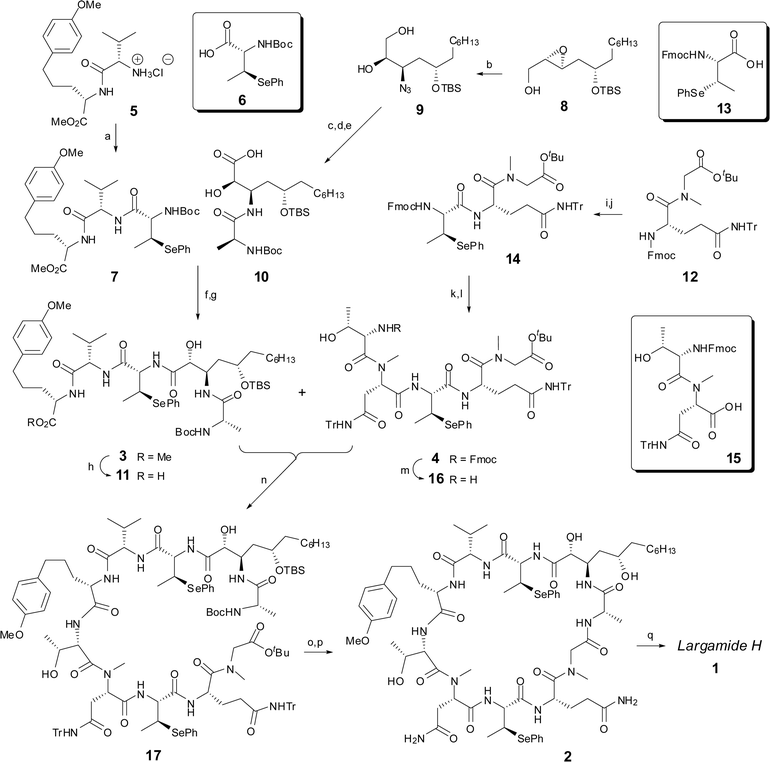 | ||
| Scheme 1 Reagents and conditions: (a) 6, PyAOP, DIPEA, CH2Cl2, 59%; (b) TMSN3, Ti(OiPr)4, benzene, reflux, 76%; (c) Pd/C, H2; (d) Boc-L-Ala-OH, PyAOP, DIPEA, 81% (two steps); (e) TEMPO, NaClO2, NaClO, phosphate buffer pH = 6.5, 35 °C; (f) 7, TFA, CH2Cl2; (g) 10, PyAOP, DIPEA, CH2Cl2, 54% (three steps); (h) n-Bu4NOH, MeOH–THF–H2O, 100%; (i) Et2NH, MeCN, 100%; (j) 13, PyAOP, collidine, HOAt, CH2Cl2, 91%; (k) Et2NH, MeCN, 100%; (l) 15, PyAOP, collidine, HOAt, CH2Cl2, 79%; (m) Et2NH, MeCN, 69%; (n) HATU, DIPEA, DMF–CH2Cl2, 58%; (o) TFA, CH2Cl2; (p) HATU, DIPEA, DMF, 46%; (q) NaIO4, CH2Cl2–H2O, 60%. | ||
The synthesis commenced with the coupling of dipeptide ester 56 with (2S,3S)-2-N-Boc-3-(phenylseleno)butanoic acid (6)7 to afford tripeptide 7 in 59% yield (Scheme 1). The synthesis of synthon 10 was achieved in four steps from the chiral epoxide intermediate 8.8 Thus, treatment of alcohol epoxide 8 with diisopropoxytitanium diazide under the condition as described by Sharpless and co-workers9 led to epoxide-opening products regioselectively (C-3 to C-2 ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Azido diol 9 was obtained as a single diastereomer in 76% yield after silica gel chromatography. The azide moiety was readily reduced to the primary amine with hydrogen and Pd/C followed by a PyAOP-mediated condensation10 with Boc-L-Alanine to afford the corresponding amide in 81% yield. Selective oxidation of the primary hydroxyl group by the action of TEMPO/NaClO/NaClO211 produced the corresponding acid 10 and set the stage for a fragment condensation. Hence, removal of the N-Boc protecting group of 7 with TFA produced the free amine which was coupled with hydroxy acid 10 to afford the key intermediate 3 in 54% yield. Hydrolysis of the methyl ester was then carried out with n-Bu4NOH12 to give the free acid 11. With acid 11 in hand, we next turned our attention to the synthesis of the lower fragment 4, which is composed of three subunits. N-terminal Fmoc protective group of 1213 was cleanly removed with diethyl amine, and the resulting free amine was coupled with (2R,3S)-2-N-Fmoc-3-(phenylseleno)butanoic acid (13)14 to afford tripeptide 14 in 91% yield. N-Fmoc deprotection of 14 followed by a PyAOP-mediated condensation with dipeptide 1515 furnished the pentapeptide 4 in 79% yield. At this juncture, the time had arrived to explore the assembly of two key fragments leading to linear peptide precursor 17 for the final macrocyclization reaction. Thus, treatment of 4 with diethylamine effected the removal of the Fmoc protecting group to give 16 in 69% yield, which underwent a HATU16-mediated coupling reaction with fragment 11 to provide 17 in 58% yield. Simultaneous deprotection of the tert-butyl ester, trityl and Boc-protecting groups afforded the desired amino acid which was immediately activated by HATU to afford cyclopeptide 2 in 46% overall yield.17 Gratifyingly, upon treatment with sodium periodate, both phenylselenide groups in 2 were converted into their corresponding selenoxides that underwent concomitant syn β-eliminations to afford largamide H in 60% yield. The spectral data (1H and 13C NMR) of synthetic largamide H ([α]20D −79.2, c 0.34, MeOH) were identical to that of natural largamide H (lit. ([α]20D −80.6, c 0.36, MeOH).3 With the synthetic largamide H in hand, the screening of cytotoxic activities toward cancer cell lines other than HCT-116 is currently under investigation and will be reported in due course.
:1). Azido diol 9 was obtained as a single diastereomer in 76% yield after silica gel chromatography. The azide moiety was readily reduced to the primary amine with hydrogen and Pd/C followed by a PyAOP-mediated condensation10 with Boc-L-Alanine to afford the corresponding amide in 81% yield. Selective oxidation of the primary hydroxyl group by the action of TEMPO/NaClO/NaClO211 produced the corresponding acid 10 and set the stage for a fragment condensation. Hence, removal of the N-Boc protecting group of 7 with TFA produced the free amine which was coupled with hydroxy acid 10 to afford the key intermediate 3 in 54% yield. Hydrolysis of the methyl ester was then carried out with n-Bu4NOH12 to give the free acid 11. With acid 11 in hand, we next turned our attention to the synthesis of the lower fragment 4, which is composed of three subunits. N-terminal Fmoc protective group of 1213 was cleanly removed with diethyl amine, and the resulting free amine was coupled with (2R,3S)-2-N-Fmoc-3-(phenylseleno)butanoic acid (13)14 to afford tripeptide 14 in 91% yield. N-Fmoc deprotection of 14 followed by a PyAOP-mediated condensation with dipeptide 1515 furnished the pentapeptide 4 in 79% yield. At this juncture, the time had arrived to explore the assembly of two key fragments leading to linear peptide precursor 17 for the final macrocyclization reaction. Thus, treatment of 4 with diethylamine effected the removal of the Fmoc protecting group to give 16 in 69% yield, which underwent a HATU16-mediated coupling reaction with fragment 11 to provide 17 in 58% yield. Simultaneous deprotection of the tert-butyl ester, trityl and Boc-protecting groups afforded the desired amino acid which was immediately activated by HATU to afford cyclopeptide 2 in 46% overall yield.17 Gratifyingly, upon treatment with sodium periodate, both phenylselenide groups in 2 were converted into their corresponding selenoxides that underwent concomitant syn β-eliminations to afford largamide H in 60% yield. The spectral data (1H and 13C NMR) of synthetic largamide H ([α]20D −79.2, c 0.34, MeOH) were identical to that of natural largamide H (lit. ([α]20D −80.6, c 0.36, MeOH).3 With the synthetic largamide H in hand, the screening of cytotoxic activities toward cancer cell lines other than HCT-116 is currently under investigation and will be reported in due course.
In summary, we have developed a convergent synthesis of largamide H. The key to success of our synthetic route was the selection of L- and L-allo-threonine derived selenide precursors and the use of oxidative elimination processes to control the stereochemistry of 2,3-dehydro-2-aminobutanoic acid units presented in the natural product.
We acknowledge financial support from the Hong Kong Research Grants Council (Project: PolyU 5407/06M) and financial support from the Shenzhen Bureau of Science, Technology and Information (08systs-01, JC200903160367A). Z. S. X. would like to thank the support from Shenzhen Foundation for R&D (SZKJ-2007011, SY2008063 00179A), Nanshan Science & Technology (NANKEYUAN2007019) and NSF of GuangDong Province (8451805704000656).
Notes and references
- A. Méjean, S. Mann, T. Maldiney, G. Vassiliadis, O. Lequin and O. Ploux, J. Am. Chem. Soc., 2009, 131, 7512 CrossRef CAS; T. L. Simmons, N. Engene, L. D. Ureña, L. I. Romero, E. Ortega-Barría, L. Gerwick and W. H. Gerwick, J. Nat. Prod., 2008, 71, 1544 CrossRef; R. G. Linington, J. González, L.-D. Ureña, L. I. Romero, E. Ortega-Barría and W. H. Gerwick, J. Nat. Prod., 2007, 70, 397 CrossRef CAS; T. Sano, K. A. Beattie, G. A. Codd and K. Kaya, J. Nat. Prod., 1998, 61, 851 CrossRef CAS; V. Admi, U. Afek and S. Carmeli, J. Nat. Prod., 1996, 59, 396 CrossRef CAS; S. Hanessian, M. Tremblay and J. F. W. Petersen, J. Am. Chem. Soc., 2004, 126, 6064 CrossRef CAS.
- S. Li, S. Liang, W. F. Tan, Z. S. Xu and T. Ye, Tetrahedron, 2009, 65, 2695 CrossRef CAS; Q. Ren, L. Dai, H. Zhang, W. F. Tan, Z. S. Xu and T. Ye, Synlett, 2008, 2379 CAS; S. Li, S. Liang, Z. S. Xu and T. Ye, Synlett, 2008, 569 CAS; Z. Chen and T. Ye, New J. Chem., 2006, 30, 518 RSC; H. W. Pang, Z. S. Xu, Z. Y. Chen and T. Ye, Lett. Org. Chem., 2005, 2, 699 Search PubMed; Y. G. Peng, H. W. Pang, Z. S. Xu and T. Ye, Lett. Org. Chem., 2005, 2, 703 Search PubMed; Z. S. Xu and T. Ye, Tetrahedron: Asymmetry, 2005, 16, 1905 CrossRef CAS; H. L. Chen, Z. S. Xu and T. Ye, Tetrahedron, 2005, 61, 11132 CrossRef CAS; Z. Y. Chen and T. Ye, Synlett, 2005, 2781 CAS; Z. S. Xu, Z. Chen and T. Ye, Tetrahedron: Asymmetry, 2004, 15, 355 CrossRef CAS; Y. G. Peng, H. W. Pang and T. Ye, Org. Lett., 2004, 6, 3781 CrossRef CAS; Z. Y. Chen, J. G. Deng and T. Ye, ARKIVOC, 2003, 268 CAS , Part VII; Z. S. Xu, Y. G. Peng and T. Ye, Org. Lett., 2003, 5, 2821 Search PubMed.
- A. Plaza and C. A. Bewley, J. Org. Chem., 2006, 71, 6898 CrossRef CAS.
- U. Schmidt, A. Lieberknecht and J. Wild, Synthesis, 1988, 159 CrossRef CAS; J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef CAS; C. Bonauer, T. Walenzyk and B. König, Synthesis, 2006, 1 CAS.
- K. B. Sharpless, M. W. Young and R. F. Lauer, Tetrahedron Lett., 1973, 14, 1979 CrossRef; H. Reich, J. M. Renga and I. L. Reich, J. Am. Chem. Soc., 1975, 97, 5434 CrossRef CAS.
- Dipeptide 5 was prepared from L-valine and L-2-amino-5-(p-methoxyphenyl)pentanoic acid: P. N. Collier, A. D. Campbell, I. Patel and R. J. K. Taylor, Tetrahedron Lett., 2000, 41, 7115 Search PubMed; H. Aoyagi, F. Horike, A. Nakagawa, S. Yokote, N. Park, Y. Hashimoto, T. Kato and N. Izumiya, Bull. Chem. Soc. Jpn., 1986, 59, 323 CrossRef CAS.
- T. Mori, S. Higashibayashi, T. Goto, M. Kohno, Y. Satouchi, K. Shinko, K. Suzuki, S. Suzuki, H. Tohmiya, K. Hashimoto and M. Nakata, Chem.–Asian J., 2008, 3, 984 CrossRef CAS; S. Higashibayashi, M. Kohno, T. Goto, K. Suzuki, T. Mori, K. Hashimoto and M. Nakata, Tetrahedron Lett., 2004, 45, 3707 CrossRef CAS.
- Epoxide alcohol 8 was prepared in six steps and 49% overall yield from octanal (for details see ESI†).
- M. Caron, P. R. Carlier and K. B. Sharpless, J. Org. Chem., 1988, 53, 5185 CrossRef CAS.
- F. Albericio, M. Cases, J. Alsina, S. A. Triolo, L. A. Carpino and S. A. Kates, Tetrahedron Lett., 1997, 38, 4853 CrossRef CAS.
- M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 1999, 64, 2564 CrossRef CAS.
- U. M. Reinscheid, B. D. Zlatopolskiy, C. Griesinger, A. Zeeck and A. de Meijere, Chem.–Eur. J., 2005, 11, 2929 CrossRef CAS; M. Fischer and S. Höger, Tetrahedron, 2003, 59, 9441 CrossRef CAS.
- Dipeptide ester 12 was prepared in 87% yield by a condensation between N2-Fmoc-N-(triphenylmethyl)-L-glutamine and tert-butyl N-methylglycinate (for details see ESI†).
- (2R,3S)-2-N-Fmoc-3-(phenylseleno)butanoic acid was prepared from (2R,3S)-2-N-Boc-3-(phenylseleno)butanoic acid, which was in turn synthesized according to the procedure shown in ref. 7.
- Dipeptide acid 15 was prepared from N-(9-fluorenylmethoxycarbonyl)-L-threonine and N2-methyl-N2-[(2-nitrophenyl)sulfonyl]-N-(triphenylmethyl)-L-asparagine methyl ester in three steps and 66% overall yield (for details see ESI†).
- L. A. Carpino and A. El-Faham, J. Org. Chem., 1994, 59, 695 CrossRef CAS; L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397 CrossRef CAS; L. A. Carpino and A. El-Faham, J. Org. Chem., 1995, 60, 3561 CrossRef CAS.
- Macrolactamization of the linear peptide (1–2 mM, DMF) using HATU (2 equiv.) in the presence of DIPEA (10 equiv.) afforded 2 in 46% yield. Head-to-tail cyclizations were also performed using DEPC and DPPA, respectively, but resulted in lower yields (10–14%).
Footnote |
| † Electronic supplementary information (ESI) available: Full details for experimental procedures for compounds 1, 3–5, 7–10, 12, 14, 15 and 17, and 1H and 13C NMR spectra for compounds 1, 3–4, 5b, 7–9, 10a, 12, 14, 15b and 17. See DOI: 10.1039/b921000h |
| This journal is © The Royal Society of Chemistry 2010 |