Ortho-C–H borylation of benzoate esters with bis(pinacolato)diboron catalyzed by iridium–phosphine complexes†
Tatsuo
Ishiyama
*,
Hironori
Isou
,
Takao
Kikuchi
and
Norio
Miyaura
*
Division of Chemical Process Engineering, Graduate School of Engineering, Hokkaido University, Sapporo, 060-8628, Japan. E-mail: ishiyama@eng.hokudai.ac.jp; Fax: +81 11 706 6562; Tel: +81 11 706 6562
First published on 17th November 2009
Abstract
Iridium complexes generated from [Ir(OMe)(COD)]2 and tris[3,5-bis(trifluoromethyl)phenyl]phosphine efficiently catalyzed the ortho-C–H borylation of benzoate esters with bis(pinacolato)diboron in octane at 80 °C to produce the corresponding arylboronates in high yields with excellent regioselectivities.
Increasing attention has been focused on transition metal-catalyzed activation and the functionalization of unreactive C–H bonds of hydrocarbons.1 An extension of this methodology to aromatic C–H borylation of arenes with bis(pinacolato)diboron (B2pin2, pin = Me4C2O2) or pinacolborane (HBpin) is of significant value for the preparation of synthetically useful aromatic boron compounds2 from the viewpoint of economy, efficiency, and environmental benignity.3–6 The most efficient catalyst for this type of transformation is the combination of [IrCl(COD)]2 or [Ir(OMe)(COD)]2 with 2,2′-bipyridine or 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy).6 The high level of catalytic activity of 1/2[Ir(OMe)(COD)]2–dtbpy allows room temperature borylation of arenes with a stoichiometric amount of substrate to produce the corresponding arylboron compounds in high yields. The functional group tolerance of the borylation is quite high. The reaction occurs selectively at the aromatic C–H bond for arenes bearing MeO, Me, I, Br, Cl, F3C, MeO2C, and NC. The regioselectivity of this C–H borylation of arenes is primarily controlled by steric effects; the functionalization occurs at the least hindered aromatic C–H bond. Thus, 1,2-disubstituted arenes having identical substituents and 1,3-disubstituted arenes even having distinct substituents produce borylated products as single isomers. A drawback of this method is therefore difficulty in achieving ortho-C–H borylation. One of the most reliable protocols would be a process involving the use of chelation-assisted C–H bond cleavage.1
We disclose herein the ortho-C–H borylation of benzoate ester derivatives (1) with bis(pinacolato)diboron (B2pin2) (2) catalyzed by iridium complexes comprised of easily available [Ir(OMe)(COD)]2 and commercial tris[3,5-bis(trifluoromethyl)phenyl]phosphine in octane at 80 °C to give the corresponding aromatic boron compounds (3) in high yields with excellent regioselectivities (Scheme 1).7,8
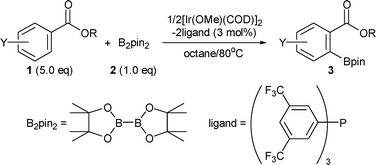 | ||
| Scheme 1 Ortho-selective C–H borylation of benzoate esters. | ||
To achieve the ortho-C–H borylation of benzoate esters 1, effects of ligands (0.03 or 0.06 mmol) were investigated for the reaction of methyl benzoate (5.0 mmol) with B2pin22 (1.0 mmol) by using [Ir(OMe)(COD)]2 (0.015 mmol) as a catalyst precursor in octane (6 ml) at 80 °C for 16 h (Table 1). Iridium complexes bearing dtbpy effectively catalyzed the aromatic C–H borylation, but they did not give any ortho-borylated products at all (Entry 1). Probably, the high coordinating ability of dtbpy retards the coordination of a carbonyl oxygen in the substrate to the iridium centre. The reaction afforded an ortho-borylated product in the absence of ligands; however, catalytic activity and regioselectivity were low (Entry 2). These results prompted us to examine monodentate ligands. Among iridium complexes having pyridine (Entry 3), triphenylphosphine (Entry 4), and triphenylarsine (Entry 5), the latter two complexes exhibited moderate ortho-selectivities but displayed low catalytic activities. Since a wide variety of triarylphosphines are commercially available, we decided to investigate them as ligands of iridium. Although catalysts having electron-rich phosphines such as tris(4-methoxyphenyl)phosphine exhibited low activity and regioselectivity (Entry 6), those having electron-poor phosphines such as tris(4-trifluoromethylphenyl)phosphine displayed high ortho-selectivity and moderate catalytic activity (Entry 7). Complexes bearing more electron-poor tris(pentafluorophenyl)phosphine improved the catalytic activity while maintaining high regioselectivity (Entry 8). Finally, high yield (95%) and highest ortho-selectivity (98%) were achieved when tris[3,5-bis(trifluoromethyl)phenyl]phosphine (P(3,5-2F3C–C6H3)3) was used as a ligand of iridium (Entry 9).
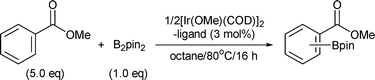
| Entry | Ligand | Yield (%)b |
o-![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m-:p- (%)c :m-:p- (%)c |
|---|---|---|---|
| a Reactions were carried out at 80 °C for 16 h by using methyl benzoate (5.0 mmol), 2 (1.0 mmol), [Ir(OMe)(COD)]2 (0.015 mmol), ligand (0.03 or 0.06 mmol), and octane (6 ml). b GC yields based on 2. c Isomer ratios were determined by 1H NMR. | |||
| 1 | dtbpy | 145 | 0:58:42 |
| 2 | None | 13 | 38:38:24 |
| 3 | 2 pyridine | 7 | 14:58:28 |
| 4 | 2 P(C6H5)3 | 11 | 64:18:18 |
| 5 | 2 As(C6H5)3 | 9 | 67:22:11 |
| 6 | 2 P(4-MeO–(C6H4)3 | 3 | 34:33:33 |
| 7 | 2 P(4-F3C–C6H4)3 | 45 | 96:2:2 |
| 8 | 2 P(C6F5)3 | 82 | 90:7:3 |
| 9 | 2 P(3,5-2F3C–C6H3)3 | 95 | 98:2:0 |
The choice of iridium precursor was crucial for the borylation. Although the combination of [IrCl(COD)]2 and P(3,5-2F3C–C6H3)3 gave borylated products in good yield (62%), ortho-selectivity was low (55%). No borylated product was obtained when the combination of [Ir(COD)2]BF4 and the phosphine was used. The choice of inert solvent was also important for efficient borylation. The reactions using 1/2[Ir(OMe)(COD)]2–2P(3,5-2F3C–C6H3)3 were faster in non-polar solvents such as octane than in more polar and coordinating solvents. The order of reactivity in different solvents was octane (95%) > mesitylene (3%) > diglyme (0%) = DMF (0%).
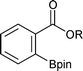
Representative results of ortho-C–H borylation of benzoate esters 1 with B2pin22 catalyzed by the combination of 1/2[Ir(OMe)(COD)]2 and 2P(3,5-2F3C–C6H3)3 in octane at 80 °C for 16 h are shown in Table 2. Not only methyl but also ethyl, isopropyl and tert-butyl benzoates were all viable substrates for producing the corresponding ortho-borylated products in high yields with excellent regioselectivities, whilst their reactivity slightly decreased in the above order (Entries 1–4). The reactions were suitable for substrates possessing various functional groups, such as Me2N, Br, and F3C as well as for substrates with potentially more reactive benzylic C–H bonds (Entries 5–16).9 Although some transition metal complexes exhibit high reactivity toward oxidative addition of Ar–Br bonds,10 methyl 2-, 3-, and 4-bromobenzoates underwent borylation at the C–H bond (Entries 7, 11, and 15). Reactions of substrates having a substituent at the 3-position only occurred at the 6-position, presumably due to steric reasons (Entries 9–12).
| Entry | Productb | Yield (%)c | Entry | Productb | Yield (%)c | ||
|---|---|---|---|---|---|---|---|
|
a All reactions were carried out at 80 °C for 16 h by using 1 (5.0 mmol), 2 (1.0 mmol), [Ir(OMe)(COD)]2 (0.015 mmol), tris[3,5-bis(trifluoromethyl)phenyl]phosphine (0.06 mmol), and octane (6 ml).
b Isomeric purities over 98% were determined by 1H NMR.
c GC yields based on 2.
d The reaction was conducted in a mixture of octane and mesitylene (1:1).
|
|||||||
| 1 |
|
R = Me | 95 | 9 |
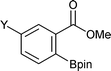
|
Y = Me2N | 99 |
| 2 | R = Et | 92 | 10 | Y = Me | 98 | ||
| 3 | R = i-Pr | 89 | 11 | Y = Br | 64 | ||
| 4 | R = t-Bu | 83 | 12 | Y = F3C | 94 | ||
| 5 |
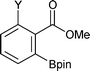
|
Y = Me2N | 97 | 13 |
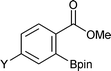
|
Y = Me2N | 93 |
| 6 | Y = Me | 92 | 14 | Y = Me | 99 | ||
| 7 | Y = Br | 60 | 15 | Y = Br | 57d | ||
| 8 | Y = F3C | 98 | 16 | Y = F3C | 98 | ||
In summary, ortho-borylated products were obtained with excellent regioselectivities by the reaction of benzoate esters with bis(pinacolato)diboron in the presence of a catalytic amount of iridium complexes generated from [Ir(OMe)(COD)]2 and commercial tris[3,5-bis(trifluoromethyl)phenyl]phosphine in octane at 80 °C. Further investigations to survey the scope and limitations of this C–H borylation, including C–H borylation of other aromatic carbonyl compounds such as ketones and amides, as well as to elucidate the reaction mechanisms are in progress.
This work was partially supported by Grant-in-Aid for Scientific Research on Priority Areas (No. 17065001, “Advanced Molecular Transformations of Carbon Resources”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Notes and references
- (a) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS; (b) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS; (c) Handbook of C–H Transformations, ed. G. Dyker, Wiley-VCH, Weinheim, 2005 Search PubMed; K. Godula and D. Sames, Science, 2006, 312, 67 Search PubMed; (d) A. L. Dick and S. Stanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS; (e) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (f) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS.
- (a) M. Vaultier and B. Carboni, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon Press, Oxford, 1995, vol. 11, p. 191 Search PubMed; (b) K. Ishihara and H. Yamamoto, Eur. J. Org. Chem., 1999, 527 CrossRef CAS; (c) S. Shinkai, M. Ikeda, A. Sugasaki and M. Takeuchi, Acc. Chem. Res., 2001, 34, 494 CrossRef CAS; (d) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927 CrossRef CAS; (e) A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515 CrossRef CAS; (f) W. Yang, X. Gao and B. Wang, Med. Res. Rev., 2003, 23, 346 CrossRef CAS; (g) Boronic Acids, ed. D. G. Hall, Wiley, Weinheim, 2005 Search PubMed.
- (a) C. N. Iverson and M. R. Smith, III, J. Am. Chem. Soc., 1999, 121, 7696 CrossRef CAS; (b) J.-Y. Cho, C. N. Iverson and M. R. Smith, III, J. Am. Chem. Soc., 2000, 122, 12868 CrossRef CAS; (c) M. K. Tse, J.-Y. Cho and M. R. Smith, III, Org. Lett., 2001, 3, 2831 CrossRef CAS; (d) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka, Jr and M. R. Smith, III, Science, 2002, 295, 305 CrossRef CAS; (e) R. E. Meleczka, Jr, F. Shi, D. Holmes and M. R. Smith, III, J. Am. Chem. Soc., 2003, 125, 7792 CrossRef CAS; (f) G. A. Chotana, M. A. Rak and M. R. Smith, III, J. Am. Chem. Soc., 2005, 127, 10539 CrossRef CAS; (g) S. Paul, G. A. Chotana, D. Holmes, R. C. Reichle, R. E. Maleczka, Jr and M. R. Smith, III, J. Am. Chem. Soc., 2006, 128, 15552 CrossRef CAS; (h) G. A. Chotana, V. A. Kallepalli, R. E. Maleczka, Jr and M. R. Smith, III, Tetrahedron, 2008, 64, 6103 CrossRef CAS.
- (a) H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391 CrossRef CAS; (b) H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS; (c) T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263 CrossRef CAS; (d) J. M. Murphy, C. C. Tzschucke and J. F. Hartwig, Org. Lett., 2007, 9, 757 CrossRef CAS; (e) C. C. Tzschucke, J. M. Murphy and J. F. Hartwig, Org. Lett., 2007, 9, 761 CrossRef CAS; (f) J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS.
- (a) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem. Int. Ed., 2001, 40, 2168 CrossRef CAS; (b) D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172 RSC; (c) I. A. I. Mkhalid, D. N. Coventry, D. Albesa-Jove, A. S. Batsanov, J. A. K. Howard, R. N. Perutz and T. B. Marder, Angew. Chem. Int. Ed., 2006, 45, 489 CrossRef CAS; (d) P. Harrisson, J. Morris, P. G. Steel and T. B. Marder, Synlett, 2009, 247.
- (a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS; (b) J. Takagi, K. Sato, J. F. Hartwig, T. Ishiyama and N. Miyaura, Tetrahedron Lett., 2002, 43, 5649 CrossRef CAS; (c) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem. Int. Ed., 2002, 41, 3056 CrossRef CAS; (d) T. Ishiyama, J. Takagi, Y. Yonekawa, J. F. Hartwig and N. Miyaura, Adv. Synth. Catal., 2003, 345, 1103 CrossRef; (e) T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924 RSC; (f) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2003, 680, 3 CrossRef CAS; (g) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271 CrossRef CAS; (h) T. Ishiyama, J. Takagi, Y. Nobuta and N. Miyaura, Org. Synth., 2005, 82, 126 CAS; (i) T. Ishiyama and N. Miyaura, Pure Appl. Chem., 2006, 78, 1369 CrossRef CAS.
- For ortho-borylation directed by a Me2HSi group, see: T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534 Search PubMed.
- For ortho-borylation of benzoate esters catalyzed by Silica-SMAP-Ir, see: S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058 Search PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS.
- (a) G. J. Leigh, R. L. Richards, in Comprehensive Organometallic Chemistry, ed. G. Wilkinson, F. G. A. Stone and E. W. Abel, Pergamon Press, Oxford, 1982, vol. 5, p. 541 Search PubMed; (b) J. D. Atwood, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon Press, Oxford, 1995, vol. 8, p. 303 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral analyses. See DOI: 10.1039/b910298a |
| This journal is © The Royal Society of Chemistry 2010 |