
A facile and eco-efficient additive strategy enables high-performance aqueous zinc-ion batteries†
Kai
Yang
*,
Jiuzhou
Yu
 ,
Xu
Zhang
,
Shaoting
Wang
,
Qiang
Wang
,
Ci
Gao
,
Yiyao
Song
and
Xingxing
Guo
,
Xu
Zhang
,
Shaoting
Wang
,
Qiang
Wang
,
Ci
Gao
,
Yiyao
Song
and
Xingxing
Guo
State Grid New Energy Cloud Technology Co., Ltd., Beijing, 100077, China. E-mail: Ninejoe_1st@163.com
First published on 27th May 2025
Abstract
High-performance aqueous zinc-ion batteries as one of the most promising battery systems have attracted much attention in the development of next-generation advanced energy storage systems. However, the development of AZIBs has been plagued by inherent issues, such as growth of zinc dendrites, irreversible dissolution of manganese-based cathode materials, hydrogen precipitation reaction, and other side reactions. In this work, we developed a facile and eco-efficient strategy that adopted an optimized electrolyte additive of Al2(SO4)3 in the ZnSO4-based electrolytes for AZIBs. The electrochemical, spectroscopic, and microscopic characterization results demonstrated that the optimized aluminum sulfate additives can inhibit the Zn dendrite formation, reduce the generation of dead manganese and stabilize the crystalline structure of the manganese-based cathode. The Zn||MnO2 full cell exhibits a high discharge plateau of ∼1.65 V and excellent cycling stability with a high-capacity retention of 78% even after 1200 cycles at 1C with a high active material loading (∼8.0 mg cm−2). This facile and eco-efficient strategy provides a promising and high-efficient strategy to design high-performance AZIBs for energy storage systems.
1. Introduction
High-performance lithium-ion batteries (LIBs) have been widely used in energy storage systems due to their high energy density and long cycle lifetime.1–3 However, the global lithium source shortage and security issues have limited the further large-scale application of LIBs.3–6 Therefore, innovating and developing advanced electrical energy storage (EES) systems has become one of the top priorities.7 Recently, aqueous ion battery systems with high safety and high capacity have been largely developed. Especially, the high-performance aqueous zinc-ion batteries (AZIBs) have unique practical application potential using the Zn metal as the anode and MnO2 as the cathode. The Zn anode has high theoretical specific capacity (820 mA h g−1) and volumetric specific capacity (5855 mA h cm−3), high safety, and low redox potential (−0.76 V vs. SHE.), which enables the design of high-capacity and high-volumetric energy-density AZIBs.8–10 However, the electrochemical performance of AZIBs is still hindered by the issues with the Zn anodes, such as uncontrolled dendrite growth, interface instability and hydrogen evolution.11–13In recent years, many effective and practical strategies, including electrode surface coating, alloy layer modification and electrolyte additive design have been proposed to improve the electrochemical performance of AZIBs.14–19 Among these methods, the electrolyte additive strategy is a simple and effective method to enable high-performance AZIB chemistry.20–25 In recent years, different electrolytes, particularly Zn(CF3SO3)2 and ZnSO4, have been generally adopted for AZIBs rather than alkaline electrolytes due to their superior electrochemical performance.26–28 However, the Zn(CF3SO3)2 electrolyte is expensive and environmentally unfriendly due to the toxic fluorine.26 While the low-cost and environment-friendly ZnSO4 electrolyte is more widely used in AZIBs, the ZnSO4-based electrolyte also faces the challenges of the side reactions and the formation of the Zn4SO4(OH)6·nH2O by-products, which cause lowered coulombic efficiency and the operation failure of the cells.28,29 To tackle these issues, researchers have made great effects on developing various electrolytes via a additive strategy, such as Na+,30,31 Mg2+,32 Mn2+,8,33–37 Al3+,38–40 Ni2+,41 and La2+ (ref. 42) ions and organic polymers as an electrolyte additive.43–46 In addition, the selection of cathode material is also a key factor affecting the electrochemical performance of AZIBs.47,48 Vanadium-based oxide and manganese-based oxide are typical cathode materials that have been utilized in recent research.49–52 Compared with vanadium-based oxide cathode materials, manganese-based oxide cathode materials exhibit promising potential for practical application owing to their low cost, environment-friendliness and higher discharge potential.53–56 It is well-known that the electrochemical performance of AZIBs with high-loading cathode materials has gradually become a significant concern for researchers who aim to meet the requirements of a practical battery and achieve its commercialization.57,58 However, there have been few reports on AZIBs with a high-loading manganese-based cathode, which may be related to their lower capacity, susceptibility to irreversible dissolution and lattice distortion during cycling.49,52 The effective additive strategies have also been reported to improve the electrochemical performance of AZIBs. For instance, 1 M Al(CF3SO3)3 with an additive of aluminum sulfate (0.1 M Al2(SO4)3, or 0.2 M Al2(SO4)3·18H2O) has been used as electrolyte in Zn(CF3SO3)2-based electrolyte.38–40 However, this electrolyte strategy has to face big challenges such as high cost and low ionic conductivity, which hinder the practical application in AZIBs.
In this work, we designed a novel method by using an optimized electrolyte additive of Al2(SO4)3 into the conventional 2 M ZnSO4-based electrolyte to prepare a high-performance bi-cation electrolyte (2 M ZnSO4 + 0.5 M Al2(SO4)3). This alternate electrolyte solution is economically viable, eco-friendly, and effectively suppresses dendrite formation, minimizes hydrogen precipitation, and promotes uniform zinc ion deposition on the anode. Using the bi-cation electrolyte (2 M ZnSO4 + 0.5 M Al2(SO4)3), the Zn||MnO2 full cell with high-loading (8.0 mg cm−2) cathode exhibits a high discharge plateau at 1.65 V and maintains good cycling stability with a high capacity retention of 78% even after 1200 cycles at 1C. The electrochemical performance, and spectroscopic and microscopic characterization results confirmed that the 0.5 M Al2(SO4)3 used as an additive can effectively inhibit the occurrence of side reactions, reduce by-product generation, and avoid the irreversible dissolution of positive electrode materials. Furthermore, combined with qualitative and quantitative analysis, the calculated results confirm that the addition of Al3+ ions affects the discharge plateau and enhances the electrochemical performance. This work provides a highly efficient and practical method for designing high-voltage, ultra-stable and high-capacity AZIBs.
2. Results and discussion
In conventional ZnSO4 electrolytes, the dendrites will inevitably occur on the surface of zinc anodes because of a “tip effect”.59 Considering the lower standard equilibrium potential for Al3+/Al (−1.66 V versus SHE) than Zn2+/Zn (−0.76 V versus SHE), the rational choice of the Al2(SO4)3 additive was made, which would induce an electrostatic shield effect on Zn deposition.60 Therefore, we optimized the amount of Al2(SO4)3 additive to tune the electrostatic shield. The mechanism of the effect of the Al2(SO4)3 additive in the electrolyte on the Zn anode is demonstrated in Fig. 1a. At room temperature, the 2 M ZnSO4 electrolyte (denoted as ZS) was chosen as the basic electrolyte. The bi-cation electrolyte (2 M ZnSO4 + 0.5 M Al2(SO4)3, denoted as AS) was prepared and optimized by dissolving anhydrous aluminum sulfate in the basic electrolyte (Fig. 1b). Another advantage of the modified electrolyte is attributed to the low cost compared with other common zinc salts and electrolyte additives (Table S1, ESI†). | ||
| Fig. 1 (a) Illustration of the Zn deposition process according to the electrostatic protection mechanism. (b) Optical photograph of 2 M ZnSO4 electrolyte with different concentrations of anhydrous aluminum sulfate additive. (c) Cycling performance of the Zn||Zn cells with a current density of 0.5 mA cm−2 and an areal capacity of 0.05 mA h cm−2. (d) Enlarged comparison of the polarization curves of Zn||Zn symmetric cells in ZS and AS electrolyte at a constant current density of 0.5 mA cm−2 and an areal capacity of 0.05 mA h cm−2; (e) coulombic efficiency of the Zn||Cu cells with a current density of 5 mA cm−2 and a cut-off charging voltage of 0.5 V. (f) 50th cycling performance of Zn||Cu cells in ZS/AS electrolyte. (g) XRD patterns of electrodeposited Zn electrodes in ZS/AS electrolyte after cycling at 5 mA cm−2, 1 mA h cm−2 for 10 cycles. | ||
The cycling performances of the assembled Zn||Zn symmetric cells using ZS and AS electrolyte were tested under a current density of 0.5 mA cm−2 (areal capacity: 0.05 mA h cm−2). As shown in Fig. 1c, the polarization voltage of the Zn||Zn cell using ZS electrolyte increased after several cycles and eventually failed after 1400 h. In contrast, the Zn||Zn cell using AS electrolyte maintained a stable polarization voltage curve after 100 h with smaller voltage fluctuations and exhibited a small polarization voltage of 54.6 mV after cycling for 400 h, much lower than 73.2 mV of the Zn||Zn cell using ZS electrolyte (Fig. 1d). Meanwhile, the Zn||Zn cell using AS electrolyte remained stable after cycling for 2000 h, and is superior to that of the Zn||Zn symmetric cell using ZS electrolyte (1400 h). Besides, the effect of different concentrations of the anhydrous aluminum sulfate additive in ZS electrolytes of Zn||Zn cells on the electrochemical properties was also investigated to find out the optimal concentration of the anhydrous aluminum sulfate additive (Fig. S1, ESI†). Results showed that among the Zn||Zn cells with different anhydrous aluminum sulfate additive concentrations, the Zn||Zn cells using 0.5 M anhydrous aluminum sulfate additive showed superior cycling stability. Therefore, the AS electrolyte (2 M ZnSO4 + 0.5 M Al2(SO4)3) was chosen as the electrolyte for AZIBs.
To further investigate the deposition and stripping behavior on the Zn anode, we assembled asymmetric Zn||Cu cells by plating 1.0 mA h cm−2 Zn on the Cu foil at a current density of 5.0 mA cm−2. Fig. 1e shows the coulombic efficiency (CE) of both Zn||Cu asymmetric cells. When AS electrolyte is used, the cell can obtain a higher average CE of 98.64% compared with ZS electrolyte (96.44%) during 120 plating/stripping cycles. Fig. 1f shows the capacity–voltage curves of Zn||Cu cells using ZS electrolyte and AS electrolyte at the 50th cycle. There is a 10 mV decrease in polarization voltage of the cells using AS electrolyte than that of ZS electrolyte after 50 cycles. X-ray diffraction (XRD) tests were performed to characterize the deposition behavior of the Zn electrode in ZS and AS electrolyte after cycling. As shown in Fig. 1g, the relative intensity ratio of peak (002) to that of peak (101) decreases from 0.96 to 0.57, suggesting the reduced (002) planes for Zn deposition in AS electrolyte. All the above cell testing performances indicate that AS electrolytes could be in favor of improving the stability and reversibility of Zn electrodes.
To gain a better understanding of the effects of AS electrolyte on the deposition morphology of the Zn electrode, we disassembled cycled Zn||Cu cells and analyzed the deposition morphology of the Zn anode surface using SEM. The SEM pictures of the bare Zn anode in the initial state are presented in Fig. 2a as a blank control sample. There are irregular and disordered dendrites on the surface of the cycled Zn electrode in the ZS electrolyte, showing an uneven Zn deposition (Fig. 2b). The dendrite formation could disrupt the surface electric field, affecting zinc deposition and battery performance. In comparison, the Zn electrode in AS electrolyte shows a dense and smooth surface without visible dendrites (Fig. 2c), indicating uniform Zn plating/striping during cycling. The same section could also be observed in SEM images of the Zn electrode under a current density of 1 mA cm−2 for 0.5, 1, and 2 hours, respectively (Fig. S2, ESI†).
 | ||
| Fig. 2 SEM images of (a) bare Zn. SEM images of the Zn electrodes of Zn||Cu cells in (b) ZS and (c) AS electrolyte after 10 cycles under a current density of 5.0 mA cm−2 for 0.2 h. (d) In situ optical microscope observation of the Zn deposition process in ZS and AS electrolyte at the current density of 5 mA cm−2. The right side and cross-section were observation surfaces. (e) A graph of Zn deposition in ZS/AS electrolyte. | ||
In situ optical microscopy setup as shown in Fig. S3 (ESI†) was used to visually understand the deposition behavior of Zn2+ ions at the electrolyte/anode interface (Fig. 2d). In conventional ZS electrolytes, zinc protrusions appeared after 10 min of plating on the Zn anode and section, then the thickness of the zinc deposition layer increased significantly with time increasing to 60 minutes, and eventually, these protrusions developed into large dendrites resulting in degraded cell performance. In contrast, during the initial 10 minutes on the Zn anode surface in the AS electrolyte, no significant changes were observed. Only a few uniform deposition patterns and bubbles due to acidic electrolyte conditions were observed at the cross-section during the subsequent 50 minutes of deposition. Based on the observed results above, the Zn deposition could be depicted in Fig. 2e: there is a Zn electrode with a dense and smooth surface in the AS electrolyte compared with a surface with dendrites in the ZS electrolyte. In conclusion, the designed AS electrolytes could induce an even and smooth Zn deposition behavior.
The effect of the metal–electrolyte interface on Zn metal corrosion can be quantified by the linear polarization experiments. As shown in Fig. 3a and Table S2 (ESI†), the corrosion current density of 2.573 mA cm−2 for Zn anode in AS electrolyte is lower than that in ZS electrolyte (3.101 mA cm−2). Additionally, the corrosion potential of the Zn anode becomes more positive, suggesting that the Zn anode in the AS electrolyte has better corrosion resistance than that in the ZS electrolyte.42 Also, chronoamperometry (CA) measurements demonstrate that AS electrolytes increase nucleation sites (Fig. 3b). Under an applied 200 mV overpotential, AS electrolyte shows a smaller response current for all 300 s during testing. More experiments have been conducted to investigate the effect of Al3+ ions on the polarization of Zn nucleation. As displayed in Fig. 3c, the overpotential of 29.4 mV for Zn2+ ion nucleation on the Zn anode in AS electrolyte is notably lower than that in ZS electrolyte (74.2 mV) under a current density of 1.0 mA cm−2. Besides, the same results can also be found under a current density of 2 mA cm−2 and 5.0 mA cm−2 (Fig. S4, ESI†). Generally, a lower Zn2+ ions nucleation overpotential on the Zn anode implies a faster and easier Zn deposition. Besides, the results were also observed in the cyclic voltammetric curves of Zn||Cu cells in different electrolytes (Fig. 3d). The cell in AS electrolyte revealed a significant decrease in the polarization of Zn nucleation (34 mV). In addition, the CV tests of Zn||Cu cells with varying concentrations of aluminum sulfate additives (0.1 M, 0.2 M, 0.5 M Al2(SO4)3) also showed a rightward shift of the Zn2+ ion deposition point, which also suggests that Al3+ ions can reduce the deposition potential of Zn2+ ions during cycling (Fig. S5, ESI†).
 | ||
| Fig. 3 (a) Linear polarization curves of the fresh Zn electrodes (the commercial Zn foils delivering the same surface conditions and the effective surface area) collected with a scanning rate of 1 mV s−1 in ZS and AS electrolytes using a three-electrode system. (b) Chronoamperometry (CA) curves of ZS and AS electrolytes using a three-electrode cell. (c) Voltage profiles of galvanostatic Zn deposition on the Zn anode at 1 mA cm−2. (d) Cyclic voltammograms (CV) of Zn||Cu cells in ZS and AS electrolytes. (e) Hydrogen evolution reaction (HER) performance of the Zn anode in ZS and AS electrolyte at a scan rate of 1 mV s−1. (f) Electrochemical impedance spectroscopy (EIS) plots of Zn||Zn coin cells with ZS and AS electrolyte. | ||
Furthermore, linear scan voltammetry (LSV) of the Zn anode in different electrolytes was used to investigate the hydrogen evolution reaction (HER) in different electrolytes. Fig. 3e shows that the Zn anode in AS electrolyte exhibits a lower overpotential. Overall electrochemical windows of ZS and AS electrolytes by the three-electrode system further reveal that AS electrolyte has a wider electrochemical window compared with ZS electrolyte (Fig. S6, ESI†). The metal–electrolyte interface activity can be demonstrated by the results of cyclic voltammetry (CV) tests on Zn||Zn coin cells.44 As shown in Fig. S7 (ESI†), compared to the ZS and AS electrolyte, the Zn||Zn symmetric cell in the AS electrolyte exhibits larger integrated peak areas. This suggests improved interfacial activity facilitated by the metal–electrolyte interface. Additionally, the charge transfer resistance of the Zn||Zn symmetric cell in AS electrolyte was significantly reduced (Fig. 3f and Table S3, ESI†), implying lower electrolyte resistance and ion diffusion resistance. Besides, EIS tests on Zn||Zn cells in different concentrations of aluminum sulfate additives were also carried out (Fig. S8, ESI†). The results show that Zn||Zn cell in AS electrolyte (2 M ZnSO4 electrolyte with 0.5 M Al2(SO4)3 additive) has the lowest impedance, which is consistent with the electrolyte concentration optimization results. These results show that AS electrolyte not only reduces the deposition potential of Zn2+ ions but also diminishes the corrosion behavior on the Zn anode.
To investigate the electrochemical performance of AS electrolyte, a MnO2 cathode was chosen as the cathode material (Fig. S9, ESI†).61Fig. 4a presents a comparison of cyclic voltammetry (CV) curves of Zn||MnO2 full cell in ZS and AS electrolyte at a scan rate of 0.1 mV s−1. There is a cathodic peak at around 1.65 V for the AS electrolyte group, while the peak voltage for the ZS group is 1.32 V. The Zn||MnO2 full cells in electrolytes with different concentrations of anhydrous aluminum sulfate additive all exhibited cathodic peaks (Fig. S10 and S11, ESI†). These electrochemical results suggest that AS electrolytes may introduce a series of new chemical reactions, which in turn increases the discharge plateau of the Zn||MnO2 full cell. The increased discharge plateau was also observed in charge/discharge curves (Fig. 4b). At 0.1C, the AS electrolyte group exhibited a more pronounced discharge plateau at around 1.65 V. Moreover, the cell in AS electrolyte shows a favorable rate capability, and discharge capacities of 155, 135, 107, and 80 mA h g−1 were recorded at rates of 0.1, 0.2, 0.5, and 1C, respectively (Fig. 4c). As shown in Fig. 4d, discharge capacities change with the increase of the current density from 0.1C to 1C. When the current density is switched back to 0.1C, a dramatic capacity loss is noticed in the Zn||MnO2 cell in conventional ZS electrolyte compared with the cell in AS electrolyte (Fig. 4d). Surprisingly, the full cell in AS electrolyte was able to maintain good cycling stability even at a high cathode loading level (8.0 mg cm−2 and 16.0 mg cm−2). The cycling test of full cells was carried out at 0.1C (Fig. S12, ESI†). The full cell in AS electrolyte with cathode loading of 8.0 mg cm−2 maintained better cycling stability at a rate of 0.1C (initial discharge specific capacity of 92.0 mA h g−1 and still maintained 79% capacity retention after 150 cycles) compared with full cell in ZS electrolytes. Also, with a higher cathode loading mass of 16.0 mg cm−2, the full cell in AS electrolyte can get an initial discharge specific capacity of 71.0 mA h g−1at 0.1C and remain stable for 70 cycles (Fig. S13, ESI†). Besides, in a long cycle test at 1C, full cells in AS electrolyte also demonstrated improved cycling stability following the activation stage of 0.1C for 5 cycles and 0.5C for 10 cycles. The full cell with a high-loading MnO2 (8.0 mg cm−2) cathode by using AS electrolyte exhibits a high discharge specific capacity of 84.0 mA h g−1 at the 1st cycle. It can maintain a high capacity retention rate of 78% and have a high coulombic efficiency of ∼100% even after 1200 cycles, as shown in Fig. 4e. And the full cell with a higher-loading MnO2 (11.0 mg cm−2) cathode still displays a high cycling stability for 3000 cycles (Fig. S14, ESI†).
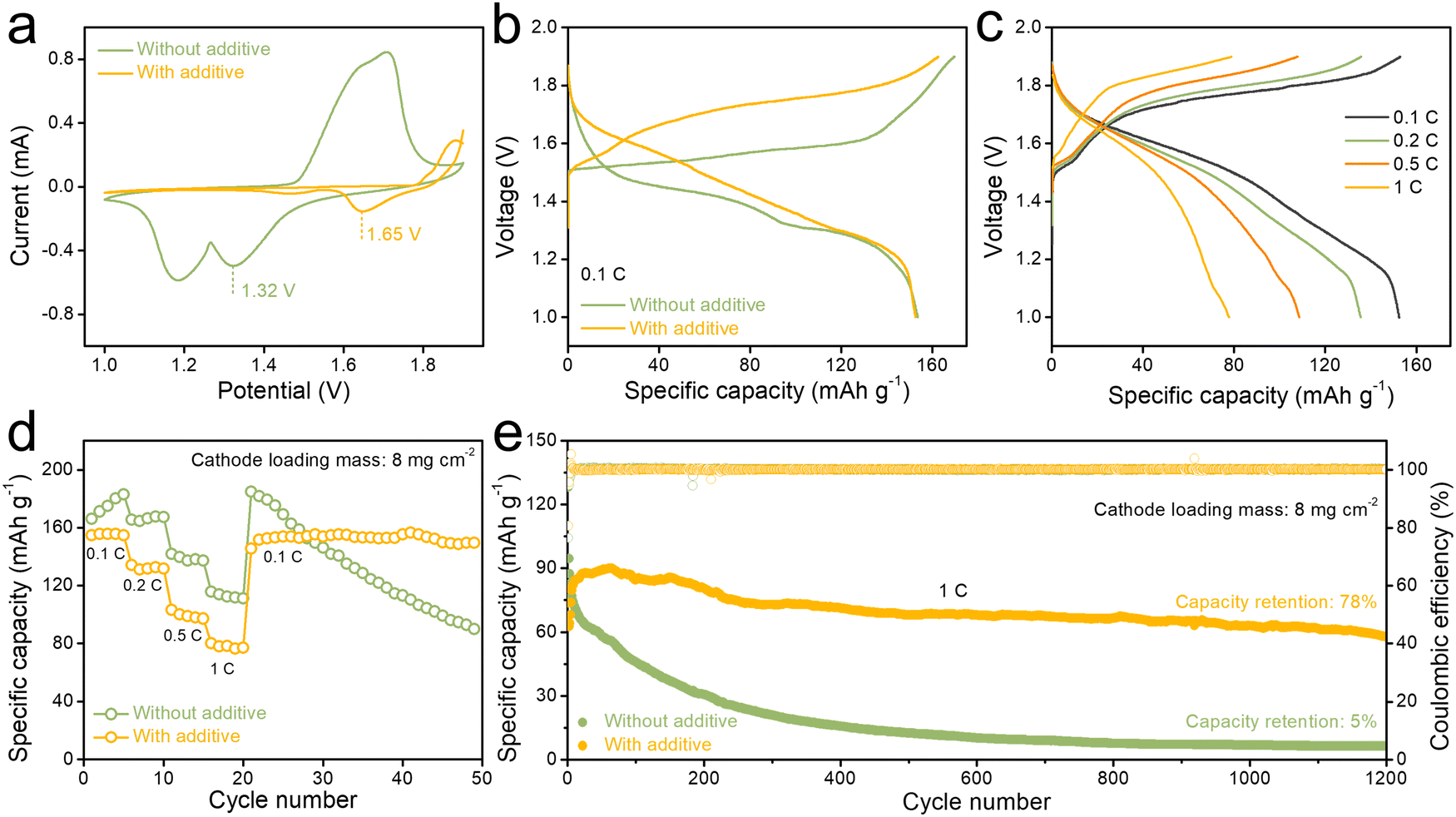 | ||
| Fig. 4 (a) CV curves of Zn||MnO2 coin cells in ZS and AS electrolyte. (b) GCD profiles of Zn||MnO2 coin cells in ZS and AS electrolyte at 0.1C (1C = 308 mA g−1). (c) GCD profiles of Zn||MnO2 coin cell in AS electrolyte from 0.1C to 1C rates. (d) Rate capability profiles from 0.1C to 1C rates. (e) Long-term cycling stability with the corresponding coulombic efficiency of Zn||MnO2 coin cells with a Zn anode and a high-loading MnO2 cathode (8.0 mg cm−2) by using ZS and AS electrolyte at 1C, respectively. | ||
Moreover, the reaction kinetics were further investigated to understand how the ions/protons intercalate into the structure of the MnO2 cathode via cyclic voltammetry (CV) testing under different scan rates (Fig. S15, ESI†). The b value of the MnO2 cathode was found to be 0.61 for the oxidation peak and 0.22 for the reduction peak, all close to 0.5. These results suggest that electrochemical charge storage in electrode material is mainly controlled by solid-state diffusion of active ions.30 In summary, the Zn||MnO2 battery in AS electrolyte displays long cycling stability under the conditions of high-loading cathode materials, with a high discharge plateau at 1.65 V. Consequently, the Zn||MnO2 full cell displays a superior cycling performance with a high-loading MnO2 cathode (Table S4, ESI†).
Ex situ XRD tests were conducted to characterize the cathode sheets in different states (Fig. 5a and b). A strong diffraction peak of the MnOOH phase at 26.3° can be clearly observed in curve A of Fig. 5b due to the MnOOH phase (MnO2 + H+ + e− → MnOOH, JCPDF:74-1049).38 During the next discharge process (A–B), the diffraction peak of MnOOH at 26.3° is elevated and no other diffraction peaks are generated. In the following charge processes (B–C–D), no obvious phase change occurred on the anode side. The composition was still dominated by the MnO2 and MnOOH with the metal cations. At 1.9 V, the anode became a mixture mainly of MnO2 and MnOOH. When discharged to 1.7 V in the second cycle (D–E), a new phase of AlOOH (JCPDF:72-1475) appeared (Fig. 5c). It may be related to the cathodic peak of Zn||MnO2 full cell at ∼1.65 V. Fig. 5c shows that the diffraction peak of the AlOOH phase disappears during the discharge of full cell from 1.7 V to 1.6 V, and the diffraction peak intensity of the MnOOH phase at 26.3° increases, suggesting that the reduction peak of ∼1.65 V may be accompanied by the transformation of the AlOOH phase into the MnOOH phase. In the discharge process (F–G), the diffraction peak of the by-product disappears and the diffraction peak of the MnOOH phase at 26.3° diminishes. The phase change in the subsequent second charging process (G–H–I) was the same as that in curve A.
 | ||
| Fig. 5 (a) The vertical galvanostatic charge and discharge profiles correspond to the XRD pattern of different stages. (b) XRD patterns of the MnO2 cathode in AS electrolyte during charge/discharge process at 0.1C (c) XRD patterns of the MnO2 cathode in AS electrolyte discharge to 1.7 V and 1.6 V during 2nd cycling. (d) XRD patterns of the MnO2 cathode at a fully charged state to 1.9 V in AS electrolyte after 30 cycles. XPS spectra of (e) Mn 2p and (f) Al 2p after 30 cycles and charge to 1.9 V for the cycled MnO2 cathode. SEM images of MnO2 cathode before (g) and after 30 cycles (h). (i) EDS mapping of MnO2 cathode after 30 cycles. | ||
To confirm the reversibility of the electrode reaction during the cycling process, we performed ex situ XRD (Fig. 5d) and XPS (Fig. 5e, f and Fig. S16, ESI†) characterization on the positive electrode after 30 cycles. The XRD results of the cathode after 30 cycles and charging to 1.9 V displayed a diffraction pattern which is consistent with curve E in Fig. 5c, suggesting the presence of MnO2 and MnOOH. In addition, two peaks of Mn at 654.6 eV and 642.8 eV can be clearly observed in the ex situ XPS test results after 30 cycles, suggesting the presence of Mn3+ and Mn4+ and corroborating the XRD results. Also, the peak of Al element at 74.4 eV in the XPS spectra further confirmed the presence of Al3+ ions in the cathode material even after all active ions were removed from the cathode. The peak of Al 2p at 74.4 eV is smaller in the XPS spectrum, indicating the Al element in the cathode, and there are no obvious diffraction peaks attributed to the Al element in the XRD test results. These results indicate that a small amount of Al3+ ions could be inserted into the cathode lattice during cycling, which can stabilize the cathode crystal structure and reduce the solubility of the MnO2 cathode material.
The Al2(SO4)3 additive effect on the side reactions in Zn||MnO2 batteries was further verified by monitoring the self-discharge behavior (Fig. S17, ESI†).62 After charging to 1.9 V and resting for 12 hours, the full battery in AS electrolyte could still maintain a high voltage of 1.70 V (vs. 1.50 V in ZS electrolyte) and complete the cycling process with a higher capacity retention ratio (83% in AS electrolyte compared with 65% in ZS electrolyte). Ex situ SEM and EDS mapping were also characterized. Compared with the loose nanowire-like MnO2 cathode structure before cycling (Fig. 5g), the surface of the circulated MnO2 cathode showed a dense monolithic structure and maintained the characteristics of the original nanowire structure (Fig. 5h). EDS mapping indicated the presence of Zn2+ and Al3+ ions in the MnO2 cathode (Fig. 5i).
Ex situ XPS survey for the MnO2 electrode after the second cycle was investigated (Fig. S18, ESI†). The signal peaks for Zn 2p were different from the others, with two characteristic peaks appearing at the discharge state (2p3/2: 1045.4 eV and 2p1/2: 1022.3 eV), suggesting the intercalation behavior of Zn2+ ions. A small amount of the intercalated Zn2+ ions remained in the cathode side after being fully charged to 1.9 V, indicating less structural stress and bonding rearrangement. The intensity of two characteristic peaks of Zn 2p weakened significantly when discharged to 1.0 V, corresponding to the deuteration of Zn2+ ions. The Al 2p spectra showed one characteristic peak of Al 2p at 74.8 eV with almost the same intensity of the two peaks, indicating few Al3+ ions embedded and detached from the cathode during the charging and discharging process. It can be demonstrated that a small amount of Al3+ ions enter the cathode side during cycling and stabilize the crystal structure of the cathode. The surface of the cathode before and after cycling exhibits a completely different morphology, suggesting that Al3+ ions mainly insert into the cathode surface to stabilize the crystal structure and thus avoid the dissolution of the cathode.
It is well known that the cations in electrolytes can easily form a solvation sheath.63,64 The solvation sheath structure can influence the active ions (de)intercalation and activation process significantly.65 Thus, DFT calculations were carried out to identify the possibly existent form of Zn2+ and Al3+ ions and the binding energy of solvation sheath in the electrolyte (Fig. 6 and Table S5, ESI†). Thermodynamically, Zn2+ and Al3+ ions are octahedrally solvated with six water molecules in the primary hybrid aqueous electrolyte. Besides, the Zn2+ solvation structure [Zn(H2O)6]2+ delivers a lower binding energy (−297.7 kcal mol−1) compared with [Al(H2O)6]3+ (−628.6 kcal mol−1). It demonstrated that Al3+ ions can combine with water molecules to form solvated molecules more easily, and then reduce the number of active water molecules in electrolyte and the irreversible dissolution of cathode material.66–68 Besides, [Zn(H2O)6]2+ could be desolated with two water molecules to form a planar [Zn(H2O)4]2+ complex which is intercalated into the Mn-based electrode according to previous research.30 Based on the above testing results and discussion, it is conjectured that during the cycling process of the Zn||MnO2 full cell in AS electrolyte, the electrode reaction is related to proton and Al3+ ion insertion. A small amount of Al3+ ions can enter the MnO2 lattice to stabilize the cathode crystal structure, forestall structure collapse, and reduce the dissolution of the MnO2 cathode.
 | ||
| Fig. 6 DFT calculation studies of ion solvation binding energy in the electrolyte. (a) The binding energy as a function of representative Zn2+ ion cluster models. (b) The binding energy as a function of representative Al3+ ion cluster models. | ||
3. Conclusions
In conclusion, the optimized addition of 0.5 M Al2(SO4)3 into aqueous Zn electrolyte has been proven to homogenize the nucleation and deposition of Zn. The symmetrical Zn cells in AS electrolyte demonstrate an ultra-stable cycling performance even after 2000 h. The Zn||MnO2 full cells show a high discharge voltage plateau of 1.65 V and maintain superior cycling stability. The Zn||MnO2 full cells using the AS electrolyte have a good capacity retention of 78% even after 1200 cycles. Furthermore, through the electrochemical tests, XRD and XPS analysis, the ion embedding and de-embedding mechanism and structural evolution of the cathode during the cycling process were elucidated. The electrochemical results confirm that AS electrolyte introduced a high discharge plateau (1.65 V). The AS-based electrolyte can also change the crystal structure of the cathode during cycling, inhibit the formation of zinc-manganese-based by-products on the positive side, and reduce the occurrence of side reactions and irreversible dissolution of the cathode. This work provides a strategy to design low-cost, high-energy density, high-safety AZIBs.4. Experimental section
4.1. The preparation of ZS and AS electrolyte
The ZS electrolyte (2 M ZnSO4) was prepared by completely dissolving 23 g of ZnSO4 (99.97%, Aladdin) into DI water to make a 40 mL solution. The 40 mL AS electrolyte (2 M ZnSO4 and 0.5 M Al2(SO4)3) was prepared by dissolving 23 g of ZnSO4 (99.97%, Aladdin) and 6.84 g Al2(SO4)3 (99%, AR, Jiuding Chemistry) into DI water then fixing the volume to 40 mL after complete dissolution. For comparison, electrolytes with 2 M ZnSO4 and 0.05 M, 0.1 M, or 0.2 M Al2(SO4)3 were also prepared by using the same method.4.2. Cathode preparation of aqueous Zn-ion batteries
MnO2 nanosheets were prepared via a hydrothermal reaction. During the chemical reaction, 0.03 M MnSO4·H2O and 2 mL 0.5 M H2SO4 were added into 100 mL DI water and stirred until a clear solution A was obtained. Afterward, 0.316 g KMnO4 was completely dissolved into 20 mL DI water to make solution B, which was slowly added into the clear solution A while stirring for 6 h. The as-prepared solution was transferred to a Teflon-lined PTFE autoclave vessel and heated at 120 °C for 12 h. Then, MnO2 nanosheets were collected, washed with DI water several times, and dried at 60 °C in a vacuum oven overnight. Cathodes were prepared using the traditional method. First, MnO2 nanosheets, super P carbon and polyvinylidene fluoride (PVDF) were mixed in N-methyl pyrrolidone (NMP) solvent in a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 and formed into a slurry. Then the mixed slurry was coated onto carbon paper (CC) and dried at 80 °C in a vacuum oven overnight to get the MnO2@CC electrode. The loading mass of the MnO2 can be controlled at 2–16 mg cm−2.
:2:1 and formed into a slurry. Then the mixed slurry was coated onto carbon paper (CC) and dried at 80 °C in a vacuum oven overnight to get the MnO2@CC electrode. The loading mass of the MnO2 can be controlled at 2–16 mg cm−2.
4.3. Electrochemical properties testing
Electrochemical properties were tested using CR2032 coin batteries with the MnO2@CC electrode as a cathode and a 99.995% Zn foil as an anode. Both 2 M ZnSO4 and 2 M ZnSO4 with 0.5 M Al2(SO4)3 aqueous solution were used as electrolytes and absorbed using a GF/D-type glass fiber separator. The amount of electrolytes (100 μL) is the same in each cell. The galvanostatic charge/discharge performance (GCD) was tested using LANHE CT3002A equipment (Voltage: 1.0–1.9 V) based on the active material. Electrochemical impedance spectroscopy (EIS) measurements were conducted over a frequency range of 0.01–100 kHz. CV and EIS curves were tested by a CHI 660E electrochemical work station and the scan rate was set at 0.1, 0.2, 0.4, 0.6, 0.8 and 1.0 mV s−1.4.4. Materials characterization
The XRD profiles were tested with X-ray X’Pert3 Powder equipped with Cu Kα radiation with a 2θ step of 2° min−1 in the range of 10°–90° at 40 kV and 40 mA and analyzed using software Jade 6.5. The XPS spectra were tested by VGESCALAB250 X-ray photoelectron spectroscopy diffractometer equipped with Al radiation 1486.6 eV and analyzed using software Avantage. The microstructure and morphology were observed through SEM (Hitachi S-4800). Contact angle tests were carried out using a DSA25S Droplet shape analyzer, KRUSS, Germany.4.5. Theoretical methods of DFT calculations
The Gaussian 09 suite of ab initio programs was employed to perform DFT calculations using the hybrid meta-GGA level M06 functional in conjugation with 6-31G(d) basis set for small atoms, and the Stuttgart relativistic effective core potential basis set for Os (ECP60MDF). The accuracy of numerical integrations is at the ultrafine grid (99590) level. Geometric optimizations were performed without restriction in zinc sulfate. The integral equation formalism polarizable continuum (IEFPCM) solvation model with the SMD radii was used for solvent effect corrections. Thermal corrections were obtained by frequency calculations on optimized structures within the harmonic potential approximation under 298.15 K and 1 atm pressure. The optimized structures were confirmed to have no imaginary vibrational mode for all equilibrium structures.
Author contributions
K. Y. conceived the idea for the project and supervised the project. J. Y., X. Z., and S. W. wrote the manuscript. Q. W. and C. G., helped K. Y., prepare the cathode materials. Y. S. and X. G. helped S. W., analyzed the eletrodes in this work. All the authors agreed upon the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors are thankful for support from the 2024 Technology Project of State Grid (4000-202419077A-1-1-ZN).References
- M. Winter, B. Barnett and K. Xu, Before Li Ion Batteries, Chem. Rev., 2018, 118(23), 11433–11456 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, 30 Years of Lithium-Ion Batteries, Adv. Mater., 2018, e1800561 CrossRef PubMed.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich and S. Lambert, et al., Recycling lithium-ion batteries from electric vehicles, Nature, 2019, 575(7781), 75–86 Search PubMed.
- X. Fu and W. H. Zhong, Biomaterials for High-Energy Lithium-Based Batteries: Strategies, Challenges, and Perspectives, Adv. Energy Mater., 2019, 9(40), 1901774 Search PubMed.
- J. B. Goodenough and Y. Kim, Challenges for Rechargeable Li Batteries, Chem. Mater., 2009, 22(3), 587–603 Search PubMed.
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Aqueous rechargeable Li and Na ion batteries, Chem. Rev., 2014, 114(23), 11788–11827 Search PubMed.
- J. Lee, D. A. Kitchaev, D. H. Kwon, C. W. Lee, J. K. Papp, Y. S. Liu, Z. Lun, R. J. Clement, T. Shi and B. D. McCloskey, et al., Reversible Mn2+/Mn4+ double redox in lithium-excess cathode materials, Nature, 2018, 556(7700), 185–190 CrossRef CAS PubMed.
- X. Shen, X. Wang, Y. Zhou, Y. Shi, L. Zhao, H. Jin, J. Di and Q. Li, Highly Reversible Aqueous Zn-MnO2 Battery by Supplementing Mn2+-Mediated MnO2 Deposition and Dissolution, Adv. Funct. Mater., 2021, 31(27), 2101579 CrossRef CAS.
- Z. Tie and Z. Niu, Design Strategies for High-Performance Aqueous Zn/Organic Batteries, Angew. Chem., Int. Ed., 2020, 59(48), 21293–21303 CrossRef CAS PubMed.
- Z. Li, Z. Gong, X. Wu, K. Ye, J. Yan, G. Wang, Y. Wei, K. Zhu, J. Yi and D. Cao, et al., Dendrite-free and anti-corrosion Zn metal anode enabled by an artificial layer for high-performance Zn ion capacitor, Chin. Chem. Lett., 2022, 33(8), 5 CrossRef.
- S. Guo, L. Qin, T. Zhang, M. Zhou, J. Zhou, G. Fang and S. Liang, Fundamentals and perspectives of electrolyte additives for aqueous zinc-ion batteries, Energy Storage Mater., 2021, 34, 545–562 CrossRef.
- Q.-N. Zhu, Z.-Y. Wang, J.-W. Wang, X.-Y. Liu, D. Yang, L.-W. Cheng, M.-Y. Tang, Y. Qin and H. Wang, Challenges and strategies for ultrafast aqueous zinc-ion batteries, Rare Met., 2020, 40(2), 309–328 CrossRef.
- S. Wu, Z. Hu, P. He, L. Ren, J. Huang and J. Luo, Crystallographic engineering of Zn anodes for aqueous batteries, eScience, 2023, 3(3), 100120 CrossRef.
- T. T. Wang, P. J. Wang, L. Pan, Z. X. He, L. Dai, L. Wang, S. D. Liu, S. C. Jun, B. A. Lu and S. Q. Liang, et al., Stabling Zinc Metal Anode with Polydopamine Regulation through Dual Effects of Fast Desolvation and Ion Confinement, Adv. Energy Mater., 2023, 13(5), 2203523 CrossRef CAS.
- Y. Shang, P. Kumar, T. Musso, U. Mittal, Q. J. Du, X. Liang and D. Kundu, Long-Life Zn Anode Enabled by Low Volume Concentration of a Benign Electrolyte Additive, Adv. Funct. Mater., 2022, 32(26), 2200606 CrossRef CAS.
- S. C. Bai, Z. D. Huang, G. J. Liang, R. Yang, D. Liu, W. Wen, X. Jin, C. Y. Zhi and X. Q. Wang, Electrolyte Additives for Stable Zn Anodes, Adv. Sci., 2024, 11(4), 2304549 CrossRef CAS PubMed.
- Y. Song, P. C. Ruan, C. W. Mao, Y. X. Chang, L. Wang, L. Dai, P. Zhou, B. A. Lu, J. Zhou and Z. X. He, Metal-Organic Frameworks Functionalized Separators for Robust Aqueous Zinc-Ion Batteries, Nano-Micro Lett., 2022, 14(1), 218 CrossRef CAS PubMed.
- M. Shimizu, K. Hirahara and S. Arai, Morphology Control of Zinc Electrodeposition by Surfactant Addition for Alkaline-based Rechargeable Batteries, Phys. Chem. Chem. Phys., 2019, 21(13), 7045–7052 RSC.
- N. Guo, Z. Peng, W. J. Huo, Y. H. Li, S. D. Liu, L. Kang, X. W. Wu, L. Dai, L. Wang and S. C. Jun, et al., Stabilizing Zn Metal Anode Through Regulation of Zn Ion Transfer and Interfacial Behavior with a Fast Ion Conductor Protective Layer, Small, 2023, 19(47), 2303963 CrossRef CAS PubMed.
- F. Wan and Z. Niu, Design Strategies for Vanadium-based Aqueous Zinc-Ion Batteries, Angew. Chem., Int. Ed., 2019, 58(46), 16358–16367 CrossRef CAS PubMed.
- J. Hao, J. Long, B. Li, X. Li, S. Zhang, F. Yang, X. Zeng, Z. Yang, W. K. Pang and Z. Guo, Toward High-Performance Hybrid Zn-Based Batteries via Deeply Understanding Their Mechanism and Using Electrolyte Additive, Adv. Funct. Mater., 2019, 29(34), 1903605 CrossRef.
- X. Zeng, J. Hao, Z. Wang, J. Mao and Z. Guo, Recent progress and perspectives on aqueous Zn-based rechargeable batteries with mild aqueous electrolytes, Energy Storage Mater., 2019, 20, 410–437 CrossRef.
- Y. Qi and Y. Xia, Electrolyte Regulation Strategies for Improving the Electrochemical Performance of Aqueous Zinc-ion Battery Cathodes, Acta Phys.-Chim. Sin., 2022, 0(0), 2205045 Search PubMed.
- L. Li, Y. H. Xie, M. L. Yao, R. Cao, X. Y. Mai, Y. Ji, L. Chen, X. L. Dong and Y. Y. Xia, Dual-additive-based Electrolyte Design for Aqueous Zinc Ion Batteries with High Plating/Stripping Efficiency, Chem. Commun., 2024, 60(53), 6809–6812 RSC.
- Z. Zhu, L. Wang, X. Tang, L. Li, Y. Shi and J. Shao, Application of Poly(vinyl alcohol) Conductive Hydrogel Electrolytes in Zinc Ion Batteries, Chin. J. Inorg. Chem., 2025, 41(5), 893–902 CAS.
- C. Sun, R. Miao, J. Li, Y. Sun, Y. Chen, J. Pan, Y. Tang and P. Wan, Green Environmentally Friendly “Zn(CH3SO3)2” Electrolyte for Aqueous Zinc-Ion Batteries, ACS Appl. Mater. Interfaces, 2023, 15(16), 20089–20099 CrossRef CAS PubMed.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu and M. Zhu, et al., Zn/MnO2 Battery Chemistry With H+ and Zn2+ Coinsertion, J. Am. Chem. Soc., 2017, 139(29), 9775–9778 CrossRef CAS PubMed.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Rechargeable aqueous zinc-manganese dioxide batteries with high energy and power densities, Nat. Commun., 2017, 8(1), 405 CrossRef PubMed.
- Y. Zhang, L. Zhao, Y. Liang, X. Wang and Y. Yao, Effect of electrolyte anions on the cycle life of a polymer electrode in aqueous batteries, eScience, 2022, 2(1), 110–115 CrossRef.
- Y. Xu, J. Zhu, J. Feng, Y. Wang, X. Wu, P. Ma, X. Zhang, G. Wang and X. Yan, A rechargeable aqueous zinc/sodium manganese oxides battery with robust performance enabled by Na2SO4 electrolyte additive, Energy Storage Mater., 2021, 38, 299–308 CrossRef.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Aqueous rechargeable zinc/sodium vanadate batteries with enhanced performance from simultaneous insertion of dual carriers, Nat. Commun., 2018, 9(1), 1656 CrossRef PubMed.
- Y. Zhang, H. Li, S. Huang, S. Fan, L. Sun, B. Tian, F. Chen, Y. Wang, Y. Shi and H. Y. Yang, Rechargeable Aqueous Zinc-Ion Batteries in MgSO4/ZnSO4 Hybrid Electrolytes, Nano-Micro Lett., 2020, 12(1), 60 CrossRef CAS PubMed.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li and P. Bhattacharya, et al., Reversible aqueous zinc/manganese oxide energy storage from conversion reactions, Nat. Energy, 2016, 1(5), 16039 CrossRef CAS.
- V. Soundharrajan, B. Sambandam, S. Kim, S. Islam, J. Jo, S. Kim, V. Mathew, Y.-K. Sun and J. Kim, The dominant role of Mn2+ additive on the electrochemical reaction in ZnMn2O4 cathode for aqueous zinc-ion batteries, Energy Storage Mater., 2020, 28, 407–417 CrossRef.
- C. Qiu, X. Zhu, L. Xue, M. Ni, Y. Zhao, B. Liu and H. Xia, The function of Mn2+ additive in aqueous electrolyte for Zn/δ-MnO2 battery, Electrochim. Acta, 2020, 351, 136445 CrossRef CAS.
- M. Li, Q. He, Z. Li, Q. Li, Y. Zhang, J. Meng, X. Liu, S. Li, B. Wu and L. Chen, et al., A Novel Dendrite-Free Mn2+/Zn2+ Hybrid Battery with 2.3 V Voltage Window and 11000-Cycle Lifespan, Adv. Energy Mater., 2019, 9(29), 1901469 CrossRef.
- M. Chamoun, W. R. Brant, C.-W. Tai, G. Karlsson and D. Noréus, Rechargeability of aqueous sulfate Zn/MnO2 batteries enhanced by accessible Mn2+ ions, Energy Storage Mater., 2018, 15, 351–360 CrossRef.
- N. Li, G. Li, C. Li, H. Yang, G. Qin, X. Sun, F. Li and H. M. Cheng, Bi-Cation Electrolyte for a 1.7 V Aqueous Zn Ion Battery, ACS Appl. Mater. Interfaces, 2020, 12(12), 13790–13796 CrossRef CAS PubMed.
- D. S. Liu, Z. Zhang, Y. Zhang, M. Ye, S. Huang, S. You, Z. Du, J. He, Z. Wen and Y. Tang, et al., Manipulating OH− -Mediated Anode-Cathode Cross-Communication Toward Long-Life Aqueous Zinc-Vanadium Batteries, Angew. Chem., Int. Ed., 2023, 62(5), e202215385 Search PubMed.
- K. Li, Y. Gong and J. H. Lin, Benzoquinone-intercalated vanadium oxide in the electrolyte with Al3+ for zinc-ion storage: dual-pillar effect and reversible disorder–order conversion, Chem. Eng. J., 2023, 452, 139621 Search PubMed.
- Z. Liu, T. Cui, G. Pulletikurthi, A. Lahiri, T. Carstens, M. Olschewski and F. Endres, Dendrite-Free Nanocrystalline Zinc Electrodeposition from an Ionic Liquid Containing Nickel Triflate for Rechargeable Zn-Based Batteries, Angew. Chem., Int. Ed., 2016, 55(8), 2889–2893 CrossRef CAS PubMed.
- R. Zhao, H. Wang, H. Du, Y. Yang, Z. Gao, L. Qie and Y. Huang, Lanthanum nitrate as aqueous electrolyte additive for favourable zinc metal electrodeposition, Nat. Commun., 2022, 13(1), 3252 CrossRef CAS PubMed.
- H. Yu, D. Chen, Q. Li, C. Yan, Z. Jiang, L. Zhou, W. Wei, J. Ma, X. Ji and Y. Chen, et al., In Situ Construction of Anode–Molecule Interface via Lone-Pair Electrons in Trace Organic Molecules Additives to Achieve Stable Zinc Metal Anodes, Adv. Energy Mater., 2023, 13(22), 2300550 CrossRef CAS.
- H. Qin, W. Kuang, N. Hu, X. Zhong, D. Huang, F. Shen, Z. Wei, Y. Huang, J. Xu and H. He, Building Metal-Molecule Interface towards Stable and Reversible Zn Metal Anodes for Aqueous Rechargeable Zinc Batteries, Adv. Funct. Mater., 2022, 32(47), 2206695 CrossRef CAS.
- W. Lv, J. Meng, X. Li, C. Xu, W. Yang, S. Duan, Y. Li, X. Ju, R. Yuan and Y. Tian, et al., Boosting zinc storage in potassium-birnessite via organic-inorganic electrolyte strategy with slight N-methyl-2-pyrrolidone additive, Energy Storage Mater., 2023, 54, 784–793 CrossRef.
- K. Xie, K. Ren, Q. Wang, Y. Lin, F. Ma, C. Sun, Y. Li, X. Zhao and C. Lai, In situ construction of zinc-rich polymeric solid–electrolyte interface for high-performance zinc anode, eScience, 2023, 3, 3 CrossRef.
- G. Li, L. Sun, S. Zhang, C. Zhang, H. Jin, K. Davey, G. Liang, S. Liu, J. Mao and Z. Guo, Developing Cathode Materials for Aqueous Zinc Ion Batteries: Challenges and Practical Prospects, Adv. Funct. Mater., 2023, 34(5), 2301291 CrossRef.
- J. Li, Z. Guo, J. Wu, Z. Zheng, Z. Yu, F. She, L. Lai, H. Li, Y. Chen and L. Wei, Dextran: A Multifunctional and Universal Electrolyte Additive for Aqueous Zn Ion Batteries, Adv. Energy Mater., 2023, 13(37), 2301743 CrossRef CAS.
- Y. Xu, G. Zhang, J. Liu, J. Zhang, X. Wang, X. Pu, J. Wang, C. Yan, Y. Cao and H. Yang, et al., Recent Advances on Challenges and Strategies of Manganese Dioxide Cathodes for Aqueous Zinc-Ion Batteries, Energy Environ. Mater., 2023, 6(6), e12575 CrossRef CAS.
- Y. Heng, Z. Gu, J. Guo and X. Wu, Research Progresses on Vanadium-based Cathode Materials for Aqueous Zinc-Ion Batteries, Acta Phys.-Chim. Sin., 2020, 37(3), 2005013 Search PubMed.
- Y. Niu, D. Wang, Y. Ma and L. Zhi, Cascading V2O3/N-doped carbon hybrid nanosheets as high-performance cathode materials for aqueous zinc-ion batteries, Chin. Chem. Lett., 2022, 33(5), 1430–1434 CrossRef CAS.
- X. Chen, P. Ruan, X. Wu, S. Liang and J. Zhou, Crystal Structures, Reaction Mechanisms, and Optimization Strategies of MnO2 Cathode for Aqueous Rechargeable Zinc Batteries, Acta Phys.-Chim. Sin., 2021, 38(11), 2111003 Search PubMed.
- N. Dong, F. Zhang and H. Pan, Towards the practical application of Zn metal anodes for mild aqueous rechargeable Zn batteries, Chem. Sci., 2022, 13(28), 8243–8252 RSC.
- Z. You, W. Hua, N. Li, H. Liu and J.-G. Wang, An in-depth mechanistic insight into the redox reaction and degradation of aqueous Zn-MnO2 batteries, Chin. Chem. Lett., 2023, 34(4), 107525 CrossRef CAS.
- X. Shen, X. Wang, N. Yu, W. Yang, Y. Zhou, Y. Shi, Y. Wang, L. Dong, J. Di and Q. Li, A Polypyrrole-Coated MnO2/Carbon Nanotube Film Cathode for Rechargeable Aqueous Zn-Ion Batteries, Acta Phys.-Chim. Sin., 2022, 38(5), 2006059 Search PubMed.
- F. Hua, Y. Gua, F. Cui, G. Song and K. Zhu, High-performance (NH4)2V6O16·0.9H2O nanobelts modified with reduced graphene oxide for aqueous zinc ion batteries, Chin. Chem. Lett., 2021, 32(12), 3793–3798 CrossRef.
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Rechargeable Batteries for Grid Scale Energy Storage, Chem. Rev., 2022, 122(22), 16610–16751 CrossRef CAS PubMed.
- M. Zhou, Y. Chen, G. Fang and S. Liang, Electrolyte/electrode interfacial electrochemical behaviors and optimization strategies in aqueous zinc-ion batteries, Energy Storage Mater., 2022, 45, 618–646 CrossRef.
- X. Zheng, T. Ahmad and W. Chen, Challenges and strategies on Zn electrodeposition for stable Zn-ion batteries, Energy Storage Mater., 2021, 39, 365–394 CrossRef.
- H. Tian, Z. Li, G. Feng, Z. Yang, D. Fox, M. Wang, H. Zhou, L. Zhai, A. Kushima and Y. Du, et al., Stable, high-performance, dendrite-free, seawater-based aqueous batteries, Nat. Commun., 2021, 12(1), 237 CrossRef CAS PubMed.
- H. Tian, G. Feng, Q. Wang, Z. Li, W. Zhang, M. Lucero, Z. Feng, Z. L. Wang, Y. Zhang and C. Zhen, et al., Three-dimensional Zn-based alloys for dendrite-free aqueous Zn battery in dual-cation electrolytes, Nat. Commun., 2022, 13(1), 7922 CrossRef CAS PubMed.
- G. Ma, L. Miao, Y. Dong, W. Yuan, X. Nie, S. Di, Y. Wang, L. Wang and N. Zhang, Reshaping the electrolyte structure and interface chemistry for stable aqueous zinc batteries, Energy Storage Mater., 2022, 47, 203–210 CrossRef.
- Z. Hou, M. Dong, Y. Xiong, X. Zhang, H. Ao, M. Liu, Y. Zhu and Y. Qian, A High-Energy and Long-Life Aqueous Zn/Birnessite Battery via Reversible Water and Zn2+ Coinsertion, Small, 2020, 16(26), e2001228 CrossRef PubMed.
- T. Zhang, Y. Tang, G. Fang, C. Zhang, H. Zhang, X. Guo, X. Cao, J. Zhou, A. Pan and S. Liang, Electrochemical Activation of Manganese-Based Cathode in Aqueous Zinc-Ion Electrolyte, Adv. Funct. Mater., 2020, 30(30), 2002711 CrossRef CAS.
- X. Li, X. Wang, L. Ma and W. Huang, Solvation Structures in Aqueous Metal-Ion Batteries, Adv. Energy Mater., 2022, 12(37), 2202068 Search PubMed.
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong and Z. Zhao, et al., Hydrated Eutectic Electrolytes with Ligand-Oriented Solvation Shells for Long-Cycling Zinc-Organic Batteries, Joule, 2020, 4(7), 1557–1574 CrossRef CAS.
- J. Chen, J. Vatamanu, L. Xing, O. Borodin, H. Chen, X. Guan, X. Liu, K. Xu and W. Li, Improving Electrochemical Stability and Low-Temperature Performance with Water/Acetonitrile Hybrid Electrolytes, Adv. Energy Mater., 2019, 10(3), 1902654 CrossRef.
- S. Liu, J. Mao, W. K. Pang, J. Vongsvivut, X. Zeng, L. Thomsen, Y. Wang, J. Liu, D. Li and Z. Guo, Tuning the Electrolyte Solvation Structure to Suppress Cathode Dissolution, Water Reactivity, and Zn Dendrite Growth in Zinc-Ion Batteries, Adv. Funct. Mater., 2021, 31(38), 2104281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01172h |
| This journal is © the Owner Societies 2025 |