
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported metal and metal oxide particles with proximity effect for catalysis
Subhadeep Biswas
a,
Anjali Pal
*a and
Tarasankar Pal
 *b
*b
aDepartment of Civil Engineering, Indian Institute of Technology, Kharagpur 721302, India. E-mail: anjalipal5@gmail.com
bDepartment of Chemical Sciences, University of Johannesburg, Auckland Park, South Africa. E-mail: tarasankar.pal@gmail.com
First published on 25th September 2020
Abstract
External influence is essential for any change to occur in this world. Similarly, the reaction path of a chemical reaction can be changed with the addition of a catalyst from outside. Sometimes a catalyst performs better when it remains associated with an inert substance, which is called a support material (SM). Improved catalyst accomplishment arises from the ‘proximity effect’. Even inert supports play a role in better product formulation or environmental remediation. In this review, it has been shown how the SM, as a nest, aids the catalyst particle synergistically to perform a good job in a chemical reaction. The structure–function relationship of SM helps in catalyst activation to some extent, and produces active centres that are difficult to fully ascertain. In the text, Langmuir–Hinshelwood (L–H), Mars-van Krevelen (MVK), and Eley–Rideal (E–R) mechanisms are highlighted for the adsorption processes as the case may be. Again, the importance of SM for both catalyst and substrates has been consolidated here in the text. Finally, the role of the initiator and the promoter is also discussed in this review.
 Subhadeep Biswas | Mr Subhadeep Biswas obtained his B.E. degree from Jadavpur University and M. Tech degree from IIT Kharagpur, India. He is currently pursuing for his PhD degree from IIT Kharagpur, India under the guidance of Prof. Anjali Pal. His research interests include environmental pollutant abatement by adsorptive and catalytic methods. His work on the development of spectroscopic methods for the analysis of different kinds of cationic surfactants in wastewater samples is unique and reliable. |
 Anjali Pal | Professor Anjali Pal, M.Sc, PhD graduated from Calcutta University in Chemistry and at present is teaching as a full professor at the Department of Civil Engineering, Indian Institute of Technology, Kharagpur. Prof. Pal is actively engaged in teaching and research in the field of environmental engineering and science. She has published more than 200 research papers. She has received an International Hall of Fame award (USA), and an R & D 100 award (USA) for her contribution in the field of biomedical and pollution monitoring. Her Convention Award from the Indian Chemical Society is for her independent contribution to organic chemistry. She has visited many countries such as USA, UK, Japan, France, and Taiwan as a visiting professor. She is working in the fields of adsolubilization, adsorption, advanced oxidation processes (AOPs), catalysis, and spectroscopy mainly for environmental remediation. |
 Tarasankar Pal | Professor Tarasankar Pal, MSc, PhD, DSc, FNASc, FWAST is an alumnus of the University of Burdwan, and worked at the Department of Chemistry, Indian Institute of Technology (IIT) Kharagpur from 1984 till 2017. His discoveries such as general methods for cost effective ‘alloying of metals’ in high boiling liquid, ‘arsenic detection kit’ for arsenic-affected rural people, and ‘bench marked model reaction’ to test any metal nanoparticle catalyst deserve special mention. His research interest extends over the applications of metal and semiconductor nanoparticles for catalytic, photocatalytic, electrocatalytic, and spectroscopic applications. His original contributions to surface enhanced Raman scattering (SERS), synergistic fluorescence enhancement by coinage metal nanoparticles, and supercapacitor fields are noteworthy. Professor Pal has supervised 32 PhD students. He is currently a Distinguished Visiting Professor of the University of Johannesburg, South Africa where his research focus embodies sensors and device fabrication. Some of his awards include a gold medal from the Indian Chemical Society, ISCAS gold medal, President Award from the Tokyo University of Japan, and R & D-100 Award from the USA. |
1. Introduction
With the gradual progress of the human civilization, the lifestyle of human beings has changed considerably and consequently, the stress on natural resources is increasing rapidly. Huge consumption of energy and already available resources has already pushed the present world into great danger. Moreover, in the coming days, energy consumption will increase more with the increase in the population. Not only energy consumption but also environmental pollution would be a matter of great concern to the present society. Thus, it is an urgent need to find a route through which new sources of energy can be identified. It is important to implement a less hazardous route to synthesize valuable chemical products and finally to develop easy and efficient methods for pollution abatement. To address these problems, scientists have devoted their research to find out suitable solutions. Catalysis can provide an attractive solution to this energy depletion and environmental problems. Catalysis helps to improve the process design and development, and therefore maximizes the yield.1 Some stimulating chemical reactions vis-à-vis significant developments were reported early in the 19th century and after that the ‘Catalysis’ term was coined by Berzelius (1779–1848) in the year 1836. Catalysis has become an ever-expanding subject in every branch of chemistry due to its practical importance, especially in chemical and pharmaceutical industries. The production of compounds in a short period is the main and important objective of catalysis. Some notable examples of catalytic reactions include oxygen preparation from the decomposition of potassium chlorate in the presence of MnO2 catalyst, ammonia production by Haber's process in the presence of an iron catalyst,2 sulphuric acid production by V2O5 catalyst,3 and hydrogen peroxide decomposition by the MnO2 catalyst.4 A catalyst often reduces the energy consumption and waste production. Moreover, the conversion of waste to valuable end products and recycling of the used material can be efficiently governed by the catalytic reactions. Iwaya et al. reported the use of K3PO4 as a catalyst for the depolymerization of unsaturated polyester from fiber-reinforced plastic.5In the current situation, global warming has become one of the most common challenging issues. CO2 is a well-known greenhouse gas that is responsible for global warming; therefore, its emission in excess is not desirable in the atmosphere. However, despite the discovery of many clean fuel sources, its emission has not been significantly reduced. As per Thomas and Harris, the annual anthropogenic rate of emission of CO2 is nearly 40 G-ton per year.6 Thus, managing its consumption and sorting out ways and means to recycle or reuse it is an important job. CO2 storage in various geological structures is an obvious option.7 However instead of storing, recycling it to some other form could be a better option for waste management. Methane production from CO2 using catalytic reduction is therefore a good choice.8 A series of supported and unsupported catalysts have been developed by various researchers to synthesize methane from CO2. Moreover, ethanol can also be synthesized by CO2 reduction.9 The ethanol thus formed can be converted to ethylene by catalytic dehydration using a Brønsted acid catalyst10 and can be further converted to produce ethylene oxide and ethylene glycol, which are undoubtedly valuable industrial products.11 Thus, a catalyst can produce a chain of value-added products from a waste product. Another prominent example of solving an environmental issue is the catalytic reduction of NOx to produce nitrogen and oxygen gas.12 A catalyst is a chemical substance that is usually added in a very small proportion to a chemical reaction to accelerate the rate of the reaction. A catalyst cannot start or end a reaction but can change the pathway of the reaction. This implies that it has no control over the thermodynamics of a chemical reaction but it can alter the kinetics of a reaction by lowering the activation energy. Surprisingly, catalysts have become an integral part of the current day world scenario. Their presence is everywhere starting from laundry wash to lens making. Catalysts have made our life easier. An example can be cited here regarding the manufacture of common day plastic items. Plastic is a conglomeration of polymers, which finds many uses in the current society. The discovery of the metallocene catalyst has made polymerization a very easy job for chemists.13
Various noble metals, transition metals, or metal oxide and bimetallic particles are used as catalysts. Thus, catalysts can be integrated into the reaction system in two ways: homogeneously or heterogeneously. This is a unifying concept for both and also for nanoparticle supports. In homogeneous systems, metal oxides or mixed oxides are made phase compatible and directly applied without any support, whereas in heterogeneous systems, the catalyst is suitably supported on a template called support material (SM). Supports play a very crucial role in the activation of the catalyst. The support plays two important roles for supported metal catalysts by positioning itself as a ligand, which conditions the nature of the active site and contributes straight-forwardly to the reactivity.
The SM not only stabilizes the nanoparticles but also acts cooperatively with nanoparticle surfaces. The SM also activates the substrates in different ways. The SM alters the electron density of the anchored metal nanoparticle, thus making inactive gold (Au) particle an effective catalyst, which is otherwise a noble metal. As described by van't Hoff for the 1st time in 1895, the adsorption of trace materials on the support surface extends active sites for the reaction to occur. On the other hand, very high loading leads to sintering and also results in decreased efficiency of the catalyst. Hence, loading should be optimized. Since the time of early development of catalysts, attempts have been continuously made to immobilize them on some inorganic or organic support. Heterogeneous materials have become attractive in terms of the selection since they are more practical in comparison to their homogeneous counterparts in terms of reusability, recyclability, and recovery. For example, transition metal complexes are very useful catalysts in pharmaceutical industries. But due to their high solubility, it is very difficult to separate them and hence, their use has been restricted to avoid the risk of metal contamination.14 The present review features the ‘proximity effect’ in catalysis with the underlined principles. A comprehensive list regarding some famous reactions catalyzed by supported catalysts has been compiled in Table 1. The current review emphasizes on the importance of the supports used for holding the catalysts, methods of synthesis of supported catalysts, various characterization techniques, useful reaction mechanisms such as Langmuir–Hinshelwood (L–H), Mars-van Krevelen (MVK), and Eley–Rideal (E–R) mechanisms, and kinetic studies. Most of the studies reviewed herein have been carried out during 1990–2019.
| Reaction | Substrate | Catalyst | SM | Maximum yield | Reference |
|---|---|---|---|---|---|
| Catalytic cracking | Tar | Cu | Rice husk char | 90.6% | 15 |
| Fe | 92.6% | ||||
| K | 82.7% | ||||
| Biomass tar | Ni | Char | 1.14 N m−3 gas per kg | 16 | |
| Methane | Ni | SiO2 | 34.6% | 17 | |
| Syngas production | Methane | Co–Pt | Al2O3 | 100% conversion of CO2 | 18 |
| Methane | Cu, Co, Fe, Pt, Pd, Ni | ZrO2 | 98.6% conversion for Pt catalyst | 19 | |
| Methane | Ni | SiO2 | — | 20 | |
| Fischer–Tropsch synthesis | CO | Ruthenium | TiO2 | 6.4% CO converted to hydrocarbon | 21 |
| CO | Co–Re | TiO2 | 52.8% CO conversion | 22 | |
| CO | Fe | Al2O3 and activated carbon | — | 23 | |
| Sonogashira cross coupling | Aryl iodides | Pd(0) | Cellulose | 98% | 24 |
| Aryl bromides | Pd | Charcoal | 100% | 25 | |
| Aryl iodides | Pd | Cross-linked polymer | 95% | 26 | |
| Iodobenzene and phenyl acetylene | Pd, Cu | Mixed oxides derived from hydrotalcite | 100% | 27 | |
| Aryl iodides, bromides, and phenyl acetylene | CuPd alloy | Reduced graphene oxide | 95% | 28 | |
| CO oxidation | CO | Au | Fe2O3 | — | 29 |
| CO | Au | Fe2O3, NiOx | 6.7 s−1 TOF | 30 | |
| TiOx, CoOx | |||||
| CO | Pd, Pt | Al2O3 and FeOx | 151 × 10−3 s−1 TOF | 31 |
2. General aspects of catalysis with support materials
2.1. General types of supports
To provide a suitable SM so as to improve the product yield, the metal, metal oxide, or bimetallic catalysts are supplied with base materials with distinguished properties such as: (i) chemical inertness, (ii) resistance towards acids and bases, and (iii) resistance to high temperature. It also needs to provide a large surface area, mechanical robustness, good durability, and strong ligation to the metallic nanoparticles (MNPs).32 The support may serve as a ligand to bind with the MNPs; hence, its role is very important in terms of the catalytic activity. A strong ligand can stabilize the MNPs but in doing so it often suppresses the activity of the catalyst. On the other hand, weak ligands make the MNPs very active but cannot inhibit detachment from the ligand or leaching from the surface of the support.32 The supports not only stabilize the nanoparticles but they also offer synergism with nanoparticle surfaces to activate the substrates.Generally, support materials are often composed of silica, alumina, other metal oxides, and carbon-based materials. Silica,33 alumina, and other metal oxides34 have proved to be very suitable supports even for nanosized catalysts. The catalytic activity of a material is often described by two terms, viz., turn over number (TON) and turn over frequency (TOF). TON is defined as the amount of the substrate that has undergone reaction per unit amount of the catalyst. TOF is obtained by dividing TON by the reaction time. Higher the TOF value, higher is the catalytic activity of the material.
Oxygen, being the most abundant element in the earth's crust, has the propensity to form innumerable binary compounds such as oxides. Oxides are very stable compounds in the earth's crust and find hundreds of applications. In this era, scientists have explored the use of smart oxide materials35 and have even produced band gap tuned oxide materials. At times, these are porous with changed morphology and changed band gap energy for different applications. Catalysis includes oxides and oxides authorize further promise not only as catalysts but also as SMs. Several SMs are presented in the following section.
Apart from using the pure form of carbon for supporting catalysts, it has been found to be highly interesting that the introduction of heteroatoms such as oxygen, sulfur, and nitrogen helps in achieving better anchoring property and catalytic activity. Among all the heteroatoms, nitrogen is a very attractive option.42 The introduction of heteroatoms alters the local electronic configuration and surface chemistry of the carbon support. Support materials such as graphene sheets, nanotubes, and metal oxide assemblies make room for the accommodation of the electron density available from the surface atoms of the nanoparticles, in other words, surface free/loosely bound electrons. In this electron density sharing process, the SM in turn becomes a stable platform and can accommodate the substrate so as to allow the catalytic reaction to take place. Suitable SMs may be involved in catalysis, as has been shown for complex metal–organic frameworks (MOF) also. Recently, new 1D, 2D, and 3D supports including nanotubes (1D), graphene derivatives (2D), and MOFs (3D) have provided great promise. Frelink et al. also mentioned the importance of carbon supports in holding the Pt catalyst for methanol oxidation.43 It has been highlighted that unsupported catalysts are not as active as the supported ones and this is due to the presence of active atoms on the support surface. This sometimes becomes relevant and is similar to metal alloy catalyst particles.
However, there are studies where it has been reported that the carbon surface itself acts as a catalyst. Yan et al. reported the oxidative dehydrogenation and dehydration of methanol over oxidized carbon nanotube catalyst (oCNT).44 The catalytic efficiency of oCNT is comparable to that of industrial metal catalysts.
One of the most prominent advantages offered by the silica support is that in most cases, it remains unaltered during the temperature-programmed reduction (TPR) studies. Vant Blik et al. reported that when γ-Al2O3 is used as a support for the Co–Rh catalyst, CoAl2O4 is also formed.55 Thus, the catalytic action is diminished. But, the in case of silica support, this does not happen and the bi-metallic Co–Rh catalyst remains unaltered.
Surfactants are often used as stabilizers for transition metal nanoparticles formed by the reduction of metal ions using NaBH4, KBH4, or other reducing agents.57 Nakao and coworkers reported the preparation and stabilization of various metallic nanoparticles such as Ru, Rh, Pd, Pt, Ag, or Au using quaternary ammonium sulfates.57 Kundu et al. showed the distinguished advantages of the SDS micellar support during the reduction of MB by arsine (AsH3) gas.58 However, it has been found that the introduction of metal nanoparticles such as silver or gold makes the reduction process facile and more efficient. The schematic has been shown in Fig. 1. Usually, in a micellar environment, the spectroscopic properties and intrinsic properties such as acid–base equilibrium get shifted.59 It has been suggested that micelle-binding helps to increase the collision probability between the molecules of MB and the arsine gas (Fig. 1). Although the reaction is thermodynamically favorable in the absence of the SDS environment, it was found that the reaction does not proceed in the experimental time scale. This sort of synergism in the catalytic activity is also noticed in the case of MB degradation by Co(II)-supported micelle-anchored alumina, which has already been discussed in the previous section.50 Nanoparticles in suitable dispersion media have often been found to be better catalysts for the reactions.60
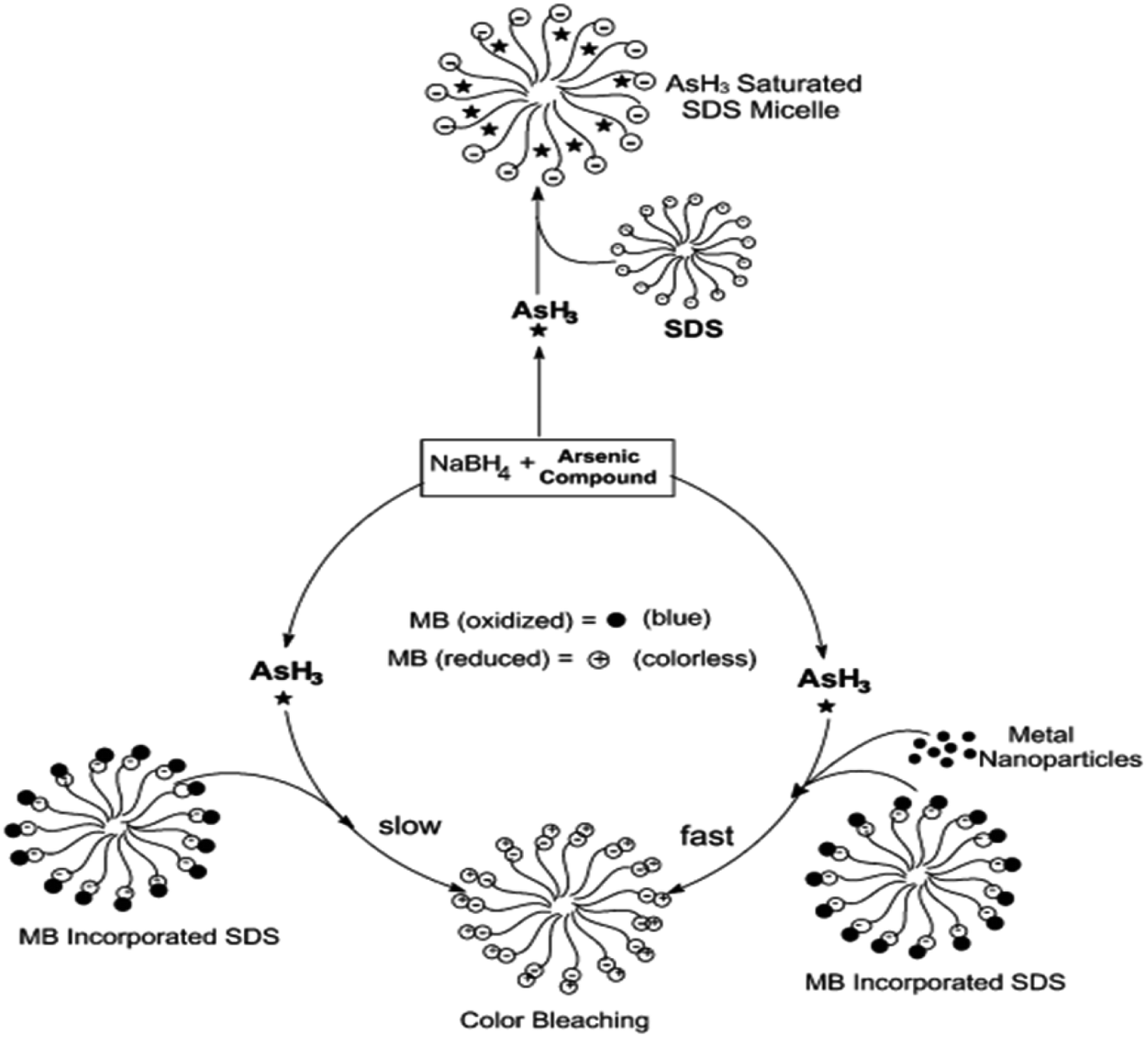 | ||
| Fig. 1 Schematic diagram of the incorporation of MB and arsine in SDS micellar environment. Reprinted with permission from the RSC journal (ref. 58). | ||
Wang et al. prepared a micellar medium in water by the self-assembled structures of block copolymer poly(N-isopropyl acrylamide)-b-poly(4-vinyl pyridine) to support gold nanoparticles.61 The diameter of the micelle formed was about 40 nm and the gold nanoparticles were of 2–4 nm diameter. The supported gold catalyst was utilized as the catalyst for 4-NP reduction in a borohydride medium. The as-prepared catalyst was very sensitive towards temperature. It was observed that above the lower critical solution temperature (LCST), the chain broke and the catalyst showed reduced activity. Semagina et al. utilized the micellar environment formed by the block copolymer poly(ethylene oxide)-block-poly-2-vinyl pyridine to support Pd nanoparticles for the catalytic selective hydrogenation of 2-butyne-1,4-diol.62 Both micelle-supported Pd and unsupported Pd (without micelle) were utilized for the reaction. However, the TOF of the supported catalysts was observed to be much higher than that of the unsupported ones.
Catalysis involving micelles (micellar catalysis) is an important field of research. Micelles can not only accommodate the catalyst in a more efficient way but can also create a specific environment to bring the substrates and catalyst together (proximity effect), thus providing suitable orientation. This is very essential in catalysis. In spite of all these advantages, it has some practical limitations for homogeneous catalysis. When catalysis occurs in homogeneous conditions, it is extremely difficult to separate the surfactants out of the system. Thus, separation becomes an inherent problem in synthetic organic chemistry, environmental remediation, and many catalytic reactions. In this regard, heterogeneous catalysis plays an important role. This solves not only the separation problem but also increases the stability of the catalyst, and in some cases, it accommodates the catalyst and the substrate in a higher amount, which in turn increases the efficiency.
Any surfactant, when present in low concentration in water medium, acts as a normal electrolyte. But, with the increase in the concentration, the behavior changes. Above a certain concentration, which is the critical micelle concentration (CMC), it forms a dynamic aggregate called micelle. The solubilizing property of micelles makes use of its hydrophobic core to remove organic (hydrophobic) molecules from water medium. The recovery of these organic molecules is otherwise very difficult when the surfactant is in the homogeneous condition. Keeping this in mind, a typical surfactant-anchored solid surface has been engineered as briefly mentioned earlier. The principle behind this is that under properly selected conditions, the surfactant molecules are allowed to adsorb on solid surfaces either as a monolayer (called hemimicelle) or a bilayer (called admicelle) (Fig. 2). The hydrophobic zone of the micellar layer can accommodate the organic molecules easily in large quantities and this is termed as “adsolubilization”. On the other hand, depending on the nature of the surfactant (anionic or cationic), the surface acquires pH-dependent charge, which can attract oppositely charged molecules. Thus, it is possible to modify the alumina surface with an anionic surfactant to form surfactant-modified alumina (SMA) so as to attract cationic dyes such as methylene blue (MB), malachite green (MG), or crystal violet (CV) (Fig. 3a).50,63,64
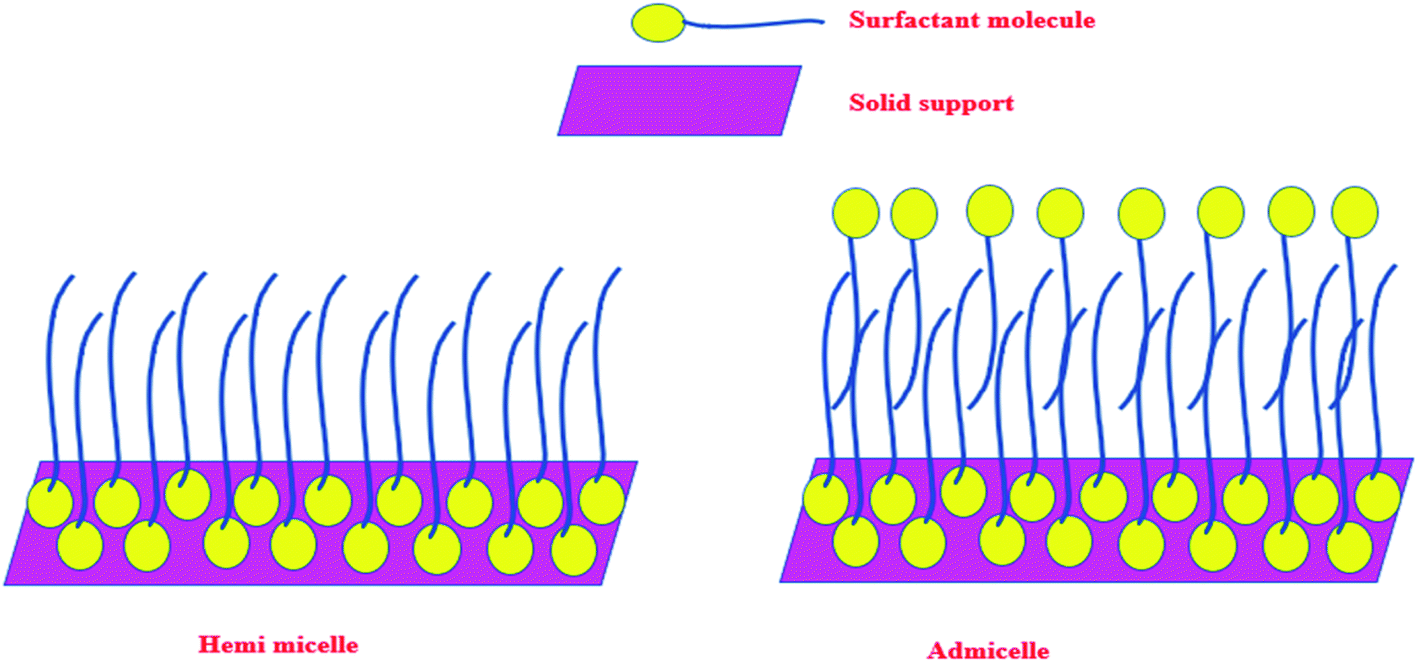 | ||
| Fig. 2 Scheme showing the formation of hemimicelles and admicelles on a solid support. | ||
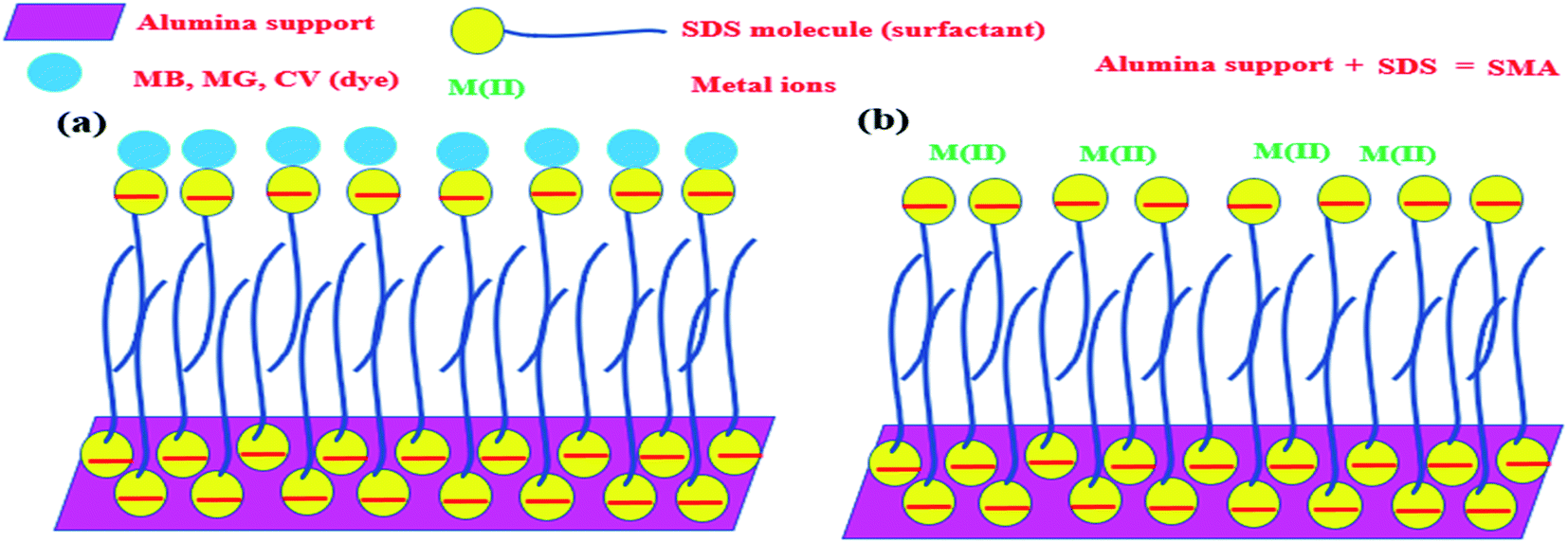 | ||
| Fig. 3 Scheme showing the adsolubilization of dyes (a) and metal ions (b) on SMA. | ||
In a similar manner, the anionic surfactant-modified alumina (SMA) can accommodate metal ions in a better way. It has been noticed that compared to alumina, metal ions can be adsolubilized/adsorbed onto SMA at a much higher concentration and in a better way (Fig. 3b).65,66
By taking advantage of the adsolubilization technique, a good modified Fenton-type catalyst was designed, where Co(II) was placed on SMA at a dose of 1.38 mg g−1. It is to be noted that this loading of Co(II) on SMA was much higher (>6 times) as compared to that of normal Al2O3 (0.205 mg g−1). This as-prepared Co(II)–SMA catalyst served well for the degradation of MB and methyl orange (MO) in the presence of H2O2 and visible light. The interesting phenomenon observed in these cases was that, being a cationic dye, MB was fully (100%) adsorbed on the Co(II)–SMA surface throughout the entire reaction, whereas MO, being an anionic dye, was partially (∼30%) adsorbed on the Co(II)–SMA surface.50,67,68 The true solid-phase catalyzed Fenton reaction of MB in the presence of Co(II)–SMA was thus reflected in the ‘zero-order’ kinetics, whereas MO degradation was observed to follow the ‘first order’ reaction rate. The advantage of the surfactant layer present on the alumina surface in the adsorption of dyes (MB or MO) is clearly visualized when Co–SMA is compared with Co–Al2O3. It was observed that while the MB adsorption was 100% on the Co–SMA surface, it was only <5% in the case of normal alumina surface.50 Thus, it is obvious that the surfactant bilayer is responsible not only for holding the catalyst [in this case Co(II)] but also the pollutant (dye) in a larger quantity and stronger way with closer contact, which led to very efficient catalytic performance (>97% degradation of MB at 20 mg L−1).
The photo-Fenton degradation of MB on the Co(II)–SMA surface was proposed to follow the Langmuir–Hinshelwood mechanism, where it was assumed that the adsolubilized MB reacted with the adsorbed radical formed in the presence of Co(II) and H2O2 on the Co(II)–SMA surface. When H2O2 was added to the reaction mixture, it reacted with Co(II) and produced the OH˙ radical and Co(III). This process is a slow process. As soon as the OH˙ radical was produced, it oxidized the SMA-adsorbed MB in a fast way and finally, the degraded products were desorbed from the solid surface. Because the slower step (or steps) did not involve MB, so the overall reaction followed ‘zero-order’ with respect to MB. The detailed mechanism has been described in our earlier publication.50
On the contrary, the MO degradation following the photo-Fenton process in the presence of the same catalyst Co(II)–SMA followed the L–H mechanism but did not follow the ‘zero-order’ kinetics, rather it followed the ‘first-order’ kinetics. The reason behind this is that MO was partially adsorbed on the Co(II)–SMA surface. Thus, during the degradation of the adsolubilized MB dye, OH˙ formation is the slowest step (i.e., rate determining step), whereas, in the case of the MO dye, the reaction between OH˙ and the MO molecule is the slowest step (i.e., rate determining step).50,67,68 Thus, adsolubilization (or solid substrate immobilization) might have a significant effect on the rate of the reaction.
Nanoparticle-catalyzed 4-nitrophenol (4-NP) reduction in excess aqueous borohydride to form 4-aminophenol (4-AP) is a well-recognized reaction. The presence of nanoparticles (gold, silver, copper, etc.) both in homogeneous and heterogeneous conditions can enhance the rate of 4-NP reduction to a large extent. Thus, a thermodynamically feasible but kinetically restricted reaction becomes viable in an experimental time scale. Micelles anchored on the solid substrate can have a synergistic effect on such a reaction. Commercially available column silica (pHzpc = 1.7), under appropriate conditions, may adsorb cationic surfactants (CSs), for e.g., cetyltrimethylammonium bromide, to form either a monolayer or a bilayer. The monolayer (called hemimicelle) or bilayer (called admicelle) are formed when the concentration of CS is below or above the CMC, respectively. This admicellar layer is capable of adsolubilizing the hydrophobic organic molecules inside its hydrophobic core; also, its cationic head group can easily attract anionic molecules very strongly.69,70 Similarly, this surfactant-modified silica (SMS) can adsolubilize negatively charged AuCl4− easily. It was observed that while normal silica can adsorb AuCl4− at a capacity of 10.36 mg g−1, SMS can hold (or adsolubilize) the same AuCl4− at a capacity of 23.61 mg g−1.53 This AuCl4− can be easily reduced to Au(0), which can be used as a catalyst for 4-NP reduction. Thus, the micellar layer on SMS can hold an increased amount of catalyst, which increases the efficiency of the reaction, as reported earlier.53
Recently, commercial ion exchangers have proved to be versatile platforms for metal and metal oxide nanoparticle synthesis.72 Strongly acidic ion exchangers have been exploited to produce nanoparticles from the reduction reaction. Simple solutions of CuSO4, AgNO3, NiSO4, FeCl3, etc., were used for the easy immobilization of metal ions onto strongly acidic cation exchangers. Sarkar et al. utilized Seralite-SRC 120 cation exchange resin for the trapping of Ni(II).73 As a precursor, NiCl2 was taken and H+ of the resin material was allowed to exchange with the Ni(II) solution. The adsorption process was visible with the transformation of the color of the resin material from pale yellow to Kelly green. Later on, the loaded Ni(II) was reduced by the ice-cold aqueous solution of NaBH4, which changes Ni(II) to Ni(0). The scheme has been shown in Fig. 4. Sinha et al. used the glass surface and the resin surface for the formation of various shapes of nano TiO2.74 Using a strongly acidic ion exchange resin, TiO2 was deposited on the resin surface. Then, the pure form of TiO2 was produced from commercially available TiO2 in steps by eluting the impurities from the resin column. Thus, the process offers an easy means of purification of commercial TiO2. Thus, dangerous chemicals such as TiCl4 and costly materials such as titanium isopropoxide or titanium(IV) isobutoxide can be avoided to obtain the TiO2 nanoparticle catalyst. Wang et al. synthesized resin-supported organoselenium catalyst75 and elaborated a very attractive catalyst synthesis procedure for obtaining hexavalent selenium on the solid support. Hexavalent Se on the polymer has been found to be a very effective catalyst, which accelerates the reaction between cyclohexanol and H2O2 to form the industrially important intermediate compound trans-1,2-cyclohexane diol. Jansson et al. prepared resin-supported palladium catalyst for the hydrogenation reaction.76 The catalyst has been found to be very successful in the hydrogenation of double and triple bond and the hydrogenolysis of the benzyl protecting group. Ford and Premecz reported the preparation of resin-immobilized rhodium-phosphine hydroformylation catalysts to obtain 1-octene with minimal leaching of rhodium.77 Huirong and Yingde reported the preparation of salicylic acid resin-supported FeCl3 Lewis acid catalyst and utilized the same for organic synthesis.78 The catalyst was judiciously utilized for the esterification of alcohol and carboxylic acid. The catalyst was reported to be non-corrosive in nature and could be used repeatedly without significantly losing its efficiency. Moreover, the catalyst was easily separable from the reaction mixture.
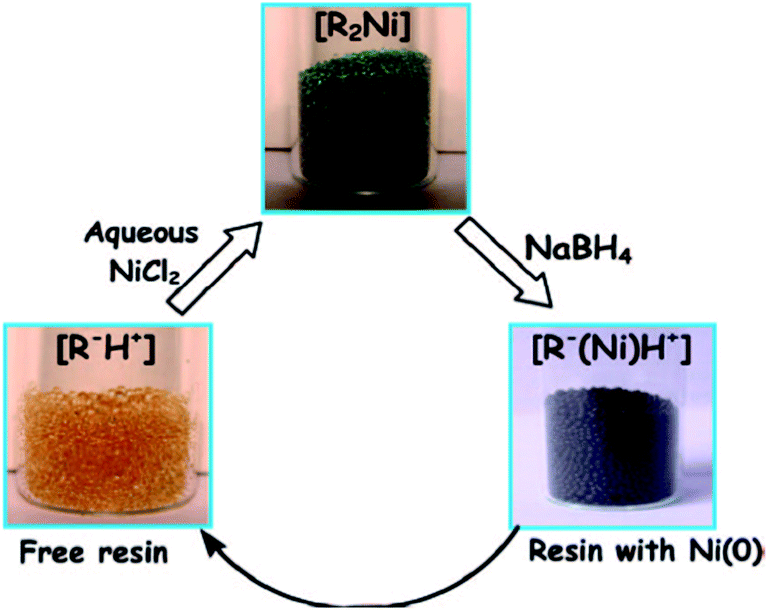 | ||
| Fig. 4 Schematic representation of the synthesis of Ni nanoparticles coated resin beads and their isolation using a laboratory magnet. Reprinted with permission from the American Chemical Society (ref. 73). | ||
Although polymer resins have emerged as one of the most suitable templates for supporting catalyst metal oxide nanoparticles, however, instability has also been reported in some cases. Instability may arise due to high temperature and chemical reactions with substrates. In some cases, regeneration is also a problem.79 Vaerenbergh et al. studied the leaching of Pd from strongly acidic and basic resins. It was observed that leaching occurred during the initial cycles. However, after consecutive cycles, redeposition was observed.80
There are several reports in the literature where biomaterials have also been chosen as suitable supports for nesting the catalyst particles. Hees et al., in one of their recent reports, described nanocellulose-supported iron catalysts for the in situ formation of polyethylene nanocomposites.88 Previously Saha et al.89 and Kongarapu et al.90 reported the catalytic reduction of 4-NP employing borohydride medium and the catalysts were Ag loaded on calcium alginate beads in one case and Ni-loaded surfactant (SDS)-modified chitosan beads in the other.
Nanoparticles may serve as a recyclable scaffold after functionalization. Thus, functionalized nanomaterials can be easily separated using centrifugation, precipitation–flocculation, nanofiltration, or magnetic decantation. Many reports have concluded that cooperative catalysis and interphase effect have made the grafted catalyst more active in comparison to their homogeneous counterparts.91 Various promising examples of particle-supported nanocatalysts have been reported, for e.g., the monolayer-protected gold clusters, gold nanoparticles with mixed monolayers, ferrite nanoparticles, dopamine-capped ferrite nanoparticles, silica-coated ferrite nanoparticles, and carbon-coated metal nanoparticles. The supported Pd catalyst has been extensively used as a semi heterogeneous catalyst for cross-coupling reactions. Hu et al. synthesized Pd and Fe3O4 nanoparticles supported on sulfonated graphene and used the same as semi-heterogeneous catalysts for the Suzuki Miyaura reaction.92
It has already been mentioned that catalysts cannot start a reaction but they can alter the reaction path just by lowering the activation energy of the reaction (uncatalyzed). The goal of the use of a catalyst is to mostly increase the yield in a cost-effective way; thus, catalysts are used to undertake environmental clean-up measures.
2.2. Procedures of synthesis
Mainly, there are two principles for synthesizing catalysts over suitable supports.Horvath et al. synthesized an Au nanoparticle-based catalyst by redox co-precipitation reaction between HAuCl4 and Fe(NO3)3·9H2O and applied the same for CO oxidation.29 Miyahara et al. utilized this technique for the preparation of Cu catalyst supported on alumina for the oxidation of benzene to phenol.95 Cu catalyst has been also prepared by the co-precipitation technique by Li et al. over ceria support for yielding methanol from CO2.96 Hydrogen has also been catalytically produced by co-precipitated Ni catalysts by Vizcaino et al.97 Methane dry reforming has been conducted by Ni catalysts deposited by similar techniques on ceria-based mixed oxide. It has been observed that the leaching of metal ions is relatively less as the entrapped metal ions go into the sediment due to co-precipitation. Lower amount of Ni leaching has been observed by Miyahara et al. for phenol production by the liquid-phase oxidation of benzene. It has been described that the co-precipitated catalysts perform better than the conventional impregnation method.95 Moreover, Haruta, in his ground breaking report on Au nanocatalytic reaction, transpired the oxidation of CO. The results revealed that the co-precipitation method is much more helpful for making use of the catalytic activity of Au than that of the traditional impregnation method98 for catalyst preparation, as discussed in the preceding section. The catalyst is successful even at a temperature of −70 °C.
Fraga et al. synthesized various carbon-supported Pt catalysts using the impregnation method.101 It has been highlighted by Fraga et al. that the presence of various functional groups on the carbon surface is responsible for the strong adsorption of Pt. Pt loading depends on the pHzpc of various types of carbon materials.
In the recent times, catalyst preparation by means of pyrolysis of several metal organic frameworks (MOFs) has gained importance. MOFs are highly porous and robust materials. Different metals after being pre-impregnated in the MOF skeleton are subjected to pyrolysis in open air. The already impregnated metals are converted to their corresponding metal oxides and the whole MOF-supported catalyst system obtains a new physico-chemical characteristic. Cr2O3-supported Pt catalyst and CeO2 catalyst prepared by the pyrolysis of CeO-MOF have been reported, which provided successful toluene oxidation pathways.105,106 It is true that pure CeO2 has good oxygen storage capacity and plenty of acid sites on the surface, which is conducive for exhibiting the catalytic activity. However, due to the self-structured character and some other steric constraints, it often fails to perform as an excellent catalyst. However, the pyrolysis of CeO-MOF, which is the as-prepared catalyst, showed superior catalytic activity for toluene oxidation. It was found that CeO-MOF/350 showed the best catalytic activity.
Since the last few decades, a new trend has emerged in the field of catalyst synthesis by means of utilization of plasma. Plasma is formed by the ionization of neutral gases and is the fourth state of matter. Cold plasma is generally denoted by high electron and low gas temperature and it has been reported to be one of the fastest, facile, and most environment-friendly ways of catalyst preparation.107 Catalysts prepared by cold plasma have been observed to contain NPs with small size and high distribution ratio and hence show higher catalytic activity. Liu et al. prepared novel Ni/SiO2 catalyst for methane reformation from plasma assistance.108 The traditional long-time calcination and thermal reduction was replaced by a cold atmospheric plasma jet. The new catalyst prepared by this way exhibited an improvement in the low-temperature CO2 reforming reaction compared to the conventional catalysts synthesized without the aid of plasma.
3. Metal particles commonly used as supported catalysts
Noble metals and their innumerable compounds are widely used in chemistry for catalysis. However, their cost and leaching is a matter of great environmental concern.57,109 In general, transition metals (TMs) commonly satisfy their secondary valence with available ligands unlike non-transition metals. This is one of the reasons why most TMs form complex compounds on accepting electron density from ligands. SMs such as graphene sheets, carbon nanotubes, and metal oxide assemblies make room to accommodate the electron density available from the surface atoms (better to say surface free/loosely bound electrons) of the nanoparticles. In this process, the SM also becomes a stable platform for the accommodation of substrates. Then, SM may be involved in catalysis. The following section highlights some elements that have been mostly used together with some suitable SMs. Then, the material is called as a metal-supported catalyst, where the metal and SM co-jointly serve the catalytic reactions as supported catalysts.3.1. Gold
Generally, gold is considered to be a noble and inert metal, as already described in the previous sections. Gold, with its ground state electronic configuration of 5d9 6s2, in general, cannot not offer a vacant position to accommodate substrates. But CO2 is formed from CO while the small gold particle surface is exploited as a catalyst. However, CO covers the gold surface while gold is considered to be noble [E0 (V) + 1.50 NHE]; thus, the nobility of gold surely comes into consideration as we find it in the earth's crust as a metal. Any metal farther from gold in the periodic table is harder and the hardness of the metal increases with the distance. Furthermore, gold with its highest electron affinity (leaving aside halogens) can form the Au− ion. Again, gold is the softest acid among all the metals.110 Therefore, it was believe that gold cannot be a suitable candidate to catalyze a reaction. A breakthrough in the 20th century was announced when Haruta discovered the catalytic activity of ‘supported’ gold nanoparticles in 1987 for the oxidation of CO to CO2 by O2.111 Haruta's report gave us new insights into catalysis, as already described in the previous sections. It has also been mentioned that the rigorous efforts of various scientists during the last few decades have helped in finding that nanoclusters of gold can be a highly efficient catalyst for various activities. The findings of Hutchings and Haruta revealed for the first time the catalytic activity of gold nanoparticles.111 Due to Hutchings, it has been possible nowadays to replace toxic mercuric chloride catalyst by supported Au catalyst for industrial purposes.98,112 It has been also noted that interestingly, the Au nanoparticles retain their metallic property even at the nanoscale. As per Ishida et al., as the size of the Au nanoparticles decreases, the continuous valence band of the bulk metal starts to get separated discretely.98 Sau et al. conducted an extensive study on the size dependency of gold nanoclusters for the reduction of eosin in the presence of NaBH4.113 However, Pal and Pal concluded that the exposed facet of the nanoparticles has much more influence in reaction in comparison to the size and shape114 of the catalyst particles, presumably due to the as-produced active centers. Nanoclusters of gold have often found to be easily oxidized and the aggregation of the clusters causes the reduction in its catalytic activity.115 The most popular catalytic reactions catalyzed by AuNPs are CO oxidation, propylene epoxidation, and alcohol oxidation.98 The schematics of some reactions catalyzed by AuNPs have been shown in Fig. 5. It has been noticed that for CO oxidation, Au NPs supported on acidic or inert support show poor catalytic activity in comparison to that of metal oxide supports.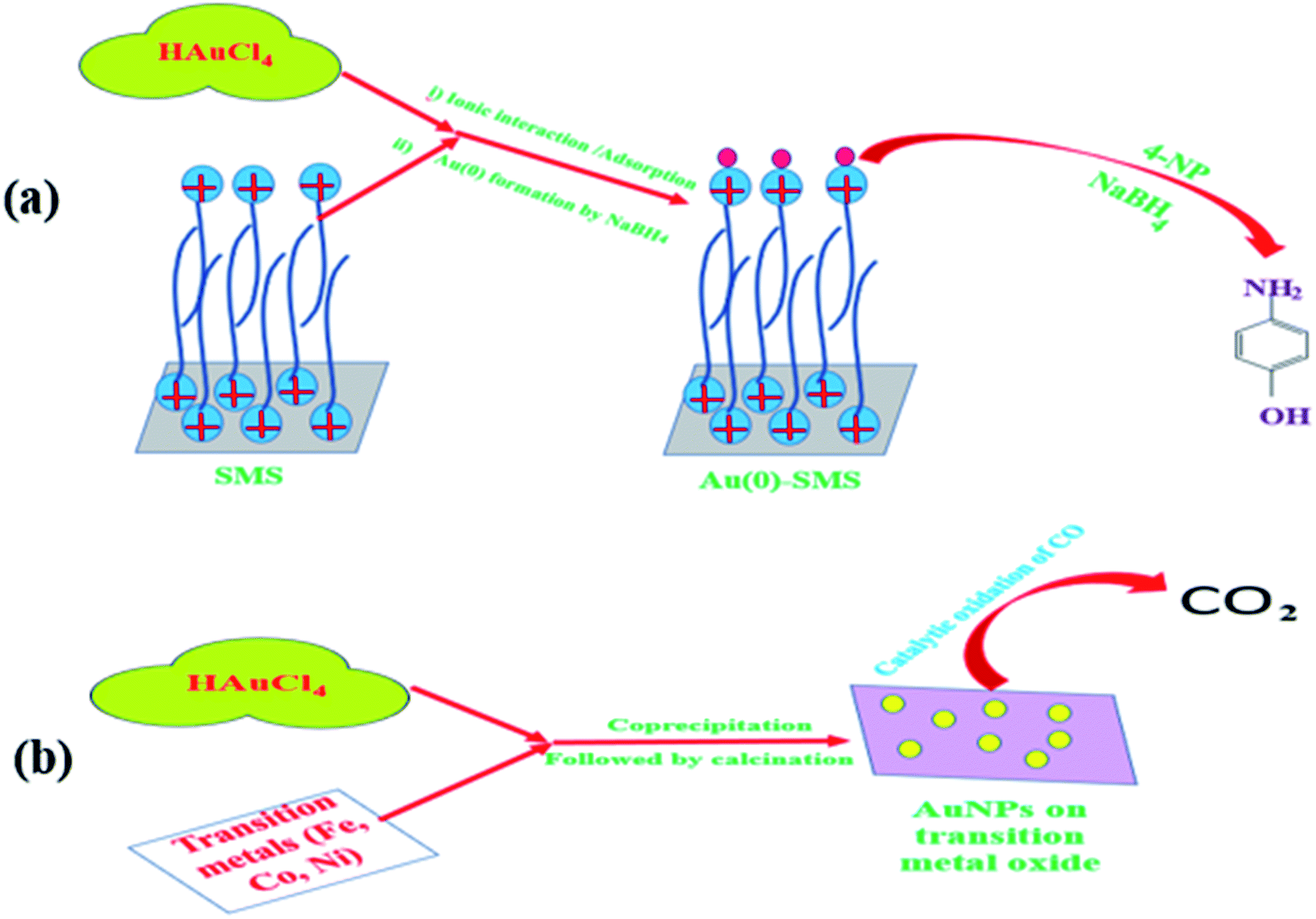 | ||
| Fig. 5 Schematic representation of the catalytic behaviour of supported Au NPs: (a) reduction of 4-NP to 4-AP by Au(0) supported on surfactant-modified silica (SMS) in the presence of NaBH4 and (b) the catalytic oxidation of CO to CO2 by Au NPs supported on transition metal oxides. | ||
Many attempts have been made to rationalize the effect of particle size towards several catalytic reactions, especially, Au-catalysed CO oxidation; however, the role of altered electronic factors of supported Au nanoparticles on semiconductor surfaces has been largely ignored. According to the ‘electron theory of catalysis’ or the ‘rigid band model’, the distribution and concentration of electrons on the catalyst surface/interface govern the catalytic processes.116 The elementary steps of a catalytic reaction are determined by the distribution of the electrons over the quantum states of the bands, i.e., the position of the Fermi level at the surface of the solid particles. When the metallic and semiconductor components are in electrical contact, there occurs a transfer of free electrons due to a difference in their work functions.117 The formation of metal–semiconductor heterojunctions can drastically modify the spatial distribution and the average free energy of the electrons near the interface. When the metal (ϕm) possesses a higher work function than the n-type (electrons as majority charge carriers) semiconductor (ϕs), as suggested by Schottky,118 the electrons diffuse from the semiconductor into the metal until the attainment of equilibrium of the corresponding Fermi levels. This results in many free electrons on the new Fermi level of the metal, which can tunnel into the vacant conduction band of the semiconductor, leading to a higher valence state of the metallic components.119 As a consequence, the metal becomes negatively charged and the semiconductor becomes positively charged, and a Helmholtz double layer is established at/near the interface due to electrostatic induction. Due to the low concentration of free charge carriers in the semiconductor, the electric field between the metal and the semiconductor interfaces cannot be effectively screened by the semiconductor. This causes the free charge carrier concentration to be depleted, which extends to a particular depth in the semiconductor, coined as the space charge region. Compared with the bulk and in essence, the conduction and valence bands are bent upwards and the distance of the Fermi level from the conduction band is increased. On the other hand, when ϕm < ϕs, the electrons are accumulated in the space charge region due to electron transfer from the metal to the semiconductor, and this region is called the accumulation layer. In general, when the Fermi level of the metal is below that of the semiconductor, the charge will flow to the metal, causing the semiconductor Fermi level to decrease and vice versa. However, when ϕm > ϕs, within the boundary of the depletion layer that is characterized by an excess positive charge, donor-type catalytic reactions, i.e., reactions where the reactant molecules donate electrons to the catalysts to form the activated state, are favored, as investigated by Haruta for catalytic CO oxidation in the presence of gold nanoparticles supported on a semiconductor matrix.120 Upon rectification of the electronic structure at the metal–semiconductor contacts, gold or silver, possessing a higher valence state, accepts the electron density from the CO molecules and the binding between Au nanoparticles and CO comes into picture to govern the activity of the catalytic reactions.
Supported Au catalysts can be classified into reducible MOx, non-reducible MOx, non-oxides, and inorganic–organic hydrides.98 On the other hand, Au nanoclusters supported on alkaline earth metal hydroxide supports are also highly active in the catalytic oxidation of CO.98 In terms of selectivity, supported Au catalysts are more promising and environment-friendly in comparison to the well-known Pt and Pd catalysts. The oxidation of alcohols to aldehydes, which is an intensive research area, has been largely explored with Au catalysts. As per the reports with respect to the yield product achievement, Au catalysts have proved to be more selective than traditional catalysts. Corma and Serna as well as Prati and Rossi reported that for the catalytic oxidation of alcohol, the TOF increased rapidly with a decrease in the size.121,122 Chen et al. also reported that oxidation by Au NPS can also be carried out in the absence of O2.123 However, the reaction has been found to be more prompt in the presence of oxygen. The support is very influential in the determination of the catalytic activity of gold NPs. Milone et al. showed that various kinds of Fe supports for Au catalysts yield different results.124
3.2. Copper
Owing to its versatility, copper has been used since a long time by human beings as a building material. It is also an essential element. Due to the increase in the concentration of greenhouse gases in the atmosphere, there is a race amongst the scientific community to find solutions so as to curb the concentration of these gases and convert them into valuable end products. CO2 storage is an option but its conversion to methanol is a much better alternative. The hydrogenation of CO or CO2 over a suitable metal catalyst helps in methanol synthesis. Methanol synthesis by supported copper catalysts either in the mixed or single form is a convenient technology. Burch et al. specified that the support plays a vital role in determining the specific activity (rate of production of methanol per unit area of the copper catalyst).125 Cu2O is the most studied semiconductor and is superior to ZnO for methanol synthesis. Apart from methanol synthesis, supported Cu catalyst has been used for other purposes as well. Shi et al. reported the homocoupling reaction of two alkenes for the synthesis of alkyne by Cu-supported polyacrylamide.126 Supported Cu catalyst has been found to be helpful in various ways, such as in the conversion of biomass to important end products. Levulinic acid (LA) is one of the most common lignocellulose-derived compounds, and γ-valerolactone (GVL) and 1,4-pentanediol are some of the intermediate value-added products.127 For example, LA conversion to GVL can be successfully carried out by Cu catalysts. Hengne and Rhode reported that Al2O3-supported Cu catalysts can convert LA to GVL with 91–100% selectivity in water medium and Cu/ZrO2 in alcohol medium at 473 K.128 Yuan et al. reported 100% efficiency concerning the product yield.129 Many researchers have reported that copper catalysts deposited over a surface have been found to be the inactive Cu(II) form in most of the cases. However, Sakata et al. reported that the coordination compound of copper phosphate is responsible for the reversible reduction–oxidation reaction, which in turn is responsible for generating catalytically active sites for the direct oxidation of benzene.130The reduction of nitrous and nitric oxide to form nitrogen gas is catalyzed by copper catalysts. From the literature survey, it is evident that in most of the cases, the reduction reaction is studied with NO over the Cu catalyst. Zhu et al. conducted the reduction of both N2O and NO by carbon material-supported Cu catalyst.131 Carniti et al. also prepared several Cu catalysts over various supports such as silica and alumina, and carried out the selective reduction of NO to produce N2 gas.132 Singoredjo et al. and Zhu et al. also reported the reduction of NO with NH3 gas with Cu catalysts supported on carbon-based materials.133,134 Thus, supported or unsupported copper-based catalysts find selective use in N2 gas production.
Various supported copper catalysts have been reported as perfect candidates for preferential CO oxidation (PROX). Yang et al. performed PROX. reaction over Ce1−xCuxO2−δ supported Cu catalysts.135 The supported CuO/Ce1−xCuxO2−δ catalyst showed excellent performance for CO PROX and was more efficient than the CuO/CeO2 catalyst. Kydd et al. reported a Cu catalyst supported over ceria support prepared by flame spray pyrolysis, which exhibited excellent catalytic activity for PROX reaction, particularly at lower temperatures.136
Water–gas shift (WGS) reaction is one of the most important reactions from the industrial point of view in terms of hydrogen production. CO and water react in the presence of a catalyst and get converted to CO2 and H2 gas. The WGS reaction deals with only 4 molecules but the reaction mechanism is quite complex. Mainly, there are two proposed mechanisms, namely, Associative and Redox.137 In the first one, a formate species is formed in the intermediate step, which gets decomposed into CO2 and H2. The supported copper catalyst is one of the notable catalysts in the WGS reaction field. Since 1966, CuO/ZnO/Al2O3 has been widely used for industrial purposes for methanol synthesis and WGS reaction. Wang et al. reported a Cu–CeO2 catalyst for WGS reaction.138 CeO2 is very useful in the WGS reaction due to its peculiar redox properties and oxygen storage capacity. The reaction did not occur with nano ceria under normal conditions and showed very little H2 conversion in the presence of only the CuO catalyst. However, maximum yield was obtained in the presence of 5% CuOx/CeO2 catalyst, which indicates that strong interaction between copper and ceria is required for the reaction to occur. Spencer explored the role of ZnO in the CuO/ZnO catalyst for the WGS reaction.139 It was deduced that epitaxial or other electronic bonding between the copper crystallite and supporting zinc oxide was the key factor behind the prevention of sintering of the copper catalyst.
3.3. Iron
Iron is the 4th most abundant element found on the earth's crust. Iron has been utilized as an effective catalyst since the first time in 1913 by the German scientist Haber for the preparation of ammonia (NH3) on an industrial scale. In this process, molybdenum (Mo) is utilized as the promoter. It has been already established that Mo model complexes can mediate the stoichiometric conversion of N2 to NH3.140 Iron is also used as a catalyst in the reaction between peroxodisulfate and iodide ions for the formation of sulfate. Apart from sulfate formation and ammonia production in the industrial sector, iron has also proved to be an efficient catalyst for other industries such as plastics and cosmetics. The conversion of hydrocarbon derivatives by Fischer–Tropsch synthesis is one of the most important reactions for the manufacture of various products. Galvis et al. prepared α-alumina and carbon nanofiber-supported iron catalysts for Fischer–Tropsch reactions.141 Jung et al. prepared a highly dispersed iron catalyst over carbon support for the hydrogenation of CO.142Apart from the above-mentioned reactions, iron has been extensively used as a catalyst for environmental purposes. The mineralization of recalcitrant pollutants by the advanced oxidation process is a good technology for pollution abatement purposes. Iron(II) was used by Fenton as a catalyst for advanced oxidation purposes in the oxidation of organic compounds, wherein it was applied as a homogeneous catalyst for the first time. However, later on, various supports have been explored to hold the iron catalyst for carrying out the Fenton reaction. A few such examples include the degradation of rhodamine B by rice hull-based silica-supported iron catalyst,143 photo-Fenton oxidation of phenols by silica-supported iron catalysts,144 and decolorization of Acid Red 1 by kaolin-supported iron catalyst.145
3.4. Palladium
Palladium has remarkable power to occlude hydrogen. Occluded hydrogen is more reactive than normal hydrogen, which superbly governs the hydrogenation reaction. Palladium has been well known for ages for its catalytic activity. It is mostly famous for its use in cross-coupling reactions. Pd-catalyzed C–C bond formation reaction is considered to be one of the most common versatile tools in the field of organic synthesis.146 Various important cross-coupling reactions such as Heck reaction, Meerwein arylation, Negishi cross-coupling, Buchwald–Hartwig reaction, and Sonogashira cross-coupling have been carried out with Pd catalysts. The schematics of some of the cross-coupling reactions are presented in Fig. 6. Usually, Pd(II) is reduced to Pd(0) in the 1st step of the catalytic reaction by a suitable reducing agent and then Pd(0) takes part in the oxidative transformation reaction. In the next step, Pd(0) reverts back to Pd(II) to restart the reaction afresh. However, the homogeneous system of the Pd catalyst is not an acceptable choice owing to its high cost, difficulty in recovery, and toxicity associated with environmental factors. Therefore, supported Pd catalysts have been used by researchers for various cross-coupling reactions.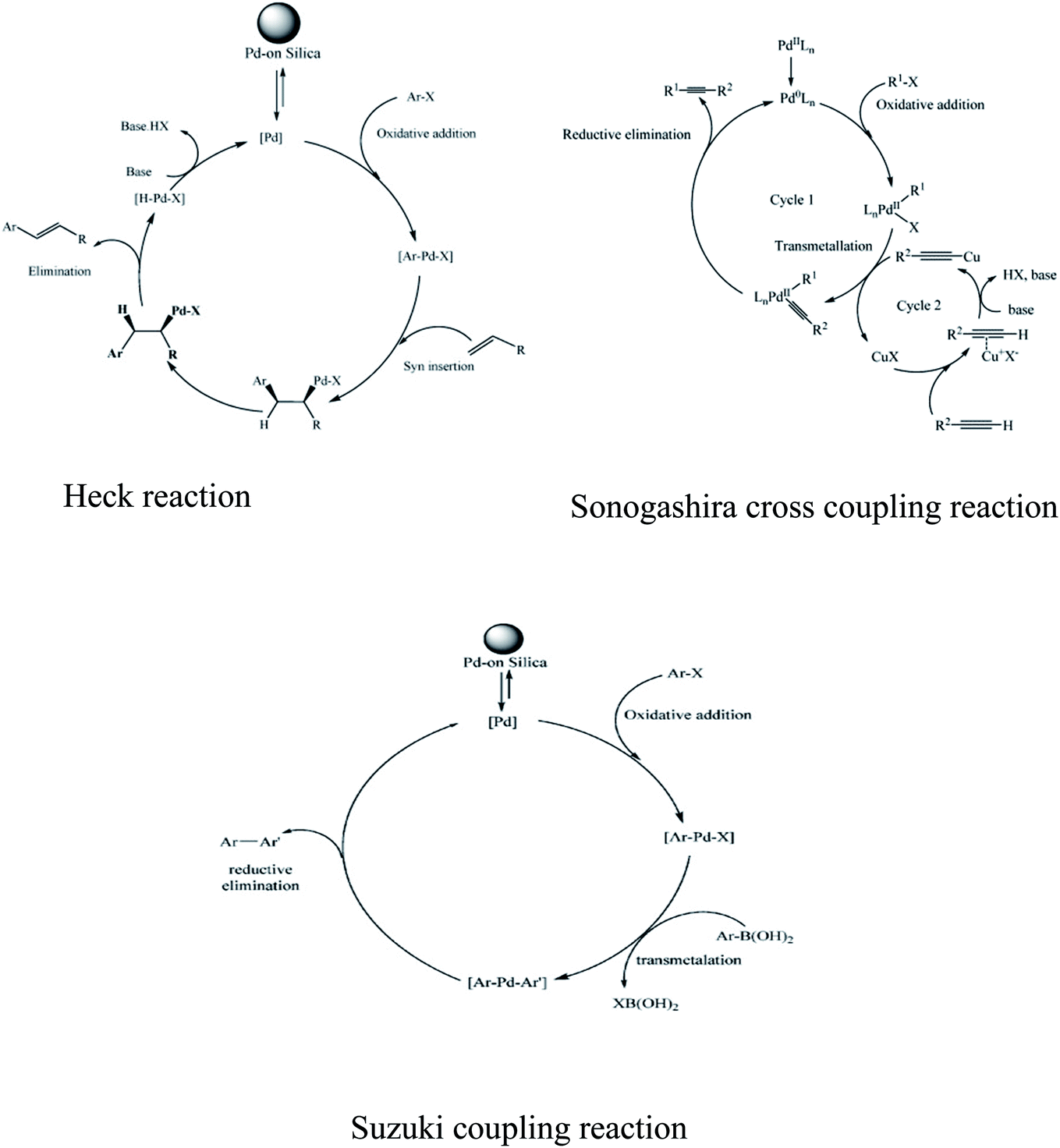 | ||
| Fig. 6 Schematic representation of Heck reaction, Sonogashira cross coupling, and Suzuki coupling reaction. Reprinted with permission from ref. 52. | ||
Li et al. showed that the Pd catalyst supported on a graphene sheet performed excellently as a catalyst towards CO oxidation.147 Esrafili et al. also concurred on the same opinion regarding the Pd catalyst.148
3.5. Platinum
Platinum is a promising oxidation catalyst as it collects the oxygen atom and is able to transfer O to a substrate. Pt is non-toxic but platinum complexes have been verified to be useful in chemotherapy. Platinum is also a well-known element due to its catalytic activity. It is worth mentioning that Pt metal, though rare, is a wonderful catalyst for carrying out innumerable reactions. Platinized asbestos, Pt with MgSO4, and silica gel support find useful applications in H2SO4 producing industries by the infamous Contact process. However, as mentioned earlier, V2O5 takes the prime lead over Pt in industries for H2SO4 production due to the obvious poisoning of rare Pt catalysts and above all, the cost of Pt. In particular, Pt catalysts are suitable for reduction processes because Pt leaches the least in a reducing environment.32 Frelink et al.43 studied the effect of particle size of the carbon-supported Pt catalyst for methanol oxidation. Pt catalysts are well known for their catalytic activity in low-temperature fuel cells.149 Various Pt supports such as TiO2, Al2O3, and SiO2 have also been reported for the successful aqueous phase reformation of ethylene glycol. Hence, it is an attractive way for hydrogen production.150 Supported Pt catalysts have also been reported in the CO2/CH4 reforming reaction.151 Pt catalyst has also been extensively used as an electrocatalyst for the proton exchange membrane fuel cell (PEMFC).152 PEMFC is one of the most important power technologies in the current day. One of the most common challenges associated with PEMFC is the commercialization of a suitable electrocatalyst. Platinum is one of the most commonly used electrocatalysts in PEMFC. However, a suitable support is one of the major criteria behind the success of the supported Pt catalyst. Miller et al. showed that with an acidic support, the TOF of propane hydrogenolysis becomes the highest.153Undoubtedly, platinum is a wonderful catalyst but it has a high tendency of getting poisoned by CO, as mentioned above. In the present day of energy crisis, various kinds of Pt electrodes are often used as suitable options. However, poisoning is a great threat to the costly Pt catalyst. Scientists have tried various options to curb CO poisoning of Pt. One method involves bringing highly oxophilic materials together with Pt so that the selective adsorption of the poisoning agent CO takes place on Ru, Ni, etc. Another way is through the prevention CO formation by alloying Pt metal, which may be called as the ensemble effect of metal alloying. In both the catalysts, Pt remains available/uncovered and catalyzes the reactions. In this respect, the SM could provide an effective way for preventing CO poisoning of the Pt catalyst. However, further studies are still required to get a direct conclusion.
3.6. Nickel
Paul Sabatier in 1912 received the Nobel Prize for hydrogenation reaction with finely divided nickel, whereby the progress of organic chemistry as well as nanochemistry has advanced in recent years. Hydrogen production is considered to be highly valuable in the present-day world in terms of clean fuel production. Ni catalyst supported on silica has been reported to be very active for the catalytic cracking of methane to produce hydrogen.154 Pd has been well known for its Suzuki coupling but Ni has slowly replaced it due to its economic value and abundance. Not only Suzuki coupling but all the other cross-coupling reactions mentioned in the previous section have also been found to be possible with Ni catalysts. From the environmental viewpoint, dry reforming of methane and CO2 is a desirable and important reaction. In this reaction, CO2 and CH4, which are two undesirable greenhouse gases, react with each other over a metal catalyst to produce CO and H2. A schematic of the dry reforming reaction has been presented in Fig. 7. One of the major aspects of suitable catalyst selection in this regard is the deactivation of the catalyst due to the carbon deposited.155 The reaction was usually conducted in the past in the presence of conventional noble metal catalysts such as Pt and Pd, where the carbon deposit cannot easily hinder the reaction. But due to the excessive cost of Pd, Pt, or Rh, transition metal elements were tried out for the purpose. Among all the transition metals, nickel is the best choice. However, the sintering of Ni due to coke deposition is again a major drawback of the Ni-bearing catalyst system.156 However, a suitable support system has been found to overcome this difficulty. For e.g., Rezaei et al. reported the use of zirconium oxide modified by P123 to support the Ni catalyst for the dry reforming reaction.157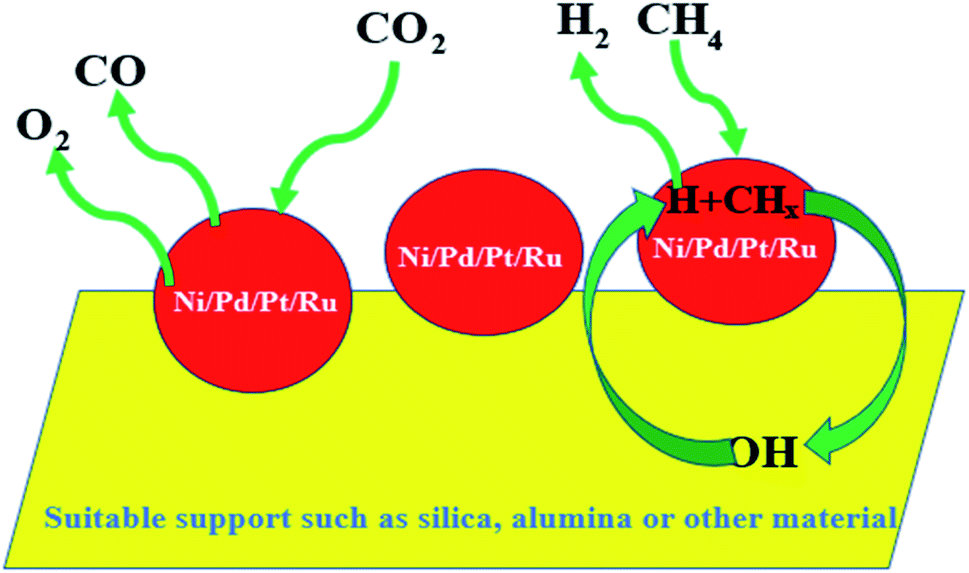 | ||
| Fig. 7 Schematic representation of dry reforming reaction. | ||
The use of Ni as a suitable catalyst was made by the American engineer Murray Raney, after which the catalyst has been named as RANEY®-Ni catalyst. The use of RANEY®-Ni catalyst is related to the hydrogenation reaction. One of the most prominent examples of the use of this catalyst is in the reduction of benzene to cyclohexane, which may be utilized for the production of adipic acid, which is an industrially important compound. However, in the synthesis of RANEY® nickel catalyst, there is a huge generation of undesirable pollution and corrosion. This is why often researchers in modern times suggest supported Ni catalysts for various hydrogenation and dehydrogenation reactions. Wang et al. reported the use of carbon nanofiber-supported Ni catalysts for the hydrogenation of chloronitrobenzene.158 Yingxin et al. also reported silica-supported Ni catalysts as the best among other synthesized supported Ni catalysts for the hydrogenation of m-dinitrobenzene to m-phenylenediamine.159 Ni has been found to be more stable than the Co catalyst in terms of CH4 decomposition.160
3.7. Supported bimetallic catalysts
The main advantages of using bimetallic particles in catalysis are: (i) cost effective measures and (ii) exposure of active metal centres due to alloying and enhancing the field effect in case of core–shell architecture. Thus, the introduction of bimetallic nanoparticles has become one of the most practical techniques for the development of new catalysts. It has been seen that the catalytic properties of a bimetallic mixture are often different from the constituent metals due to the modification of the surface geometries and the electronic configurations.161,162 Noble metal-based catalysts have been well known due to their high reactivity in hydrogen release from ammonia borane and hydrous hydrazine. However, the high costs associated with noble metals have limited their applications. In this regard, binary metallic catalysts based on noble and non-noble metals have been extensively studied.163 Among various options available for hydrogen production, the use of bimetallic catalysts is promising since it offers a stable and optimal catalytic performance.164 Ni–Fe, Cu–Co,162 and Ni–Rh combinations have been found to be suitable for this reaction. Kugai et al. reported hydrogen production from ethanol via the CeO2-supported bimetallic catalyst of Ni–Rh.165Bimetallic nanoclusters of Au and Ag have also been reported by Ganguly et al., which have superior performance in comparison to the individual Ag and Au clusters.115 The increase in the activity of the resultant species than the sum of their effort is defined as the action of synergism. It is noteworthy to mention here that the synergism of Au and Ag is more than any other combination owing to the location of both the elements in the same group of the periodic table.166 In particular, the Au–Ag alloy has a lower surface-volume ratio in comparison to monometallic gold. It has also proved to be an excellent catalyst for CO oxidation at low temperatures. Here, the authors mentioned the important role of a suitable template and also stated that such templates are very few in number.
Due to alloying in bimetallic catalysts, a change is often observed in the d-band shift for the transition metals. Yen et al. reported that the d-band centre of the CuNi alloy is in between −2.67 eV and 1.29 eV of the individual species.167
3.8. Promoters and initiators
It has already been mentioned that catalysts cannot start a reaction but alter the reaction path just by lowering the activation energy of the reaction (uncatalyzed). The goal of the use of a catalyst is to mostly increase the yield in a cost-effective way. Again, some compounds along with the catalyst are sometimes used in the reaction chamber to improve the yield further. These are called ‘promoters’. A small amount of Mo in the Haber's process for NH3 synthesis and CrO3 in the water–gas reaction for CH3OH production are popular promoters. They are used as catalyst activators in the industries to obtain higher yield of the product. On the other hand, some compounds are similar to free radicals (atoms or groups with odd electrons) that can trigger catalytic reactions. These compounds are ‘initiators’, which are not true catalysts but an integral part of the end products of a catalytic reaction. They form a weak chemical bond that evolves the active species as if they are intermediates and perform well as co-reactants, which quicken the progress of a chemical reaction. Peroxides and azo compounds act as initiators for olefin polymerization.4. Characterization techniques
4.1. Fourier transform infrared spectroscopy (FTIR) spectroscopy
To find out the functional groups present in a supported catalyst, FTIR spectroscopy and Raman spectroscopy are very useful techniques. In FTIR spectroscopy, light rays are absorbed, while in Raman spectroscopy, light rays are scattered. Vibrational spectroscopies are often considered to be extremely useful for characterizing the surface molecular species of supported oxides, which possess a strong covalent character of the metal–oxygen bond.168,169 Buciuman et al. carried out the FTIR and Raman spectroscopic study on SiO2 and γ-Al2O3 supported manganese oxides.170 It was concluded that the FTIR spectra were not very useful because the strong bands of the support overlap with those of the manganese oxides. However, the FTIR spectra support the results obtained from the Raman spectroscopic analyses of different points on the surface of the catalyst, which reveals that the surface was inhomogeneous. Li et al. conducted NO oxidation by TiO2 template-supported Pt cluster catalyst.171 They studied the mechanism of the reaction by in situ FTIR spectroscopy.Bitter et al. carried out the IR spectroscopic analyses of various supported and unsupported Pt catalysts for CH4/CO2 dry reforming.151 From the FTIR spectra, it was clear that the CO and carbonate species were linearly bound on the support. However, no carbonate was found for the Pt/SiO2 catalyst, which was proved to be active for methane decomposition. It was also shown that the catalyst with carbonate formation was much more active than the others. The routine identification of functional groups (free) and the binding motifs of the functional groups (bound) with the substrates are easily identified from the vibrational signatures.
4.2. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM)
Highly powerful microscopes and spectrometers have enriched science, delivering information with almost atomic resolutions. Thus, nanoscience and nanotechnology have become the forerunner in the miniaturization of devices, leaving aside other innumerable advancements in science. Two major instruments, TEM and SEM, are now available to the scientists since 1931 and 1937, respectively, and they have proven to be essential for the advancement of solid-state science. TEM operates on the same principles as that of a normal optical microscope but provides atomic resolution (degree of sharpness of the image) for the compounds under investigation. Most importantly, instead of a light source, in the TEM instrument, an electron beam (i.e., particle beam) is necessary, which is transmitted through the sample. This is indeed the requirement for imaging nanometer-size particles, which are much smaller than the wavelength of visible light. Therefore, electron (particle with smaller de Broglie wavelength) beams are capable of imaging nanometer-sized test particles under observation. This is conceivable because of the closeness in the sizes of the interacting wavelengths of the particles, i.e., the wavelength of the electron and the wavelength of the test materials, which is understood from wave-particle duality.SEM is also a kind of electron microscope that reveals the surface topography and composition of a sample with a focused electron beam. It scans the surface of the test substance with an electron beam and a secondary electron detector detects the secondary electrons from the excited atoms (reflected electrons).
Moreover, it helps in getting a clear picture of the structure–reactivity relationships.172 Zhang and Amiridis found from the micrographs of the spent catalyst that a filamentous structure of carbon grew on the catalyst surface with a bright tip, which was identified as the nickel particle.154 This result has been explained as an occurrence due to hydrocarbon cracking on the surface. However, some reports by Kuijpers et al. identified that fast catalyst deactivation does not lead to the formation of a filament-like structure.173
It is pertinent to mention that from the SEM image (Fig. 8), we could conclude that rod-shaped manganese oxide was formed on the surface of surfactant-modified alumina.49 Rezaei et al. showed from the SEM analysis that coke formation decreases on the catalyst surface as a result of addition of cerium and lanthanum oxide.157
 | ||
| Fig. 8 SEM images showing the SMA-MnOx nanorods. Reprinted with permission from Elsevier (ref. 49). | ||
Moreover, the structure of the potassium-promoted catalyst was found to be different from that of the unpromoted catalyst. The TEM analysis of the same work showed that the zirconia calcined at 700 °C showed sintering and resulted in an irregular shape. From the TEM image, Wang et al. concluded that the Ni nanoparticles are well dispersed over the carbon nanofiber (CNF) support.158
Apart from the X-ray diffraction result, Yen et al. also showed from the TEM result that the prepared catalyst was ordered mesoporous in nature (shown in Fig. 9 and 10).167 An idea about the size of the nanoparticles can also be obtained from the TEM images (5–10 nm for Ni and CuNi nanoparticles, whereas about 13 nm for Cu nanoparticles). The images confirm the presence of mesoporous carbon particles with metal nanoparticles uniformly dispersed on them.
 | ||
| Fig. 9 (a) TEM and STEM-HAADF images and (b) the EDS elemental mapping of Cu0.5Ni0.5/MCM-48. Reprinted with permission from the American Chemical Society (ref. 167). | ||
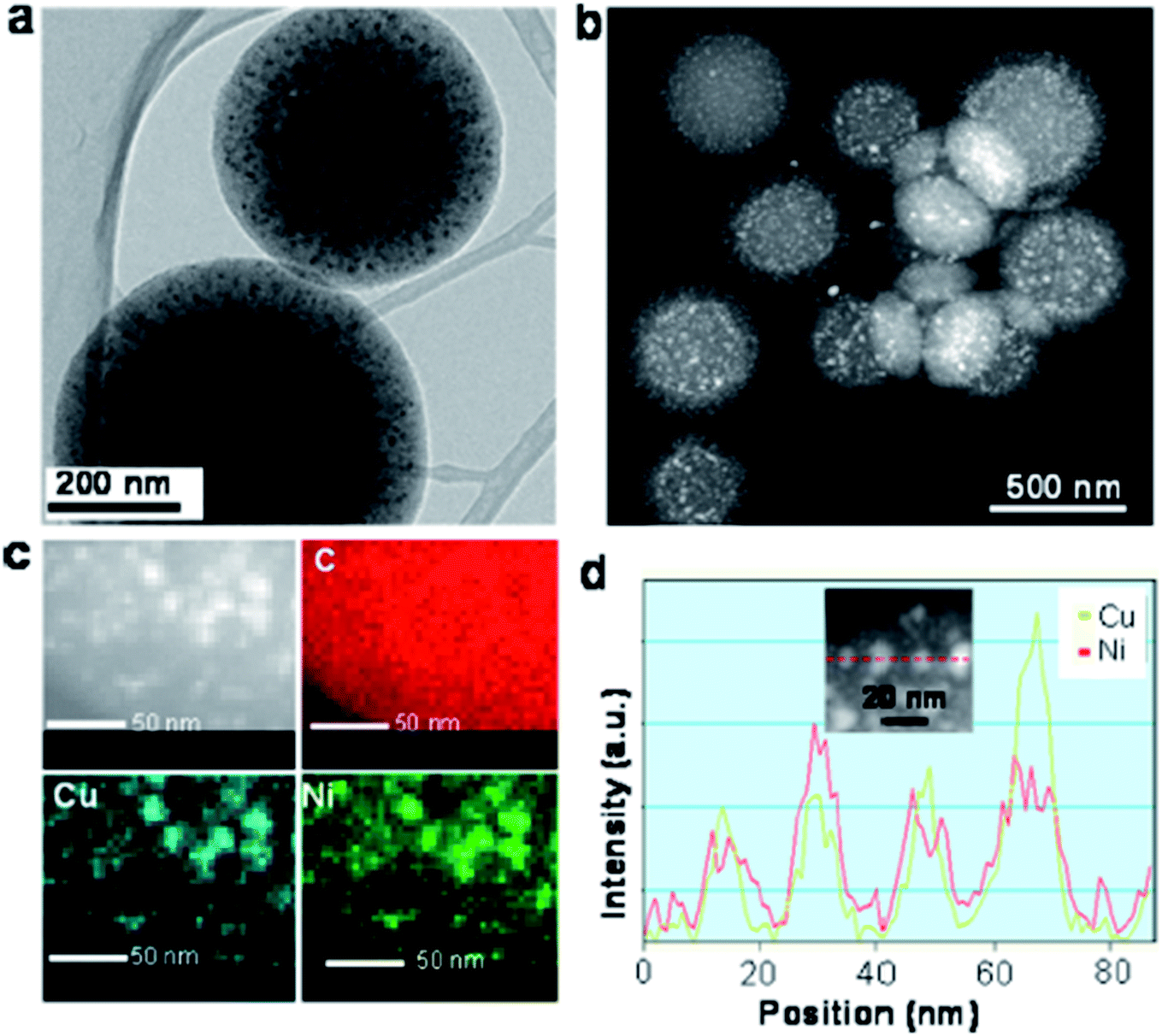 | ||
| Fig. 10 (a) TEM image, (b) Scanning TEM high angle annular dark field (STEM-HAADF) image, and (c) the corresponding energy dispersive spectroscopy (EDS) phase mapping with (d) line-scanning profiles across the metal, as indicated in the inset of the Cu0.5Ni0.5/MCNS catalyst. Reprinted with permission from the American Chemical Society (ref. 167). | ||
In addition to TEM, ETEM (environmental transmission electron microscopy) helps to get more insight about the catalytic activity of the catalyst. In normal TEMs, samples are placed under a vacuum in order to get almost zero electron scattering by residual gas molecules. On the other hand, ETEM is a dynamic observation technique, in which the samples are subjected to liquid or gas exposure and hence a clear insight can be obtained regarding the catalyst under the conditions they were formed or subjected to reaction.174 Yoshida et al.175 conducted ETEM analysis in order to get more insight regarding the interaction of the gas molecules with the supported gold catalyst. It was observed that the adsorbed CO gas molecule led to the reconstruction of the {100} facets of the gold nanoparticle during CO oxidation. Benavidez et al.176 utilised the ETEM technique in order to find out the anomalous size distribution of the supported catalyst during reaction. It was observed that at the early stage of catalyst sintering, an anomalous expansion occurred, which ultimately led to the broadening of the particle size distribution.
4.3. X-ray diffraction
The analysis of a material, particularly in the solid-state, is done by XRD studies. This is the key instrument for solid-state scientists and is now used by all the researchers. In general, it describes the phase purity of an alloy, the crystal structure, and the lattice parameters of the phases. Recorded XRD patterns provide vast information regarding the catalyst material. Thus, the oxidation state, crystallinity, and support–catalyst interaction are verified. Yingxin et al. carried out the XRD analysis for the support and the Ni-support material.159 The results suggest that the interaction with the support material was weak and NiO mainly existed in the free dispersion state. However, strong new peaks were observed in the case of alumina; hence, strong interaction was concluded. Wang et al. conducted the study on the Ni catalyst supported on CNT.158 Rezaei et al. conducted the XRD analysis on zirconia-supported Ni catalysts.157 They concluded that with the increase in the calcination temperature, the crystallinity increases but the surface area decreases. The tetragonal phase was stabilized at the room temperature without the addition of any dopant, which may be due to the nano-size effect.Wang et al. used the Scherrer equation to find out the crystallite size of the nickel catalyst.156 It was concluded that the alumina-supported catalyst was reduced in size in comparison to the nickel nanoparticles due to support interaction. This is an inference out of the XRD analysis. Furthermore, Yen et al. confirmed the ordered mesoporous structure of the mesoporous silica MCM-48 and mesoporous carbon CMK-1 with a low X-ray diffraction angle.167 The XRD pattern on the MCM support showed no visible peaks in the range of 2θ = 30–60°, which implies that the immobilized metal particles on the support are in an amorphous condition or in a very small amount so as to be detected by XRD. The prominent peaks of Cu0.5Ni0.5/CMK-1 showed an fcc structure of (111) and (200) of Cu and the CuNi alloy. Nowadays, facet selective crystallites are considered to study the differential activities of different crystal faces.
4.4. X-ray photoelectron spectroscopy
To know the oxidation state of the metal catalyst conclusively, XPS analysis is often carried out. Some important results are briefly discussed here for some SM-interacting metallic counterparts in relation to their binding energies and oxidation states. Yingxin et al. conducted the XPS analysis of the supported nickel catalyst to find out the interaction between NiO and the support and also the state of nickel present on the surface.159 The binding energy of Ni 2p3/2 for Ni/diatomite and Ni/TiO2 was the same as that of pure NiO. However, in the case of SiO2 support, it was found to be 0.8 eV higher, which implies that Ni was in the oxidative state and interacted with the support material. Carniti et al. performed NO reduction by silica-supported copper catalysts.132 To find out the oxidation state of copper, the spectra of Cu 2p3/2 were deconvoluted and a peak was observed at 933 eV, which is very close to that of CuO 937 eV. Moreover, no significant difference was observed between the fresh and used catalysts. Also, Yen et al. analysed the XPS spectra of Cu, where they found that the XPS spectra of Cu were mainly associated with Cu0 and Cu+ corresponding to the binding energy of Cu 2p3/2 and Cu 2p1/2 at 932 and 952 eV, respectively.167 The presence of a small peak at about 945 eV could be attributed to the presence of Cu2+. In addition to Cu, the XPS spectra of Ni were also analysed. From the spectra of Ni 2p, it was revealed that Ni2+ is a major component corresponding to the binding energies of 855.5 and 872.5 eV for Ni 2p3/2 and Ni 2p1/2, respectively. The XPS spectra confirmed that the oxidation states of CuNi bimetallic nanocatalysts on the two supports MCM and CMK are more or less comparable.4.5. Brunauer–Emmett–Teller (BET) surface area
The porosity of the catalysts, an important characteristic of a catalyst, is often described by the BET surface area measurement. The BET theory is an oversimplified model of physisorption.177 Based on nitrogen adsorption–desorption, mostly there are four types of isotherms that have been standardized by IUPAC. According to the degree of matching with these isotherms, catalysts are often defined as microporous and mesoporous. Yen et al. found their bimetallic Cu–Ni catalysts supported on the as a type IV isotherm with a narrow pore size distribution of ∼3 nm.167 Carniti et al. found their amorphous silica-supported copper catalyst following type IV isotherm which indicates that it is a mesoporous material and capillary condensation takes place at higher pressures.132 Thus, surface area-dependent catalytic activity is ascertained with certainty.4.6. Temperature-programmed reduction (TPR)
Temperature-programmed reduction (TPR) is a characterization technique used in heterogeneous catalysis to find out the most efficient conditions. The TPR technique is also often utilized to determine the support–catalyst interaction.168 Here, a few examples are mentioned for metal loading and doping. From the TPR profile of Cu/MCM-48, a strong reduction peak is observed at 201 °C, which can be attributed to the finely dispersed copper oxide species into copper. For Ni/MCM-48, the peak at 420 °C indicates the reduction of Ni species with the support. Weckhuysen et al. conducted the TPR study of the supported Cr catalyst on the Al2O3 surface.178 Three important conclusions were made by the authors from the TPR profile. Firstly, the intensity of the TPR peak increases with the increase in Cr loading. Secondly, the width of the peak becomes narrowed down with the decrease in Cr loading and lastly, maximum TPR peak shifts are observed towards the lower temperature with an increase in the Cr loading. From the H2-TPR study, it was deduced that only a small amount of Cu can be incorporated within the lattice of CeO2, which leads to the formation of the non-stoichiometric solid solution Ce1−xCuxO2−δ and showed much better reducibility than pure CeO2.1354.7. X-ray atomic absorption spectroscopy
X-ray absorption spectroscopy is a powerful technique that is often used for determining local geometric or electronic structure of matter. Wang et al. utilised X-ray atomic fine structure for investigating the interaction between copper oxides or ceria with CO and H2 under ambient conditions.138 Ketchie et al. monitored the size of various supported Ru catalysts under aqueous phase conditions.179 It was observed that a significant growth of metal particles occurred in the case of Ru/γ-Al2O3 and Ru/SiO2 catalysts, while in the case of Ru/TiO2 and Ru/C, no such changes occurred. Daniel et al. confirmed from the X-ray Absorption Spectroscopic analysis along with the TEM results that the supported nanocatalyst used for the hydrogenolysis of glycerol indeed has bimetallic activity of the catalyst with a particle size of less than 2 nm in diameter.180The XAFS technique is extremely useful for determining metal–support interaction and also for obtaining quantitative information regarding the metal–support interface. Koningsberger and Gates reported structural information about various supported catalysts.181 Average metal–support oxygen distances were compiled for various supported catalysts. In case of metal subcarbonyls, the average metal–support oxygen distance was about 2.1–2.2 A. On the other hand, in case of higher activity of the metal nanoparticles, the distance was 2.5–2.7 A. Guzman and Gates applied the EXAFS analysis to their MgO supported-nanogold cluster catalyst for CO oxidation.182 Based on the EXAFS results, the observed coordination number for Au was observed to vary between 1–10 for different samples.
5. Reaction mechanism and kinetic studies for surface-catalyzed reaction
The development of a suitable kinetic model is very important to determine the rate-determining step of the reaction. Three useful and popular reaction mechanisms are commonly encountered in catalysis, which are the Eley–Rideal (ER), Langmuir–Hinshelwood (LH), and Mars-van Krevleen (MVK) mechanisms. The schematics of the ER and LH mechanism are shown in Fig. 11.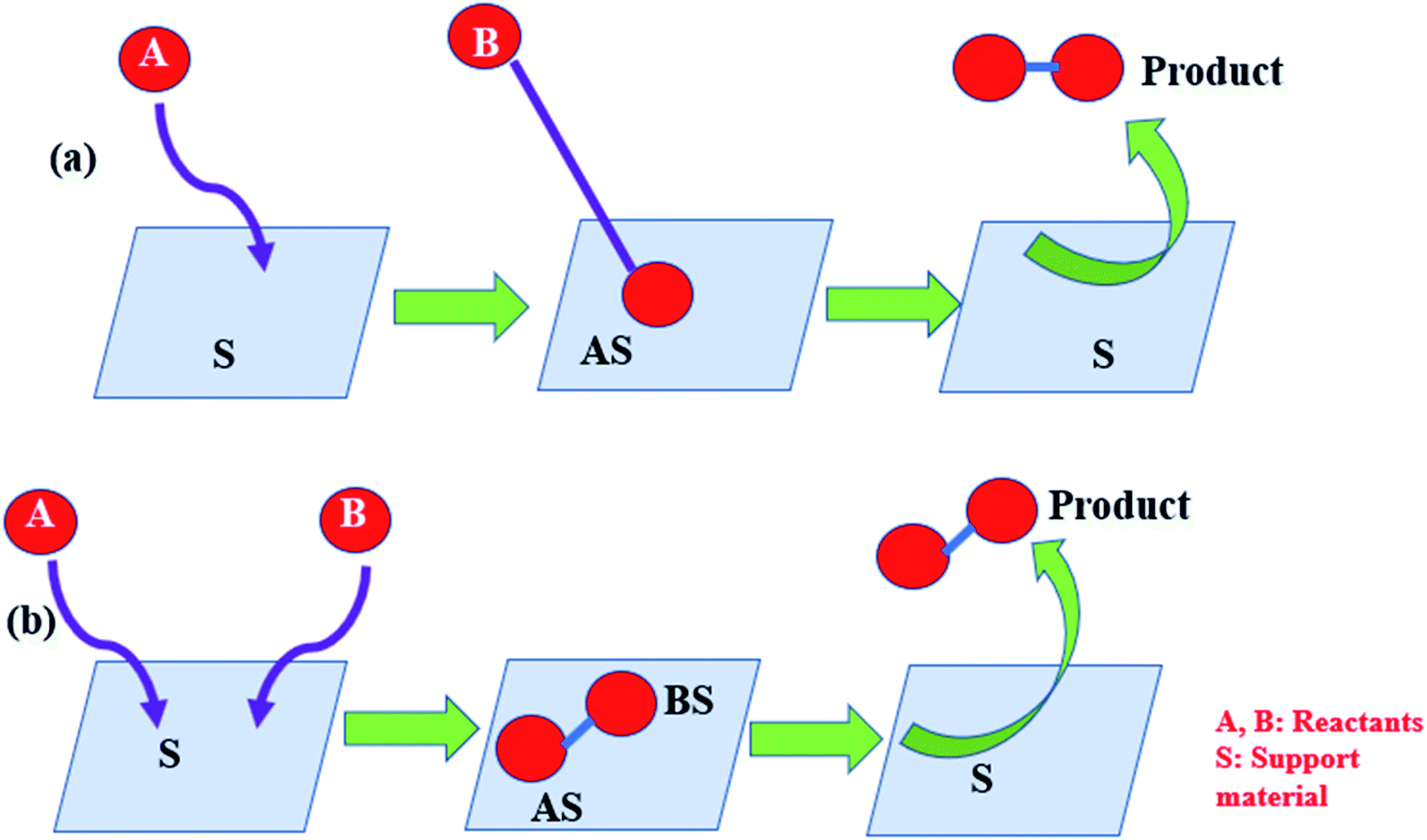 | ||
| Fig. 11 Schematic of (a) ER mechanism and (b) LH mechanism. | ||
Before the discussion of the reaction mechanism, it would be pertinent to unveil the background of the development of the surface reaction. Surface catalysis was demonstrated by van't Hoff by taking the AsH3 decomposition reaction. He realized for the first time that vessels with various sizes and shapes have different surface to volume ratio. These vessels catalyze the decomposition of AsH3 into As and H2 at different rates. For example, the decomposition proceeds more rapidly in a vessel with a larger proportion of the surface. The important advancement in surface catalysis for the adsorption of gases on surfaces was conclusively explained by Langmuir in 1921 with the idea of a chemisorption process rather than physical adsorption or van der Waals adsorption. The interpretation is now known as the Langmuir adsorption isotherm. This relates the fraction of the solid surface covered to the pressure of the gas. His adsorption kinetics work was extended by Hinshelwood, which yielded the Langmuir–Hinshelwood (LH) mechanism (Fig. 11). Almost at the same time, Rideal pointed out that the surface reaction between two substances sometimes involves the adoption of only one substance, leaving the other in the gas phase. The other substance reacts with the adsorbate from the gas phase. Thus, it becomes the Eley–Rideal (ER) mechanism (Fig. 11).
In the ER mechanism, one of the reacting species is adsorbed first onto the support surface. Then, the other species attacks it and the final product is formed, and finally gets desorbed from the surface. In the LH mechanism, two molecules get adsorbed onto the surface site and the final product is formed there. Chen and Yang reported that NO reduction with NH3 takes place on the TiO2-supported SO42− catalyst, as observed from the ER mechanism.183 XPS and chemisorption measurement confirmed the mechanism. In a study conducted by Liu et al., it has been shown that both ER and LH mechanisms are suitable for the interpretation of SCR (selective catalytic reduction) reaction depending on the reaction temperature.184 They found that the LH mechanism was well fitted at <200 °C, while the ER mechanism followed at a temperature >200 °C. Semagina et al. showed that the selective hydrogenation of 2-butyne-1,4-diol by the Pd catalyst supported over micelles followed the LH mechanism.62 Apart from these two traditional reaction theories, some new theories regarding catalytic reaction have emerged recently. For example, Xu et al. proposed that CO oxidation by the Pd-mediated single-atom graphene defect proceeded via a new termolecular ER mechanism, which is different from the traditional ER theory.185 The termolecular reaction is relatively rare in chemistry, where three atoms are required to collide to form a new species. In this recent theory, it has been proposed that oxygen is activated by two pre-adsorbed CO species, leading to the oxidation of CO on the supported catalyst (schematically shown in Fig. 12) and CO2 is desorbed from the surface.
 | ||
| Fig. 12 CO oxidation on single Pd atom embedded defect-graphene via a new termolecular Eley–Rideal mechanism. Reprinted with permission from Elsevier (ref. 185). | ||
During the oxidation of n-hexane by silica-supported cobalt and manganese catalyst, different mechanisms have been found suitable for different catalysts.100 In the presence of Co–Mn catalysts, the reaction was found to follow the Mars-van Krevelen model and in the presence of Co, it followed the LH mechanism. As per the LH concept, the oxidation reaction takes place between adsorbed oxygen and n-hexane, and the surface reaction is the rate-determining step. On the other hand, as per the ER mechanism, the reaction should follow first-order kinetics concerning oxygen, which is not probable in the above-mentioned case. But according to Vannice, the Mars-van Krevelen model is irrelevant for redox reactions on the solid support.166 Wootsch et al. also described CO oxidation by zirconia and alumina-supported Pt catalyst by the competitive and non-competitive LH reaction model.186 As per the assumption, oxygen got adsorbed onto the Pt surface and reacts alternately with CO and hydrogen. Putra et al.51 applied all of the ER, LH, and MVK models for the oxidative dehydrogenation (ODH) of propane by various alumina supported catalysts. As per the ER model, propane first gets adsorbed on the surface of the catalyst and then oxygen from the gaseous phase reacts with it. As per the LH model, both oxygen and propane get adsorbed onto the surface of the catalyst and they react with each other.
6. Recyclability and regeneration of the supported catalysts
To synthesize a catalyst for practical applications, economic viability should be looked into. The catalyst should be recyclable for repetitive use. Moreover, in various catalytic processes, the catalyst gets poisoned and therefore demands regeneration. Thus, there must be some easy regeneration procedure. Osada et al. reported about the poisoning of the supported ruthenium catalyst by sulphur and the regeneration of the same for subcritical water treatment.187 One of the most important reactions in organic chemistry is the dehydration of glycerol to acrolein by the acid catalyst. But in many cases, it has been observed that the catalysts suffer from the formation of carbonaceous materials on their surface. In this regard, Katryniok et al. mentioned that the support has an important role in the regeneration of the catalyst.188 They have found that the zirconia support is the most suitable template for stabilizing silico-tungstic acid. As we have already discussed in our previous sections, it is to be noted that in methane decomposition, the catalyst gets covered by carbon. To regenerate it, Li et al. allowed the deposited carbon to react with oxygen or carbon dioxide.160The traditional regeneration of catalysts used for hydrogenation purposes involves the stripping of the hydrocarbon from the surface by using either nitrogen, hydrogen, or steam, followed by an oxidation step, and finally reduction with hydrogen to reactivate the catalyst. Two traditional mechanisms of metal sintering are ripening and coalescence, which occur above the Tamman temperature.91
In case of the supported nanoparticles, the support/ligand plays a very crucial role in the recyclability of the catalyst. Wan et al.32 reported a polyHIPE-supported Pt catalyst that is relevant for reduction studies for more than 1500 cycles.
Metal colloids are very active catalysts at times owing to the presence of a large ratio of exposed atoms on the catalyst surface. However, one of the major issues with soluble metal colloids as a catalyst is their recovery from the reaction mixture. Therefore, it is often suggested to immobilize them by using aqueous/organic biphasic conditions or any fluorinated solvents or using any porous SM.57
7. Conclusions
The importance of the ‘proximity effect’ and the subsequent explanations are still to be discussed in the literature. Exposed facets provide active atoms synergistically and improve the catalytic performance. The published literature largely reports the increased yield of the products to meet our never-ending demand but the reasons for the choice of SM have not been studied in depth. Solid supports and organized assemblies interact with the catalyst particles, which not only improves the catalyst performance, proving the significance of heterojunction or stairs, kinks, etc., but also increases the TON even for environmental remediation.189 At times, the aggregation of catalyst particles is held responsible for the loss of catalyst activity and hence support materials are introduced.190 Cleaning efficacy of the catalyst surface vis-à-vis the abstraction of the by product from the catalyst may be carried forward by the SM. Here, two types of catalysts are mainly discussed: metal particles (MPs) and metal oxide particles, especially in their nano dimensions. Though the interaction between the SM and the catalyst has revolutionized the industrial processes (Haber process for ammonia production using Fe catalyst with a small amount of Mo, Contact process for sulphuric acid production using V2O5 catalyst with silica granules support, etc.); at large, the SM-catalyst binding motif is difficult to describe. Thus, different SMs are being tried in a ‘hit-or-miss’ manner. However, electrostatic field force, hydrophobic effect, van der Waals force, different modes of particle stabilization, solvent effect, etc., have to be considered as future endeavors.Conflicts of interest
There are no conflicts of interest.References
- S. Dey, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC
.
- F. Haber, Nobel Lecture, June 2, 1920 Search PubMed
- E. K. Rideal, Chem. Age, 1921, 4, 245–246 CAS
- A. Cimino and V. Indovina, J. Catal., 1974, 33, 493–496 CrossRef CAS
- T. Iwaya, S. Tokuno, M. Sasaki, M. Goto and K. Shibata, J. Mater. Sci., 2008, 43, 2452–2456 CrossRef CAS
- J. M. Thomas and K. D. M. Harris, Energy Environ. Sci., 2016, 9, 687–708 RSC
- A. Busch, S. Alles, Y. Gensterblum, D. Prinz, D. N. Dewhurst, M. D. Raven, H. Stanjek and B. M. Krooss, Int. J. Greenhouse Gas Control, 2008, 2, 297–308 CrossRef CAS
- Y. Zhu, S. Zhang, Y. Ye, X. Zhang, L. Wang, W. Zhu, F. Cheng and F. Tao, ACS Catal., 2012, 2, 2403–2408 CrossRef CAS
- Y. Liu, B. Huang, Y. Dai, X. Zhang, X. Qin, M. Jiang and M. Whangbo, Catal. Commun., 2009, 11, 210–213 CrossRef CAS
- I. Takahara, M. Saito, M. Inaba and K. Murata, Catal. Lett., 2005, 105, 249–252 CrossRef CAS
- Y. Kim, N. Park, J. Shin, S. R. Lee, Y. J. Lee and D. J. Moon, Catal. Today, 2003, 87, 153–162 CrossRef CAS
- M. Iwamoto and H. Hamada, Catal. Today, 1991, 10, 57–71 CrossRef CAS
- T. Kitagawa, T. Uozumi, K. Soga and T. Takata, Polymer, 1997, 38, 615–620 CrossRef CAS
- A. Schatz, O. Reiser and W. J. Stark, Chem.–Eur. J., 2010, 16, 8950–8967 CrossRef
- F. Guo, X. Li, Y. Liu, K. Peng, C. Guo and Z. Rao, Energy Convers. Manage., 2018, 167, 81–90 CrossRef CAS
- M. Hu, M. Laghari, B. Cui, B. Xiao, B. Zhang and D. Guo, Energy, 2018, 145, 228–237 CrossRef CAS
- R. Aiello, J. E. Fiscus, H. Loye and M. D. Amiridis, Appl. Catal., A, 2000, 192, 227–234 CrossRef CAS
- S. S. Itkulova, G. D. Zakumbaeva, Y. Y. Nurmakanov, A. A. Mukazhanova and A. K. Yermaganbetova, Catal. Today, 2014, 228, 194–198 CrossRef CAS
- M. E. S. Hegarty, A. M. O'Connor and J. R. H. Ross, Catal. Today, 1998, 42, 225–232 CrossRef CAS
- F. Looij, J. C. Giezen, E. R. Stobbe and J. W. Geus, Catal. Today, 1994, 21, 495–503 CrossRef
- E. Kikuchi, M. Matsumoto, T. Takahashi, A. Machino and Y. Morita, Appl. Catal., 1984, 10, 251–260 CrossRef CAS
- S. Storsaeter, O. Borg, E. A. Blekkan and A. Holmen, J. Catal., 2005, 231, 405–419 CrossRef CAS
- E. I. Mabaso, E. Steen and M. Claeys, Syn. Gas Chem., Dresden, Germany, October 4–6, 2006 Search PubMed
- K. R. Reddy, N. S. Kumar, P. S. Reddy, B. Sreedhar and M. L. Kantam, J. Mol. Catal. A: Chem., 2006, 252, 12–16 CrossRef CAS
- Z. Novak, A. Szabo, J. Repasi and A. Kotschy, J. Org. Chem., 2003, 68, 3327–3329 CrossRef CAS
- Y. Wang, J. Liu and C. Xia, Tetrahedron Lett., 2011, 52, 1587–1591 CrossRef CAS
- A. Corma, H. Garcia and A. Primo, J. Catal., 2006, 241, 123–131 CrossRef CAS
- S. Diyarbakir, H. Can and O. Metin, ACS Appl. Mater. Interfaces, 2015, 7, 3199–3206 CrossRef CAS
- D. Horvath, L. Toth and L. Guczi, Catal. Lett., 2000, 67, 117–128 CrossRef CAS
- M. M. Schubert, S. Hackenberg, A. C. Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS
- L. Liu, F. Zhou, L. Wang, X. Qi, F. Shi and Y. Deng, J. Catal., 2010, 274, 1–10 CrossRef CAS
- Y. Wan, Y. Feng, D. Wan and M. Jin, RSC Adv., 2016, 6, 109253–109258 RSC
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652–9656 CrossRef CAS
- A. Gniewek, J. J. Ziolkowski, A. M. Trzeciak, M. Zawadzki, H. Grabowska and J. Wrzyszcz, J. Catal., 2008, 254, 121–130 CrossRef CAS
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS
- I. C. Gerber and P. Serp, Chem. Rev., 2020, 120, 1250–1349 CrossRef CAS
- H. P. Boehm, E. Diehl, W. Heck and R. Sappok, Angew. Chem., Int. Ed., 1964, 3, 669–677 CrossRef
- R. Huang, J. Wang, B. Zhang, K. Wu, Y. Zhang and D. S. Su, Catal. Sci. Technol., 2018, 8, 1522–1527 RSC
- G. Saianand, A. Gopalan, J. Lee, C. Satish, K. Gopalakrishnan, G. E. Unni, D. Shanbhag, V. D. B. C. Dasireddy, J. Yi, S. Xi, A. H. Al-Muhtaseb and A. Vinu, Small, 2020, 16, 1903937 CrossRef CAS
- B. Lin, Y. Zhang, Y. Zhu, Y. Zou, Y. Hu, X. Du, H. Xie, K. Wang and Y. Zhou, Catal. Commun., 2020, 144, 106094 CrossRef CAS
- B. Fang, N. K. Chaudhari, M. Kim, J. H. Kim and J. Yu, J. Am. Chem. Soc., 2009, 131, 15330–15338 CrossRef CAS
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS
- T. Frelink, W. Visscher and J. A. R. Veen, J. Electroanal. Chem., 1995, 382, 65–72 CrossRef
- P. Yan, X. Zhang, F. Herold, F. Li, X. Di, T. Cao, B. J. M. Etzold and W. Qi, Catal. Sci. Technol., 2020, 10, 4952–4959 RSC
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS
- K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977–9980 CrossRef CAS
- E. Heracleous, A. F. Lee, K. Wilson and A. A. Lemonidou, J. Catal., 2005, 231, 159–171 CrossRef CAS
- M. C. Kung and H. H. Kung, Top. Catal., 2000, 10, 21–26 CrossRef CAS
- S. Biswas and A. Pal, J. Water Process Eng., 2020, 36, 101272 CrossRef
- P. Mahamallik and A. Pal, RSC Adv., 2016, 6, 100876–100890 RSC
- M. D. Putra, S. M. Al-Zahrani and A. E. Abasaeed, Catal. Commun., 2012, 26, 98–102 CrossRef CAS
- V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599–2626 CrossRef CAS
- P. Mahamallik and A. Pal, RSC Adv., 2015, 5, 78006–78016 RSC
- R. L. McCormick and G. O. Alptekin, Catal. Today, 2000, 55, 269–280 CrossRef CAS
- H. F. J. Vant Blik, D. C. Koningsberger and R. Prins, J. Catal., 1986, 97, 210–218 CrossRef CAS
- Y. Min, M. Akbulut, K. Kristiansen, Y. Golan and J. Israelachvili, Nat. Mat., 2008, 7, 527–538 CrossRef CAS
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS
- S. Kundu, S. K. Ghosh, M. Mandal and T. Pal, New J. Chem., 2002, 26, 1081–1084 RSC
- T. Pal and N. R. Jana, Langmuir, 1996, 12, 3114–3121 CrossRef CAS
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 696–697 RSC
- Y. Wang, G. Wei, W. Zhang, X. Jiang, P. Zheng, L. Shi and A. Dong, J. Mol. Catal. A: Chem., 2007, 266, 233–238 CrossRef CAS
- N. Semagina, E. Joannet, S. Parra, E. Sulman, A. Renken and L. Kiwi-Minsker, Appl. Catal., A, 2005, 280, 141–147 CrossRef CAS
- A. K. Das, S. Saha, A. Pal and S. K. Maji, J. Environ. Sci. Health, Part A: Environ. Sci. Eng., 2009, 44, 896–905 CrossRef CAS
- A. Adak, M. Bandyopadhyay and A. Pal, Sep. Purif. Technol., 2005, 44, 139–144 CrossRef CAS
- M. U. Khobragade and A. Pal, J. Environ. Chem. Eng., 2014, 2, 2295–2305 CrossRef CAS
- M. U. Khobragade and A. Pal, Sep. Sci. Technol., 2016, 51, 1287–1298 CrossRef CAS
- P. Mahamallik and A. Pal, Catal. Today, 2020, 348, 212–222 CrossRef CAS
- P. Mahamallik and A. Pal, J. Environ. Chem. Eng., 2017, 5, 2886–2893 CrossRef CAS
- S. Koner, A. Pal and A. Adak, Desalin. Water Treat., 2010, 22, 1–8 CrossRef CAS
- A. Adak, A. Pal and M. Bandyopadhyay, Colloids Surf., A, 2006, 277, 63–68 CrossRef CAS
- A. Dabrowski, Z. Hubicki, P. Podkoscielny and E. Robens, Chemosphere, 2004, 56, 91–106 CrossRef CAS
- M. Basu and T. Pal, Hybrid Nanomaterials: Synthesis, Characterization, and Applications, ed. B. P. S. Chauhan, John Wiley & Sons, Inc., 2011, pp. 23–63 Search PubMed
- S. Sarkar, S. Pande, S. Jana, A. K. Sinha, M. Pradhan, M. Basu, S. Saha, S. Y. Yusuf and T. Pal, J. Phys. Chem. C, 2009, 113, 6022–6032 CrossRef CAS
- A. K. Sinha, S. Jana, S. Pande, S. Sarkar, M. Pradhan, M. Basu, S. Saha, A. Pal and T. Pal, CrystEngComm, 2009, 11, 1210–1212 RSC
- Y. Wang, L. Yu, B. Zhu and L. Yu, J. Mater. Chem. A, 2016, 4, 10828–10833 RSC
- A. M. Jansson, M. Grotti, K. M. Halkes and M. Meldal, Org. Lett., 2002, 4, 27–30 CrossRef CAS
- M. E. Ford and J. E. Premecz, J. Mol. Catal., 1983, 19, 99–112 CrossRef CAS
- Y. Huirong and L. B. C. Yingde, Synth. Commun., 1998, 28, 1233–1238 CrossRef
- P. E. Garrou, Organometallics, 1986, 5, 466–473 CrossRef
- B. V. Vaerenbergh, J. Lauwaert, J. W. Thybaut, P. Vermeir and J. Clereq, Chem. Eng. J., 2019, 374, 576–588 CrossRef
- J. C. Duchet, M. J. Tilliette, D. Cornet, L. Vivier, G. Perot, L. Bekakra, C. Moreau and G. Szabo, Catal. Today, 1991, 10, 579–592 CrossRef CAS
- K. I. Hadjiivanov and D. G. Klissurski, Chem. Soc. Rev., 1996, 61–69 RSC
- J. A. Z. Pieterse, G. Mul, I. Melian-Cabrera and R. W. Brink, Catal. Lett., 2005, 99, 41–44 CrossRef CAS
- S. Besell, Appl. Catal., A, 1995, 126, 235–244 CrossRef
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, J. Am. Chem. Soc., 2011, 133, 2378–2381 CrossRef CAS
- K. G. Nishanth, P. Sridhar, S. Pitchumani and A. K. Shukla, Fuel Cells, 2012, 12, 146–152 CrossRef CAS
- H. Wang, S. Yan, S. O. Salley and K. Y. S. Ng, Fuel, 2013, 111, 81–87 CrossRef CAS
- T. Hees, F. Zhong, T. Rudolph, A. Walther and R. Mulhaupt, Adv. Funct. Mater., 2017, 27, 1605586 CrossRef
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2010, 26, 2885–2893 CrossRef CAS
- R. J. Kongarapu, P. Mahamallik and A. Pal, J. Environ. Chem. Eng., 2017, 5, 1321–1329 CrossRef CAS
- R. J. Liu, P. A. Crozier, C. M. Smith, D. A. Hucul, J. Blackson and G. Salaita, Appl. Catal., A, 2005, 282, 111–121 CrossRef CAS
- J. Hu, Y. Wang, M. Han, Y. Zhou, X. Jiang and P. Sun, Catal. Sci. Technol., 2012, 2, 2332–2340 RSC
- A. E. Aksoylu, A. N. Akin, Z. I. Onsan and D. L. Trimm, Appl. Catal., A, 1996, 145, 185–193 CrossRef CAS
- G. Li, L. Hu and J. M. Hill, Appl. Catal., A, 2006, 301, 16–24 CrossRef CAS
- T. Miyahara, H. Kanzaki, R. Hamada, S. Kuroiwa, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2001, 176, 141–150 CrossRef CAS
- S. Li, L. Guo and T. Ishihara, Catal. Today, 2020, 339, 352–361 CrossRef CAS
- A. J. Vizcaino, M. Lindo, A. Carrero and J. A. Calles, Int. J. Hydrogen Energy, 2012, 37, 1985–1992 CrossRef CAS
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Chem. Rev., 2020, 120, 464–525 CrossRef CAS
- J. Okamura, S. Nishiyama, S. Tsuruya and M. Masai, J. Mol. Catal. A: Chem., 1998, 135, 133–142 CrossRef CAS
- S. Todorova, A. Naydenov, H. Kolev, J. P. Holgado, G. Ivanov, G. Kadinov and A. Caballero, Appl. Catal., A, 2012, 413–414, 43–51 CrossRef CAS
- M. A. Fraga, E. Jorado, M. J. Mendes, M. M. A. Freitas, J. L. Faria and J. L. Figueiredo, J. Catal., 2002, 209, 355–364 CrossRef CAS
- K. Takeishi and Y. Akaike, Appl. Catal., A, 2016, 510, 20–26 CrossRef CAS
- Y. Zeng, T. Wang, S. Zhang, Y. Wang and Q. Zhong, Appl. Surf. Sci., 2017, 411, 227–234 CrossRef CAS
- Y. Lu, J. Song, W. Li, Y. Pan, H. Fang, X. Wang and G. Hu, Appl. Surf. Sci., 2020, 506, 145000 CrossRef CAS
- X. Chen, X. Chen, S. Cai, J. Chen, W. Xu, H. Jia and J. Chen, Chem. Eng. J., 2018, 334, 768–779 CrossRef CAS
- X. Chen, X. Chen, E. Yu, S. Cai, H. Jia, J. Chen and P. Liang, Chem. Eng. J., 2018, 344, 469–479 CrossRef CAS
- A. Daliri, J. Zhang and C. H. Wang, Woodhead Publishing Series in Composites Science and Engineering, Lightweight Composite Structures in Transport, ed. J. Nijuguna, 2016, vol. 67, pp. 121–163 Search PubMed
- G. Liu, Y. Li, W. Chu, X. Shi, X. Dai and Y. Yin, Catal. Commun., 2008, 9, 1087–1091 CrossRef CAS
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS
- G. Wulfsberg, Inorganic Chemistry, University Science Books, CA, USA, Indian edn, 2002, p. 202 Search PubMed
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS
- B. Nkosi, N. J. Coville and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1988, 71–72 RSC
- T. K. Sau, A. Pal and T. Pal, J. Phys. Chem. B, 2001, 105, 9266–9272 CrossRef CAS
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC
- M. Ganguly, J. Jana, A. Pal and T. Pal, RSC Adv., 2016, 6, 17683–17703 RSC
- E. Weiss and M. Fohnan, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 2025–2041 RSC
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112(10), 5520–5551 CrossRef CAS
- W. Schottky and H. Sperrschicht, Naturwissenschaften, 1938, 26, 843 CrossRef CAS
- S. Wang, F. Jia, X. Wang, L. Hu, Y. Sun, G. Yin, T. Zhou, Z. Feng, P. Kumar and B. Liu, ACS Omega, 2020, 5(10), 5209–5218 CrossRef CAS
- M. Okumura, T. Fujitani, J. Huang and T. Ishida, ACS Catal., 2015, 5, 4699–4707 CrossRef CAS
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS
- J. Chen, W. Fang, Q. Zhang, W. Deng and Y. Wang, Chem.–Asian J., 2014, 9, 2187–2196 CrossRef CAS
- C. Milone, R. Ingoglia, L. Schipilliti, C. Crisafulli, G. Neri and S. Galvagno, J. Catal., 2005, 236, 80–90 CrossRef CAS
- R. Burch, S. E. Golunski and M. S. Spencer, Catal. Lett., 1990, 5, 55–60 CrossRef CAS
- X. Shi, Q. Hu, F. Wang, W. Zhang and P. Duan, J. Catal., 2016, 337, 233–239 CrossRef CAS
- Q. Xu, X. Li, T. Pan, C. Yu, J. Deng, Q. Guo and Y. Fu, Green Chem., 2016, 18, 1287–1294 RSC
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC
- J. Yuan, S. Li, L. Yu, Y. Liu, Y. Cao, H. He and K. Fan, Energy Environ. Sci., 2013, 6, 3308–3314 RSC
- S. Sakata, T. Nakai, H. Yahiro and M. Shiotani, Appl. Catal., A, 1997, 165, 467–472 CrossRef CAS
- Z. H. Zhu, L. R. Radovic and G. Q. Lu, Carbon, 2000, 38, 451–464 CrossRef CAS
- P. Carniti, A. Gervasini, V. H. Modica and N. Ravasio, Appl. Catal., B, 2000, 28, 175–185 CrossRef CAS
- L. Singoredjo, M. Slagt, J. V. Wees, F. Kapteijn and J. A. Moulijn, Catal. Today, 1990, 7, 157–165 CrossRef CAS
- Z. Zhu, Z. Liu, S. Liu, H. Niu, T. Hu, T. Liu and Y. Xie, Appl. Catal., B, 2000, 26, 25–35 CrossRef CAS
- L. Yang, S. Zhou, T. Ding and M. Meng, Fuel Process. Technol., 2014, 124, 155–164 CrossRef CAS
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, Q. Zeng, A. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369–377 CrossRef CAS
- N. A. Koryabkina, A. A. Phatak, W. F. Ruettinger, R. J. Farrauto and F. H. Ribeiro, J. Catal., 2003, 217, 233–239 CAS
- X. Wang, J. A. Rodriguez, J. C. Hansen, D. Gamarra, A. Martinez-Arias and M. Fernandes-Garcia, J. Phys. Chem. B, 2006, 110, 428–434 CrossRef CAS
- M. S. Spencer, Top. Catal., 1999, 8, 259–266 CrossRef CAS
- J. Chatt, J. R. Dilworth and R. L. Ricards, Chem. Rev., 1978, 78, 589–625 CrossRef CAS
- H. M. T. Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. Jong, Science, 2012, 35, 835–838 CrossRef
- H. Jung, P. L. Walker and M. A. Vannice, J. Catal., 1982, 75, 416–422 CrossRef CAS
- P. P. Gan and S. F. Y. Li, Chem. Eng. J., 2013, 229, 351–363 CrossRef CAS
- M. Hartmann, S. Kullmann and H. Keller, J. Mater. Chem., 2010, 20, 9002–9017 RSC
- N. K. Daud and B. H. Hameed, Desalination, 2011, 269, 291–293 CrossRef CAS
- A. M. Trzeciak and J. J. Ziolkowski, Coord. Chem. Rev., 2005, 249, 2308–2322 CrossRef CAS
- Y. Li, Y. Yu, J. Wang, J. Song, Q. Li, M. Dong and C. Liu, Appl. Catal., B, 2012, 125, 189–196 CrossRef CAS
- M. D. Esrafili, P. Nematollahi and R. Nurazar, Superlattices Microstruct., 2016, 92, 60–67 CrossRef CAS
- E. Antolini, J. Mater. Sci., 2003, 38, 2995–3005 CrossRef CAS
- J. W. Shabaker, G. W. Huber, R. R. Davda, R. D. Cortright and J. A. Dumesic, Catal. Lett., 2003, 88, 1–8 CrossRef CAS
- J. H. Bitter, K. Seshan and J. A. Lercher, J. Catal., 1997, 171, 279–286 CrossRef CAS
- D. He, C. Zeng, C. Xu, N. Cheng, H. Li, S. Mu and M. Pan, Langmuir, 2011, 27, 5582–5588 CrossRef CAS
- J. T. Miller, B. L. Meyers, F. S. Modica, G. S. Lane, M. Vaarkamp and D. C. Koningsberger, J. Catal., 1993, 143, 395–408 CrossRef CAS
- T. Zhang and M. D. Amiridis, Appl. Catal., A, 1998, 167, 161–172 CrossRef CAS
- J. R. Rostrup-Nielsen and J. Bak-Hansen, J. Catal., 1993, 144, 38–49 CrossRef CAS
- S. Wang and G. Q. Lu, Appl. Catal., A, 1998, 169, 271–280 CrossRef CAS
- M. Rezaei, S. M. Alavi, S. Sahebdelfar, P. Bai, X. Liu and Z. Yan, Appl. Catal., B, 2008, 77, 346–354 CrossRef CAS
- C. Wang, J. Qiu, C. Liang, L. Xing and X. Yang, Catal. Commun., 2008, 9, 1749–1753 CrossRef CAS
- L. Yingxin, C. Jixiang and Z. Jiyan, Chin. J. Chem. Eng., 2007, 15, 63–67 CrossRef
- J. Li and K. J. Smith, Appl. Catal., A, 2008, 349, 116–124 CrossRef CAS
- J. K. Norskov, T. Bliggard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS
- G. Prieto, S. Beijer, M. L. Smith, M. He, Y. Au, Z. Wang, D. A. Bruce, K. P. Jong, J. J. Spivey and P. E. Jongh, Angew. Chem., Int. Ed., 2014, 53, 6397–6401 CrossRef CAS
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS
- V. D. Santo, A. Gallo, A. Naldoni, M. Guidotti and R. Psaro, Catal. Today, 2012, 197, 190–205 CrossRef
- J. Kugai, S. Velu and C. Song, Catal. Lett., 2005, 101, 255–264 CrossRef CAS
- C. I. Richards, S. Choi, J. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef CAS
- H. Yen, Y. Seo, S. Kaliaguine and F. Kleitz, ACS Catal., 2015, 5, 5505–5511 CrossRef CAS
- G. Deo and I. E. Wachs, J. Phys. Chem., 1991, 95, 5889–5895 CrossRef CAS
- D. S. Kim, K. Segawa, T. Soeya and I. E. Wachs, J. Catal., 1992, 136, 539–552 CrossRef CAS
- F. Buciuman, F. Patcas, R. Craciun and D. R. T. Zahn, Phys. Chem. Chem. Phys., 1999, 1, 185–190 RSC
- L. Li, Q. Shen, J. Cheng and Z. Hao, Catal. Today, 2010, 158, 361–369 CrossRef CAS
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS
- E. G. M. Kuijpers, J. W. Jansen, A. J. V. Dillen and J. W. Gues, J. Catal., 1981, 72, 75–82 CrossRef CAS
- T. Kawasaki, Surf. Interface Anal., 2018, 171–175 Search PubMed
- H. Yoshida, Y. Kuwauchi, J. R. Jinscheck, K. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta and S. Takeda, Science, 2012, 335, 317–319 CrossRef CAS
- A. D. Benavidez, L. Kovarik, A. Genc, N. Agrawal, E. M. Larsson, T. W. Hansen, A. M. Karim and A. K. Datye, ACS Catal., 2012, 2, 2349–2356 CrossRef CAS
- K. Sing, Colloids Surf., A, 2001, 187–188, 3–9 CrossRef CAS
- B. M. Weckhuysen, R. A. Schoonheydt, J. Jehng, I. E. Wachs, S. J. Cho, R. Ryoo, S. Kljlstra and E. Poels, J. Chem. Soc., Faraday Trans., 1995, 91, 3245–3253 RSC
- W. C. Ketchie, E. P. Maris and R. J. Davis, Chem. Mater., 2007, 19, 3406–3411 CrossRef CAS
- O. M. Daniel, A. DeLaRiva, E. L. Kunkes, A. K. Datye, J. A. Dumesic and R. J. Davis, ChemCatChem, 2010, 2, 1107–1114 CrossRef CAS
- D. C. Koningsberger and B. C. Gates, Catal. Lett., 1992, 14, 271–277 CrossRef CAS
- J. Guzman and B. C. Gates, J. Phys. Chem. B, 2002, 106, 7659–7665 CrossRef CAS
- J. P. Chen and R. T. Yang, J. Catal., 1993, 139, 277–288 CrossRef CAS
- F. Liu, H. He, C. Zhang, W. Shan and X. Shi, Catal. Today, 2011, 175, 18–25 CrossRef CAS
- G. Xu, R. Wang, F. Yang, D. Ma, Z. Yang and Z. Lu, Carbon, 2017, 118, 35–42 CrossRef CAS
- A. Wootsch, C. Descorme and D. Duprez, J. Catal., 2004, 225, 259–266 CrossRef CAS
- M. Osada, N. Hiyoshi, O. Sato, K. Arai and M. Shirai, Energy Fuels, 2008, 22, 845–849 CrossRef CAS
- B. Katryniok, S. Paul, M. Capron, V. Belliere-Baca, P. Rey and F. Dumeignil, ChemSusChem, 2012, 5, 1298–1306 CrossRef CAS
- A. K. Sinha, M. Pradhan, S. Sarkar and T. Pal, Environ. Sci. Technol., 2013, 47, 2339–2345 CrossRef CAS
- K. Guo, Y. Ding, J. Luo, M. Gu and Z. Yu, ACS Appl. Energy Mater., 2019, 2, 5851–5861 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2020 |