DOI:
10.1039/D0RA05339B
(Paper)
RSC Adv., 2020,
10, 35473-35479
Extraction of Pd(II) and Pt(II) from aqueous hydrochloric acid with 1,3-diaminocalix[4]arene: switching of the extraction selectivity by using different extraction modes†
Received
18th June 2020
, Accepted 10th September 2020
First published on 28th September 2020
Abstract
The extraction ability of 1,3-diaminocalix[4]arene (2: H2L) toward platinum group metals (PGMs) has been investigated, which revealed that 2 is able to extract Pd(II) and Pt(II) from hydrochloric acid via different extraction modes. The extraction species for Pd(II) and Pt(II) are [PdL] and [Pt2Cl6(H3L)2], respectively, as evidenced by equilibrium analysis and X-ray crystallography. In [PdL], the two phenoxide oxygens and two amino nitrogens of L2− coordinate to the Pd ion. On the other hand, in [Pt2Cl6(H3L)2], two anionic trichloro complexes PtCl3− are sandwiched between two H3L+, in which one amino nitrogen directly coordinates to a PtCl3− species and another protonated amino group forms an ion pair with another PtCl3−. Utilizing the different extraction modes, switching of the extraction selectivity has been achieved in the competitive extraction between Pd(II) and Pt(II) by varying the concentrations of H+ and Cl− in the aqueous phase. Finally, from the extracted organic phase, back-extraction of Pd(II) and Pt(II) was easily performed, respectively.
Introduction
Although platinum group metals (PGMs) such as Pd and Pt are widely used in industry, refining them from ores is an expensive and high-energy consuming process. Therefore, efficient methods of recovering and separating PGMs from industrial waste are in demand. Solvent extraction from concentrated hydrochloric acid solutions is a widely used method for recovering and separating PGMs,1–16 which exist as chloro complexes under the conditions. This allows them to be extracted by the cation exchange, solvation, and anion exchange mechanisms, depending on the type of extractant and/or acid concentration.17 Usually, the extraction of PGMs is performed stepwise in the order of ease-of-extraction, i.e., Pd > Pt > Ir > Rh. The separation process requires a large number of steps due to the low selectivity of the extractant used for each PGM. If a PGM other than Pd could be selectively extracted from a mixture of PGMs, the extraction process could be considerably simplified. However, such examples are quite rare in the literature,18–20 because the ligand exchange and chloro complex formation of Pd is the fastest among the PGMs.
Calixarenes have been used as a platform for the construction of metal extractants, taking advantage of their specific structures and the ease of chemical modification.21–23 Various coordinating groups have been introduced to the upper and/or lower rims of calixarenes to produce multidentate ligands that can be applied to the extraction of PGMs.24–34 However, similar to other types of extractants, most reports on PGM extraction with calix-based extractants deal with the extraction and selectivity of Pd, and papers on the extraction of other PGMs are scarce. In addition, to the best of our knowledge, there is no report on the selective extraction of other PGMs with calix-based extractants in the presence of Pd. We previously reported the synthesis of hybrid-type calixarenes,35–38 in which two distal phenolic hydroxy groups of calix[4]arenes are directly replaced with other coordinating groups. We applied the novel calixarenes as metal extractants and succeeded in the selective extraction of Yb(III) from lanthanoid ions39 and Zr(IV) over Hf(IV)40 using calix[4]arenediphosphonic acid, and that of Pd(II) from PGMs using calix[4]arenedicarboxylic acid.41 One of the origin for the high metal discrimination abilities of these extractants is their rigid coordination environments that are realized by the coordinating groups directly introduced on the calix skeleton. In this study, we have investigated the PGM extraction ability of 1,3-diaminocalixarene (2),36 expecting the direct coordination of the amino groups to the soft metals and the formation of ion pairs with chloro complexes under acidic conditions.
Results and discussion
Extraction of PGMs from aqueous hydrochloric acid
Solvent extraction experiments were carried out to evaluate the extraction ability of compound 2 toward PGMs [Pd(II), Pt(II), Pt(IV), Ir(IV) and Rh(III)]. A solution of 2 in chloroform (5 cm3, [2] = 1.0 × 10−3 M) and an aqueous solution (5 cm3) containing a metal ion ([Metal]aq,init = 1.0 × 10−4 M) and HCl (0.001–7 M) were pipetted into a 30 cm3 vial. The combined phases were shaken at 300 strokes per min for 24 h at ambient temperature (20 ± 3 °C). The percentage of extraction (E%) was calculated according to the following equation:| | |
E% = ([Metal]aq,init − [Metal]aq)/[Metal]aq,init ×100%
| (1) |
where [Metal]aq is the concentration of the metal ion in the aqueous phase after the extraction.
First, the dependence of E% of PGMs on the concentration of HCl in the aqueous phase ([HCl]) was investigated. The results for Pd(II) and Pt(II) are shown in Fig. 1. Pd(II) was extracted at high E% from low [HCl] solutions (0.001–0.1 M), with E% decreasing significantly when [HCl] exceeded 0.5 M, and Pd(II) not being extracted at all at [HCl] above 3 M (Fig. 1a). On the other hand, Pt(II) could not be extracted from low [HCl] solutions (<0.01 M), with E% increasing with increasing [HCl] (0.1–1 M) (Fig. 1b). When the acid concentration was increased further, E% decreased, and Pt(II) was not extracted above 3 M. Pt(IV) was extracted with medium E% (44–57%) at a [HCl] range of 0.01–1.0 M, but E% of Rh(III) and Ir(IV) were low over the entire [HCl] range tested (Table S1†). As a comparison, the extraction ability of monoaminocalix[4]arene (3)36 was also examined (Table S2†). Compound 3 extracted PGMs with low E% (≤23%), indicating that two amino groups are necessary for the efficient extraction of PGMs.
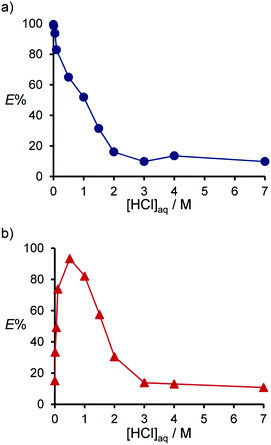 |
| | Fig. 1 Dependence of E% on [HCl] for the extraction of Pd(II) (a) and Pt(II) (b) with compound 2. Aq. phase: [Metal]aq,init = 1.0 × 10−4 M. Org. phase: CHCl3, [2]org,init = 1.0 × 10−3 M. | |
Dependence of E% on chloride ion concentration
In the extraction of Pd(II) and Pt(II) with compound 2, E% decreased and increased, respectively, with increasing [HCl] (0.001 to 0.5 M) (Fig. 1). This suggests that compound 2 forms extraction species with Pd(II) and Pt(II), accompanied by proton dissociation and addition, respectively. Considering this, the dependence of E% on [Cl−] was investigated by the addition of LiCl to the aqueous phase (Fig. 2); [H+] was fixed to 0.5 M using HCl. As [Cl−] increased, E% of Pd(II) decreased significantly (Fig. 2a), while that of Pt(II) decreased only slightly (Fig. 2b). It has been reported that Pd(II) and Pt(II) exist mainly as chloro complexes formulated as PdClx(x−2)− and PtClx(x−2)− (x = 3, 4), respectively, in HCl solutions with concentrations above 0.01 M.42 Therefore, it is concluded that Pd(II) is extracted via the cation exchange mode, with the dissociation of chloride ions from Pd(II) and the dissociation of protons from compound 2. On the other hand, Pt(II) seems to be extracted by protonated compound 2 as an anionic chloro complex via the solvation and/or anion exchange mode. This is also supported by the dependence of E% on [H+], wherein [Cl−] was fixed to 0.04 M by varying the HCl and LiCl concentrations (Fig. S1†).
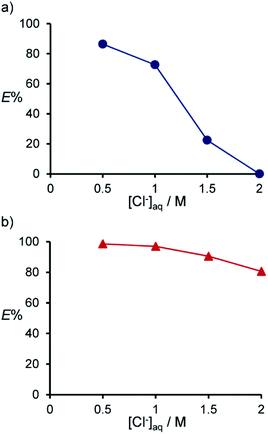 |
| | Fig. 2 Dependence of E% on [Cl−] for the extraction of Pd(II) (a) and Pt(II) (b) with compound 2. Aq. phase: [Metal]aq,init = 1.0 × 10−4 M, [HCl] = 0.5 M. [Cl−] was adjusted by the addition of LiCl. Org. phase: CHCl3, [2]org,init = 1.0 × 10−3 M. | |
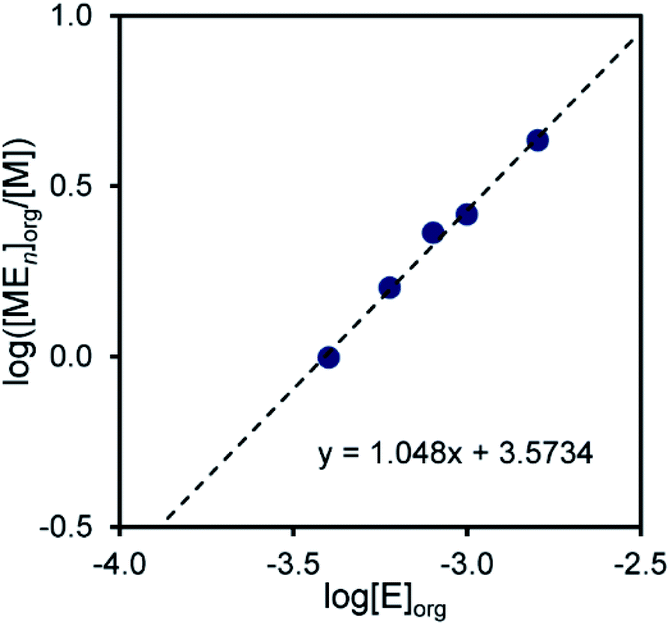
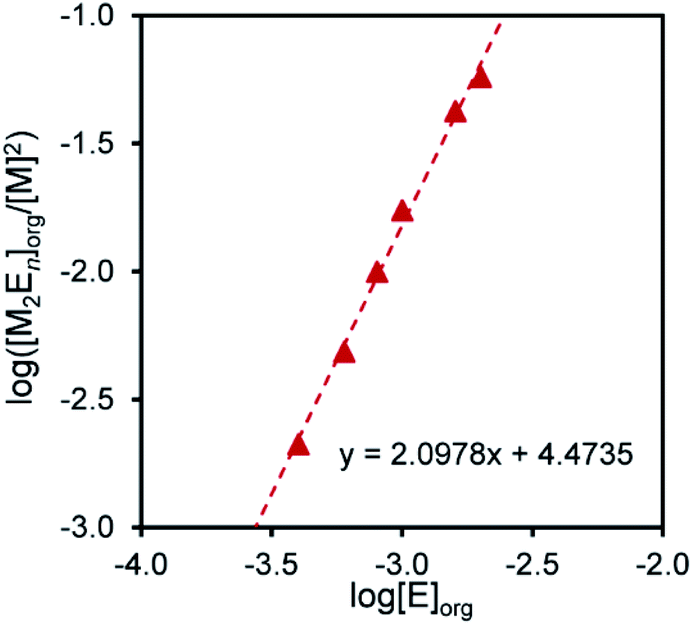
Equilibrium analysis for extraction of Pd(II) and Pt(II)
Equilibrium analysis was performed to determine the molar ratios of the extractant to the metal ions in the extraction species. When an extractant E and a metal species M form a complex with an M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E molar ratio of m:n, the extraction formula omitting the involvement of protons and chloride ions can be represented by eqn (2).where the subscript ‘‘org’’ denotes the species in the organic phase.
:E molar ratio of m:n, the extraction formula omitting the involvement of protons and chloride ions can be represented by eqn (2).where the subscript ‘‘org’’ denotes the species in the organic phase.
The extraction constant (Kex) is given by eqn (3), which can be rewritten as eqn (4).
| | |
Kex = [MmEn]org/[M]m[E]orgn
| (3) |
| | |
log([MmEn]org/[M]m) = logKex+nlog[E]org
| (4) |
Therefore, n is given as the slope of the log([MmEn]org/[M]m) vs. log[E]org plot; m is assumed to be a natural number. When m = 1 for Pd(II) and m = 2 for Pt(II), linear relationships were obtained, giving slopes of 1 and 2, respectively. This indicates that the molar ratios of compound 2 to Pd(II) and Pt(II) in the extraction species are 1:1 and 2:2, respectively (Fig. 3 and 4).
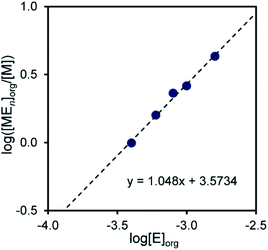 |
| | Fig. 3 Log([MEn]org/[M]) vs. log[E]org plot for the extraction of Pd2+ with compound 2. Aq. phase: [Metal]aq,init = 1.0 × 10−4 M, [HCl]aq = 1.0 × 10−3 M, [LiCl]aq = 0.05 M. Org. phase: CHCl3, [2]org,init = 4.0 × 10−4–1.6 × 10−3 M. | |
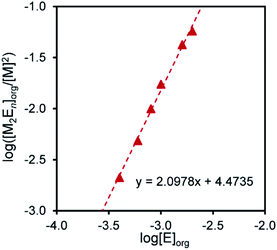 |
| | Fig. 4 Log([M2En]/[M]2) vs. log[E]org for the extraction of Pt2+ with compound 2. Aq. phase: [Metal]aq,init = 1.0 × 10−4 M, [HCl] = 1 M, [LiCl]aq = 0.5 M. Org. phase: CHCl3, [2]org,init = 4.0 × 10−4–2.0 × 10−3 M. | |
X-ray crystallography of Pd(II) and Pt(II) complexes
To gain insight into the structures of the extraction species, Pd(II) and Pt(II) complexes of compound 2 (H2L) were prepared and analyzed by X-ray crystallography. A Pd(II) complex could be prepared by the treatment of compound 2 with 1 molar equiv. of palladium acetate in chloroform. Vapor diffusion of acetonitrile into a chloroform solution of the complex gave single crystals formulated as [PdL](CH3CN)3.67. On the other hand, a Pt(II) complex could be prepared by the treatment of compound 2 with 1 molar equiv. of K2PtCl4 in a mixed solvent of chloroform, methanol and 0.5 M HCl. Vapor diffusion of ethanol to a 1,2-dichloroethane solution of the complex gave single crystals formulated as [Pt2Cl6(H3L)2](C2H5OH)5(CH2ClCH2Cl). These complexes have M:L ratios consistent with the results of the equilibrium analysis (vide supra), strongly suggesting that they are identical to the extraction species. In preparation of Pd(II) complex, however, palladium acetate was used instead of palladium chloride. Therefore, the solvent extraction of Pd(II) (1 molar equiv. for 2) from 0.001 M HCl with CDCl3 solution of compound 2 was performed. 1H NMR spectrum of CDCl3 solution after extraction indicated that [PdL] also forms as major complex from hydrochloric acid solution of Pd(II) (Fig. S2†).
The crystal of the Pd(II) complex belongs to the trigonal system with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group (Fig. 5a). A Pd ion is located on a C2 symmetry axis of deprotonated compound 2 (L2−). The two phenoxide oxygens and two amino nitrogens coordinate to the Pd ion, forming an ideal four-coordinated planar structure (Fig. 5b). On the other hand, the crystal of the Pt(II) complex belongs to the monoclinic system with the P21/c space group (Fig. 6a). In a complex, two anionic trichloro complexes PtCl3− are sandwiched between two H3L+, in which one amino group of H2L is protonated. The complex has a Ci symmetry with an inversion center at the midpoint of the two Pt centers. One amino nitrogen of a H3L+ directly coordinates to a PtCl3− species, forming a four-coordinated planar structure around the metal center (Fig. 6b). Another protonated amino group of the H3L+ forms an ion pair with another PtCl3−; hydrogen bonds are observed between two pairs of NH and Cl.
c space group (Fig. 5a). A Pd ion is located on a C2 symmetry axis of deprotonated compound 2 (L2−). The two phenoxide oxygens and two amino nitrogens coordinate to the Pd ion, forming an ideal four-coordinated planar structure (Fig. 5b). On the other hand, the crystal of the Pt(II) complex belongs to the monoclinic system with the P21/c space group (Fig. 6a). In a complex, two anionic trichloro complexes PtCl3− are sandwiched between two H3L+, in which one amino group of H2L is protonated. The complex has a Ci symmetry with an inversion center at the midpoint of the two Pt centers. One amino nitrogen of a H3L+ directly coordinates to a PtCl3− species, forming a four-coordinated planar structure around the metal center (Fig. 6b). Another protonated amino group of the H3L+ forms an ion pair with another PtCl3−; hydrogen bonds are observed between two pairs of NH and Cl.
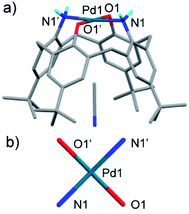 |
| | Fig. 5 X-ray structure of [PdL](CH3CN)3.67 (a) and the primary coordination sphere of Pd1 (b). Acetonitrile molecules and hydrogen atoms except for the amino groups are omitted for clarity. Selected distances (Å) and angles (°): N1–Pd1 2.0480(18), O1–Pd1 2.0242(15), N1–Pd1–O1 89.67(7), N1–Pd1–O1′ 90.32(7). | |
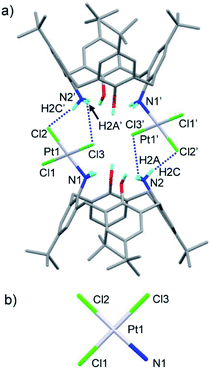 |
| | Fig. 6 X-ray structure of [Pt2Cl6(H3L)2]·(C2H5OH)3(CH2ClCH2Cl) (a) and the primary coordination sphere of Pt1 (b). Solvent molecules, hydrogen atoms except for the hydroxy and amino groups and disordered atoms are omitted for clarity. Dotted lines represent hydrogen bond. Selected distances (Å) and angles (°): N1–Pt1 2.079(10), Cl1–Pt1 2.284(4), Cl2–Pt1 2.309(4), Cl3–Pt1 2.298(4), H2A–Cl3′ 2.912, H2C–Cl2′ 2.228, N1–Pt1–Cl1 92.4(3), Cl1–Pt1–Cl2 89.51(13), Cl2–Pt1–Cl3 90.63(14), Cl3–Pt1–N1 87.4(3). | |
Mechanistic consideration for extraction of Pd(II) and Pt(II)
Based on the above observations, the extraction equilibria of Pd(II) and Pt(II) with compound 2 are shown in eqn (5) and (6), respectively, taking into account the above-mentioned report that Pd(II) and Pt(II) exist as anionic species coordinated by three or four chloride ions in aqueous HCl solutions.42| | |
H2Lorg + PdClx(x−2)− ⇄ [PdL]org + 2H+ + xCl−
| (5) |
| | |
2H2Lorg + 2PtClx(x−2)− + 2H+ ⇄ [Pt2Cl6(H3L)2]org + 2(x − 3)Cl−
| (6) |
The difference in the extraction equilibrium between Pd(II) and Pt(II) is rationalized as follows. First, the two amino groups of compound 2 along with the two hydroxy groups that are transformed into oxide groups on complexation, are pre-organized to fit the coordination space of the planar tetracoordinated structure of Pd(II). The ease of chloride ion exchange for Pd(II) also helps the formation of PdL. On the other hand, chloro complexes of Pt(II) are hard to dissociate the chloride ions, making it difficult to form a similar PtL complex. Second, the two amino groups of compound 2 are located at a distance that allows the amino groups to coordinate separately to a Pt(II) center and to form an ion pair with PtCl3−, resulting in two H3L+ molecules facing each other in a tail-to-tail manner to form a stable sandwich structure. On the other hand, since Pd(II) is a harder ion than Pt(II) in the hard and soft acids and bases (HSAB) principle,43 such a dinuclear complex is not likely to be stabilized only with a N–Pd bond and two NH–Cl hydrogen bonds between each H3L+ and two PdCl3− ions.
Switching the extraction selectivity for Pd(II) and Pt(II)
Taking advantage of the difference in the extraction mode, switching of the extraction selectivity was achieved in the competitive extraction between Pd(II) and Pt(II) (Fig. 7). Compound 2 selectively extracted Pd(II) over Pt(II) from a 0.001 M HCl solution, in which cation exchange is advantageous. On the other hand, Pt(II) was selectively extracted from a 0.5 M HCl solution containing 1.5 M LiCl, despite the fact that both Pd(II) and Pt(II) were extracted from 0.5 M HCl solutions in the noncompetitive experiments (Fig. 1). This suggests that an increasing chloride ion concentration inhibits Pd(II) extraction via the cation exchange mode. On the other hand, significant decrease in E% for both Pd(II) and Pt(II) were observed when KBr or LiNO3 was added in aqueous phase, suggesting that high concentrations of the chloride ion are required for the selective extraction of Pt(II) under the competitive conditions with Pd(II) (Fig. S3 and S4†).
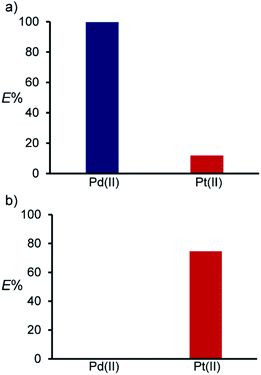 |
| | Fig. 7 Competitive extraction between Pd(II) and Pt(II) from 0.001 M HCl (a) and 0.5 M HCl with LiCl (1.5 M) (b). Aq. phase: [Metal]aq,int = 1.0 × 10−4 M (for each metal ion). Org. phase: CHCl3, [2]org,init = 1.0 × 10−3 M. | |
Back-extraction of Pd(II) and Pt(II)
Finally, back-extraction was investigated. Pd(II) and Pt(II) were extracted from 0.001 M HCl and 0.5 M HCl, respectively, using a chloroform solution of compound 2. The metal ions extracted into the organic phase were back-extracted by shaking the organic phase with an aqueous HCl solution at ambient temperature for 24–48 h. The concentration of the metal ions remaining in the aqueous phase, 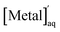 , was measured by ICP-OES, and the back-extraction percentage (E%back) was calculated according to the following equation:
, was measured by ICP-OES, and the back-extraction percentage (E%back) was calculated according to the following equation:| |
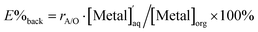 | (7) |
where rA/O indicates the volume ratio of the aqueous phase to the organic phase in the back-extraction. The results are listed in Table 1. Pt(II) was quantitatively back-extracted by the treatment with 4 M HCl in a volume ratio of 2 (entry 7). While E%back of Pd(II) with 4–6 M HCl was only moderate (entries 1–3), quantitative back-extraction could be achieved when 0.5 M thiourea was added to 4 M HCl (entry 4).
Table 1 Back-extraction of Pd(II) and Pt(II) with aqueous HCla
| Entry |
Metal |
HCl (M) |
rA/O |
Time (h) |
E%back |
| Forward extraction was conducted under the following conditions. Aq. phase: [Metal]aq,init = 1.0 × 10−4 M, [HCl] = 0.001 M for Pd(II) and 0.5 M for Pt(II). Org. phase: CHCl3, [2]org,init = 1.0 × 10−3 M. Thiourea (0.5 M) was added. |
| 1 |
Pd(II) |
4 |
1 |
24 |
44 |
| 2 |
4 |
2 |
24 |
43 |
| 3 |
6 |
2 |
24 |
45 |
| 4 |
4b |
2 |
48 |
100 |
| 5 |
Pt(II) |
4 |
1 |
24 |
89 |
| 6 |
4 |
2 |
24 |
91 |
| 7 |
4 |
2 |
48 |
100 |
Conclusions
1,3-Diaminocalix[4]arene 2 extracted Pd(II) and Pt(II) by forming complexes with completely different structures, which were impacted by the nature of the amino group, the arrangement of the two amino and two hydroxyl groups and the rigid calixarene skeleton. Owing to the difference in the extraction modes, the extraction selectivities of Pd(II) and Pt(II) could be switched by altering the extraction conditions. This allows Pt(II) to be extracted even in the presence of Pd(II), which is usually preferentially extracted over other PGMs. From the extracted organic phase, back-extraction of Pd(II) and Pt(II) was easily performed. We expect that molecular design based on our synthetic protocol for the direct replacement of the hydroxy groups of calix[4]arenes with other coordinating groups35–38 will enable the development of selective extractants for various metals including other PGMs.
Experimental
General
Inductively coupled plasma optical emission spectra (ICP-OES) were measured using a Thermo Fisher Scientific iCAP6500. 1H NMR spectra were measured on a 400 MHz spectrometer using CDCl3 and tetramethylsilane as a solvent and internal standard, respectively. Compound 2 was prepared as previously.36 Reagent-grade chemicals and solvents were used as purchased. Stock solutions of PGM [Pd(II), Pt(II), Pt(IV), Ir(IV) and Rh(III)] were prepared by dissolving corresponding metal chlorides in 0.01 M HCl, respectively.
Solvent extraction
General procedure for the extraction experiment is as follows: To a 30 cm3 vial tube were pipetted a solution of compound 2 ([2]org,init = 1.0 × 10−3 M) in chloroform (5 cm3) and an aqueous solution (5 cm3) containing HCl (0.001–7 M) and PGM ([Metal]aq,init = 1.0 × 10−4 M). The mixture was shaken for 24 h at 300 strokes per min at room temperature (20 ± 3 °C). After the aqueous phase was separated by centrifugation, the concentration of the metal species remaining in the aqueous phase, [Metal]aq, was measured by ICP-OES. The percentage of extraction (E%) was calculated according to eqn (1).
Preparation of metal complexes of compound 2 (H2L) with Pd(II) and Pt(II)
[PdL]: To a solution of compound 2 (H2L) (30.0 mg, 46.3 mmol) in chloroform (5 mL) was added palladium(II) acetate (10.4 mg, 46.2 mmol), and the mixture was stirred at ambient temperature for 18 h. Evaporation of the solvent left a residue, which was suspended in acetonitrile (3 mL). The precipitate was collected by filtration and dried in vacuo to afford complex [PdL] (13.8 mg, 40%) as an orange powder. Mp: 272.6–277.9 °C (decomp.); 1H NMR (400 MHz, CDCl3) δ 1.21 (s, 18H), 1.23 (s, 18H), 3.40 (d, 4H, J = 12.4 Hz), 4.10 (brs, 4H), 5.96 (brs, 4H) 7.00 (s, 4H), 7.10 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 31.3, 31.4, 34.0, 34.3, 34.8, 125.3, 125.3, 126.1, 129.3, 132.4, 132.7, 140.4, 147.0. IR (KBr) 2958, 1581, 1481, 1462, 1203 cm−1. HRMS (ESI) calcd for C44H56N2O2Pd ([PdL] + H+): 751.34493. Found: 751.34449.
[Pt2Cl6(H3L)2]: To a solution of compound 2 (H2L) (30.0 mg, 46.3 mmol) in chloroform/methanol (1:3, 12 mL), was added aqueous solution (3 mL) containing 0.5 M HCl and K2PtCl4 (19.4 mg, 46.8 mmol). The mixture was stirred at ambient temperature for 18 h. Precipitations were collected, washed with methanol (10 mL) and dried in vacuo to afford complex [Pt2Cl6(H3L)2] (30.1 mg, 68%) as a yellow powder. Mp: 257.1–260.7 °C (decomp.); 1H NMR (400 MHz, CDCl3) δ 1.11 (s, 9H), 1.17 (s, 9H), 1.22 (s, 18H), 3.54 (d, 2H, J = 14.2 Hz), 3.66 (d, 2H, J = 13.4 Hz), 4.67 (d, 2H, J = 14.2 Hz), 5.19 (d, 2H, J = 13.4 Hz), 6.97 (s, 2H), 7.03 (s, 2H), 7.10 (s, 2H), 7.15 (s, 2H), 7.88 (brs, 2H), 8.42 (brs, 2H), 7.10 (brs, 2H), 7.14 (brs, 2H), 7.91 (brs, 2H), 8.38 (brs, 2H), 10.10 (brs, 3H); 13C NMR (100 MHz, CDCl3) δ 30.9, 31.0, 31.4, 34.1, 34.2, 34.5, 34.5, 34.7, 125.2, 125.9, 126.5, 126.8, 132.2, 132.2, 133.1, 134.3, 136.3, 144.9, 144.9, 147.5, 148.1, 152.3. IR (KBr) 3483, 3244, 2958, 1601, 1481, 1192 cm−1. HRMS (ESI) calcd for C88H117Cl6N4O4Pt ([Pt2Cl6(H3L)2]–H+): 1897.64841. Found: 1897.64804.
Single-crystal XRD analysis of metal complexes
Vapor diffusion of acetonitrile to a solution of [PdL] in chloroform gave single crystals formulated as [PdL]·(CH3CN)3.67. Vapor diffusion of ethanol to a solution of [Pt2Cl6(H3L)2] in 1,2-dichloroethane gave single crystals formulated as [Pt2Cl6(H3L)2]·(C2H5OH)5(CH2ClCH2Cl). The X-ray diffraction data was collected on a Bruker APEX-II with a CCD diffractometer equipped with a multi-layered confocal mirror and a fine focus rotating anode, using Mo-Kα radiation (λ = 0.71073 Å). The structure was solved by the direct method by SHELXL-97 (ref. 44) and refined by the least-squares method on F2 using SHELXL-2017.45 Yadokari-XG 2009 was used as a GUI for SHELXL-97 and SHELXL-2017.46,47
Data for [PdL]·(CH3CN)3.67: C51.33H67N5.67O2Pd, fw = 901.81, trigonal, Rc, a = 20.518(3) Å, b = 20.518(3) Å, c = 60.941(13) Å, V = 22218(8) Å3, Z = 18, T = 100(2) K, 40824 reflections measured, 5690 independent reflections, 4837 reflections were observed (I > 2σ(I)), R1 = 0.0360, wR2 = 0.0976 (observed), R1 = 0.0459, wR2 = 0.1072 (all data).
Data for [Pt2Cl6(H3L)2]·(C2H5OH)5(CH2ClCH2Cl): C100H152Cl8N4O9Pt2, fw = 2228.03, monoclinic, P21/c, a = 19.698(7) Å, b = 12.374(4) Å, c = 22.309(7) Å, β = 103.164(5)°, V = 5295(3) Å3, Z = 2, T = 100(2) K, 29971 reflections measured, 12035 independent reflections, 6025 reflections were observed (I > 2σ(I)), R1 = 0.0821, wR2 = 0.1887 (observed), R1 = 0.1785, wR2 = 0.2454 (all data).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Number 15K05466 and Iketani Science and Technology Foundation.
References
- F. L. Bernardis, R. A. Grant and D. C. Sherrington, React. Funct. Polym., 2005, 65, 205–217 CrossRef CAS.
- B. Gupta and I. Singh, Hydrometallurgy, 2013, 134–135, 11–18 CrossRef CAS.
- T. H. Nguyen, C. H. Sonu and M. S. Lee, Hydrometallurgy, 2016, 164, 71–77 CrossRef CAS.
- H. Narita, T. Suzuki and R. Motokawa, Nippon Kinzoku Gakkaishi, 2017, 81, 157–167 CAS.
- C. Saguru, S. Ndlovu and D. Moropeng, Hydrometallurgy, 2018, 182, 44–56 CrossRef CAS.
- H. Narita, K. Morisaku and M. Tanaka, Chem. Commun., 2008, 5921–5923 RSC.
- P. Malik and A. P. Paiva, Solvent Extr. Ion Exch., 2010, 28, 49–72 CrossRef CAS.
- A. Cieszynska and M. Wisniewski, Hydrometallurgy, 2012, 113–114, 79–85 CrossRef CAS.
- J. Traeger, J. König, A. Städtke and H.-J. Holdt, Hydrometallurgy, 2012, 127–128, 30–38 CrossRef CAS.
- R. Poirot, D. Bourgeois and D. Meyer, Solvent Extr. Ion Exch., 2014, 32, 529–542 CrossRef CAS.
- H. Narita, K. Morisaku, K. Tamura, M. Tanaka, H. Shiwaku, Y. Okamoto, S. Suzuki and T. Yaita, Ind. Eng. Chem. Res., 2014, 53, 3636–3640 CrossRef CAS.
- V. T. Nguyen, J.-c. Lee, A. Chagnes, M.-s. Kim, J. Jeong and G. Cote, RSC Adv., 2016, 6, 62717–62728 RSC.
- M. C. Costa, R. Almedia, A. Assuncão, A. M. R. Costa, C. Nogueria and A. P. Paiva, Sep. Purif. Technol., 2016, 158, 409–416 CrossRef CAS.
- A. P. Pavia, O. Ortet, G. I. Carvalho and C. A. Nogueira, Hydrometallurgy, 2017, 171, 394–401 CrossRef.
- K. Senthil, U. Akiba, K. Fujiwara, F. Hamada and Y. Kondo, Ind. Eng. Chem. Res., 2017, 56, 1036–1047 CrossRef CAS.
- M. L. Firmansyah, F. Kubota and M. Goto, J. Chem. Technol. Biotechnol., 2018, 93, 1714–1721 CrossRef CAS.
- A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123–134 RSC.
- F. Kubota, E. Shigyo, W. Yoshida and M. Goto, Solvent Extr. Res. Dev., Jpn., 2017, 24, 97–104 CrossRef.
- K. Matsumoto, S. Yamakawa, K. Haga, K. Ishibashi, M. Jikei and A. Shibayama, Sci. Rep., 2019, 9, 12414–12421 CrossRef.
- K. Matsumoto, S. Yamakawa, Y. Sezaki, H. Katagiri and M. Jikei, ACS Omega, 2019, 4, 1868–1873 CrossRef CAS.
- B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893–962 CrossRef CAS.
- K. Ohto, Solvent Extr. Res. Dev., Jpn., 2010, 17, 1–8 CrossRef CAS.
- M. Yamada, M. Rjiv Gandhi, K. Uma Maheswara Rao and F. Hamada, J. Inclusion Phenom. Macrocyclic Chem., 2016, 85, 1–18 CrossRef CAS.
- V. J. Mathew and S. M. Khopkar, Talanta, 1997, 44, 1699–1703 CrossRef CAS.
- A. T. Yordanow, O. M. Falana, H. F. Koch and D. M. Roundhill, Inorg. Chem., 1997, 36, 6468–6471 CrossRef.
- A. T. Yordanov, B. R. Whittlesey and D. M. Roundhill, Inorg. Chem., 1998, 37, 3526–3531 CrossRef CAS.
- C. Fontàs, E. Anticó, F. Vocanson, R. Lamartine and P. Seta, Sep. Purif. Technol., 2007, 54, 322–328 CrossRef.
- V. G. Torgov, G. A. Kostin, V. I. Mashukov, T. M. Korda, A. B. Drapailo, O. V. Kas’yan and V. I. Kal’chenko, Russ. J. Inorg. Chem., 2008, 53, 1809–1815 CrossRef.
- V. G. Torgov, T. V. Us, T. M. Korda, G. A. Kostin and V. I. Kalchenko, Russ. J. Inorg. Chem., 2012, 57, 1261–1269 CrossRef.
- V. G. Torgov, G. A. Kostin, T. M. Korda, E. A. Guskova, V. I. Mashukov and V. I. Kalchenko, Russ. Chem. Bull. Int. Ed., 2012, 61, 1437–1444 CrossRef CAS.
- M. R. Gandhi, M. Yamada, Y. Kondo, A. Shibayama and F. Hamada, Hydrometallurgy, 2015, 151, 133–140 CrossRef.
- V. G. Torgov, T. V. Us, S. A. Lavrukhina, N. I. Petrova, L. N. Mazalov and A. V. Kalinkin, Russ. J. Inorg. Chem., 2017, 62, 854–861 CrossRef CAS.
- M. Yamada, M. R. Gandhi, Y. Kaneta, Y. Hu and A. Shibayama, ChemistrySelect, 2017, 2, 1052–1057 CrossRef CAS.
- M. Yamada, Y. Kaneta, M. R. Gandhi and U. M. R. Kunda, Hydrometallurgy, 2019, 184, 103–108 CrossRef CAS.
- S. Tanaka, R. Serizawa, N. Morohashi and T. Hattori, Tetrahedron Lett., 2007, 48, 7660–7664 CrossRef CAS.
- Y. Nakamura, S. Tanaka, R. Serizawa, N. Morohashi and T. Hattori, J. Org. Chem., 2011, 76, 2168–2179 CrossRef CAS.
- S. Tanaka, T. Umetsu, S. Nebuya, N. Morohashi and T. Hattori, J. Org. Chem., 2015, 80, 1070–1081 CrossRef CAS.
- K. Hirasawa, S. Tanaka, T. Horiuchi, T. Kobayashi, T. Sato, N. Morohashi and T. Hattori, Organometallics, 2016, 35, 420–427 CrossRef CAS.
- N. Morohashi, T. Hayashi, Y. Nakamura, T. Kobayashi, S. Tanaka and T. Hattori, Chem. Lett., 2012, 41, 1520–1522 CrossRef CAS.
- N. Morohashi, Y. Kato, S. Sumida, Y. Kurusu and T. Hattori, Bull. Chem. Soc. Jpn., 2019, 92, 967–972 CrossRef CAS.
- N. Morohashi, S. Iijima, K. Akasaka and T. Hattori, New J. Chem., 2017, 41, 2231–2234 RSC.
- C. Colombo, C. J. Oates, A. J. Monhemius and J. A. Plant, Geometry: Exploration, Environment, Analyst, 2008, 8, 1–11 Search PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- G. M. Sheldrick, SHELEX-97, Programs for the Refinement of Crystal structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Wakita, Yadokari-XG, Software for Crystal Structure Analyses, 2001 Search PubMed.
- C. Kabuto, S. Akine, T. Nemoto and E. Kwon, Nippon Kessho Gakkaishi, 2009, 51, 218–224 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Data of solvent extraction and crystallographic data. CCDC 2006761 and 2006762. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05339b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yu Kurusu,
Kosuke Akasaka and
Tetsutaro Hattori
*,
Yu Kurusu,
Kosuke Akasaka and
Tetsutaro Hattori





 , was measured by ICP-OES, and the back-extraction percentage (E%back) was calculated according to the following equation:
, was measured by ICP-OES, and the back-extraction percentage (E%back) was calculated according to the following equation: