
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAldehyde catalysis – from simple aldehydes to artificial enzymes
Zeqin Yuan
a,
Jun Liao
a,
Hao Jiang
b,
Peng Cao
a and
Yang Li
 *a
*a
aCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu 610068, China. E-mail: yangli@sicnu.edu.cn
bUndisclosed Pharmaceutical Company, Copenhagen, Denmark
First published on 25th September 2020
Abstract
Chemists have been learning and mimicking enzymatic catalysis in various aspects of organic synthesis. One of the major goals is to develop versatile catalysts that inherit the high catalytic efficiency of enzymatic processes, while being effective for a broad scope of substrates. In this field, the study of aldehyde catalysts has achieved significant progress. This review summarizes the application of aldehydes as sustainable and effective catalysts in different reactions. The fields, in which the aldehydes successfully mimic enzymatic systems, include light energy absorption/transfer, intramolecularity introduction through tether formation, metal binding for activation/orientation and substrate activation via aldimine formation. Enantioselective aldehyde catalysis has been achieved with the development of chiral aldehyde catalysts. Direct simplification of aldehyde-dependent enzymes has also been investigated for the synthesis of noncanonical chiral amino acids. Further development in aldehyde catalysis is expected, which might also promote exploration in fields related to prebiotic chemistry, early enzyme evolution, etc.
 Zeqin Yuan | Zeqin Yuan was born in Sichuan Province, China in 1995. In 2019, he received bachelor's degree in chemistry from Kashgar University. In September of the same year, he started his study at Sichuan Normal University for a master's degree. His research direction is organic synthesis methodology. His research interest is focused on developing new synthetic methods of chiral organophosphorus and boron compounds. |
 Jun Liao | Jun Liao received his bachelor's degree in Chemistry in 2018 at Qilu University of Technology. In the same year he started his study for a master degree at Sichuan Normal University under the supervision of professor Peng Cao. Jun Liao mainly studies organic chemistry and asymmetric catalysis. |
 Hao Jiang | Hao Jiang received his PhD degree in organic chemistry in 2011 under the supervision of Prof. Karl Anker Jørgensen at the Center for Catalysis, Aarhus University. His academic research primarily focused on the development of new methodologies in asymmetric catalysis and organic synthesis. Currently, he holds an industrial managerial position in the pharmaceutical industry primarily working with process implementation in CMC chemical pilot plants. |
 Peng Cao | Peng Cao completed his undergraduate degree at Sichuan Normal University in 2004 and received a PhD in chemistry at SIOC (Shanghai, China) with Prof. Yong Tang and Prof. Zuo-Wei Xie. He moved to a postdoctoral position at CaRLa (Heidelberg, Germany), a joint research laboratory of Heidelberg University and BASF. He then returned to China with a position as associate professor at CIB (Chengdu, China). From 2017, he started his research career at Sichuan Normal University. His research interests are focused on the cyanidation chemistry. |
 Yang Li | Dr. Yang Li received her BSc in Chemistry from Sichuan Normal University in 2011. She continued her postgraduate study on organic chemistry at Central China Normal University under the supervision of Prof. Wen-Jing Xiao. From 2012 to 2013 she was a visiting postgraduate student at Aarhus University in the group of Prof. Karl Anker Jørgensen. In 2018, she received her PhD in Chemistry from Aarhus University under the supervision of Prof. Karl Anker Jørgensen. Since 2018, she joined College of Chemistry and Materials Science, Sichuan Normal University and focused on academic research. Her current research interests include catalysis and organophosphorus chemistry. |
1 Introduction
Inspired by evolutionary optimized catalytic capabilities of enzymes, chemists have a long history of mimicking useful biocatalytic processes by application of low-molecular-weight organic compounds, trivially named organocatalysts. The field of organocatalysis plays an increasingly important role in organic synthesis,1 primarily due to its robust nature (air/water stable), low toxicity, and sustainability. Many catalytic modes have become well-established during the past decades, while several new modalities have recently surfaced. Herein, small molecule aldehydes have been revealed as competent mimics of biocatalytic processes.2 Aldehydes represent an important functionality in biological systems and are also involved in the research of prebiotic chemistry and investigation of life origination.3 Featured examples of aldehyde catalysis include their capability to mimic the modes of action of biological photocatalytic systems, proteases and PLP-dependent enzymes (Fig. 1, left).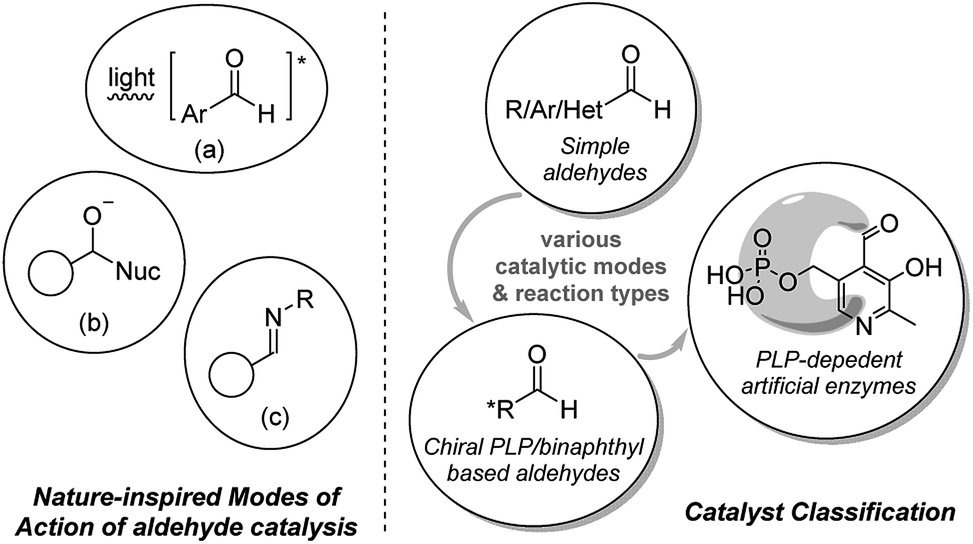 | ||
| Fig. 1 Representative examples of enzymatic mimicking of aldehyde catalysts (left); simple classification of the aldehyde catalysts (right). | ||
Metal complexes (e.g. chlorophylls) and organic molecules (e.g. carotenoids) are employed by nature for interaction with light.4 Accordingly, chemists have developed various transition metal containing compounds and organic dyes for light chemistries.1k,5 Simple low molecular weight aromatic aldehydes may serve as a green alternative. The π-conjugation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functioning and aromatic group enables aromatic aldehyde to become activated upon UV radiation (Fig. 1, left, (a)). The resulting species have been applied as useful photo-organocatalyt in important reactions such as isomerizations, atomtransfer radical additions, coupling reactions, polymerization and etc.2h
O functioning and aromatic group enables aromatic aldehyde to become activated upon UV radiation (Fig. 1, left, (a)). The resulting species have been applied as useful photo-organocatalyt in important reactions such as isomerizations, atomtransfer radical additions, coupling reactions, polymerization and etc.2h
The electrophilicity of the aldehyde group enables aldehydes to act as catalysts by interacting with substrate of nucleophilic nature, e.g. amines, alcohols, thiols, and nitriles. Upon the addition of nucleophiles, the aldehyde could transform to a tether intermediate bearing a hydroxyl group.2a,b By mimicking proteolysis6 (Fig. 1), this hydroxyl group activates either target functional groups on the substrate (e.g. amides) or nucleophiles (e.g. H2O), and facilitates the transformations of the functional groups (Fig. 1, left, (b)).
In the reaction with primary amine substrate, an aldimine intermediate could be generated.2c–g This successfully mimics PLP-dependent enzymes (PLP: pyridoxal phosphate) for the reactions of amino acid derivatives (Fig. 1, left, (c)).7 Except for simple aldehydes, relevant complex/chiral aldehydes and simplified enzymatic PLP are also worth mentioning along the developments in this field. By mimicking the mode of enzyme–substrate complementarity (“Lock-and-key” mode),8 the aldehyde catalyst function either alone or cooperate with metal which promotes an abundance of different types of reactions.
Understanding the properties of aldehydes is of great scientific significance that provides a source of inspiration for novel development of catalytic transformations. In this tutorial review, we aim to give an overview of the important synthetic applications, in which aldehydes play a role as catalysts (mainly with catalytic amount of the aldehydes). This review is structured by framing the aldehyde catalysts into the categories of structurally simple aldehydes, designed aldehydes with distinct structural framework (mainly focusing on PLP and binaphthyl-based system), and complex aldehyde-based artificial enzymes (Fig. 1, right). Modes of actions, applicability and aspects of future development will be discussed.
2 Simple aldehydes
Photo-organocatalytic reaction
Aldehydes are well known as photo-organocatalysts in organic synthesis. They are considered to be more sustainable and eco-friendly alternatives of metal-based photoredox catalysts.2h Seminal works were reported by Hammond and coworkers in early 1960s where they explored the use of aromatic aldehydes to catalyze the isomerization of cis-piperylene (Scheme 1).9 The trans to cis ratio of piperylene reached as high as 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the presence of 60 mol% 1-naphthaldehyde. The proposed mechanism involves the aldehyde catalyst being activated to the triplet stage upon UV irradiation. This in turn generates the triplet stage of the alkene through energy transfer (Paterno–Büchi reaction), which bridges the cis to trans isomerization.9b,10
:1 in the presence of 60 mol% 1-naphthaldehyde. The proposed mechanism involves the aldehyde catalyst being activated to the triplet stage upon UV irradiation. This in turn generates the triplet stage of the alkene through energy transfer (Paterno–Büchi reaction), which bridges the cis to trans isomerization.9b,10
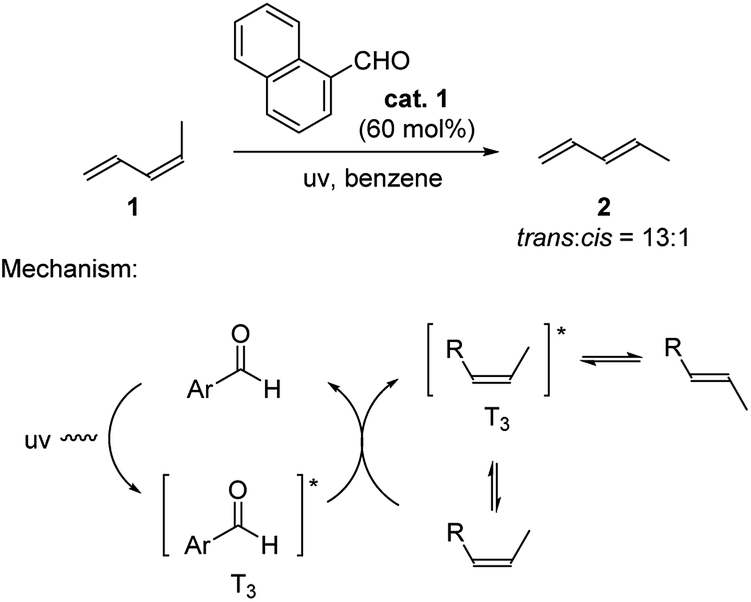 | ||
| Scheme 1 1-Naphthaldehyde catalyzed isomerization of cis-piperylene. | ||
This field caught limited attention for over 50 years. Nevertheless, it inspired the Melchiorre group to utilize p-anisaldehyde to catalyze atom-transfer radical addition (ATRA)11 of a variety of alkyl halides to olefins in 2014 (Scheme 2).12 The reaction was carried out under the irradiation by a 23 W compact fluorescent light (CFL) and giving generally high yields of the products. Examples of applying primary, secondary and tertiary halides (–Cl/–Br/–I) as substrates were provided. Terminal olefins, internal cyclic olefins, olefin containing naturally occurring compounds as well as alkynes were successfully applied in this reaction. The proposed mechanism involves the aldehyde catalyst being excited to singlet state (S1) through an n → π* transition by irradiation. Subsequently transformation to its triplet state (T1) through intersystem crossing (ISC)13 is followed by an energy transfer between the triplet aldehyde and halide substrate generating the triplet alkyl halide and reforming the ground state aldehyde. The triplet alkyl halide then undergoes a homolytic cleavage giving the alkyl radical which preformed the ATRA with the olefins.
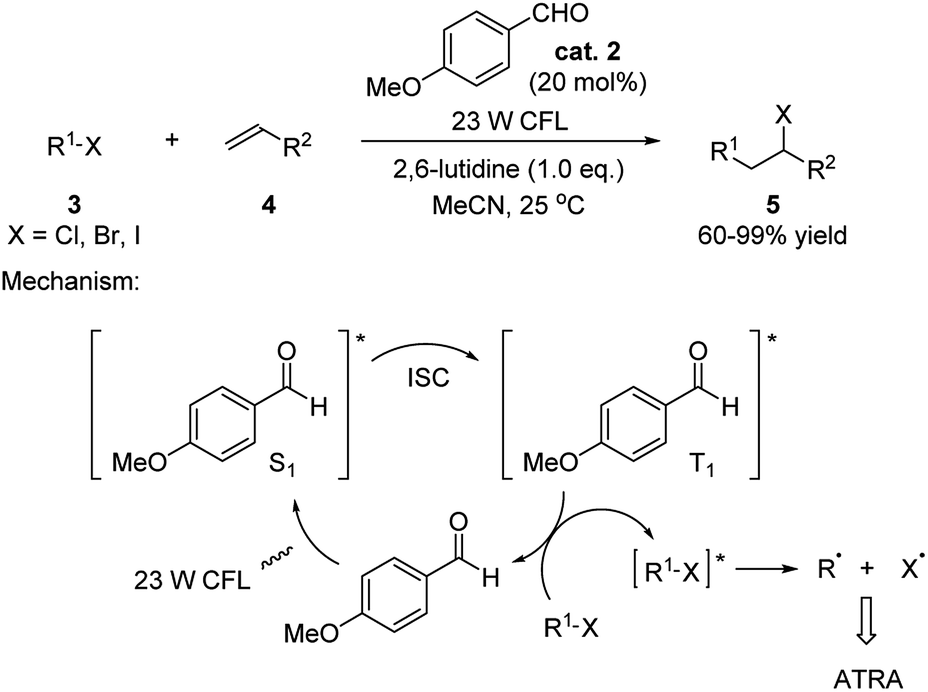 | ||
| Scheme 2 p-Anisaldehyde catalyzed ATRA of alkyl halides to olefins. | ||
In 2016, Ji and coworkers discovered that benzaldehyde largely enhanced the reactivity in the photoredox reaction for cross-dehydrogenative coupling (CDC) between amides and electron deficient heteroarenes.14 As shown in Scheme 3, the reaction of benzothiazole 6 and formamide occurred under 23 W CFL irradiation in the presence of radical initiator (NH4)2S2O8 and produced the product in 59% yield by NMR. The addition of 50 mol% benzaldehyde resulted in an increase of yield to 90%. With higher amount of benzaldehyde (1.0 eq.) and TsOH (1.0 eq.) as additives, the yield of the product was further improved to 99% yield by NMR (94% yield of isolated product). This reactivity was also extended to other heteroarenes, e.g. imidazoles and pyridines, N-alkylated amides and ethers.
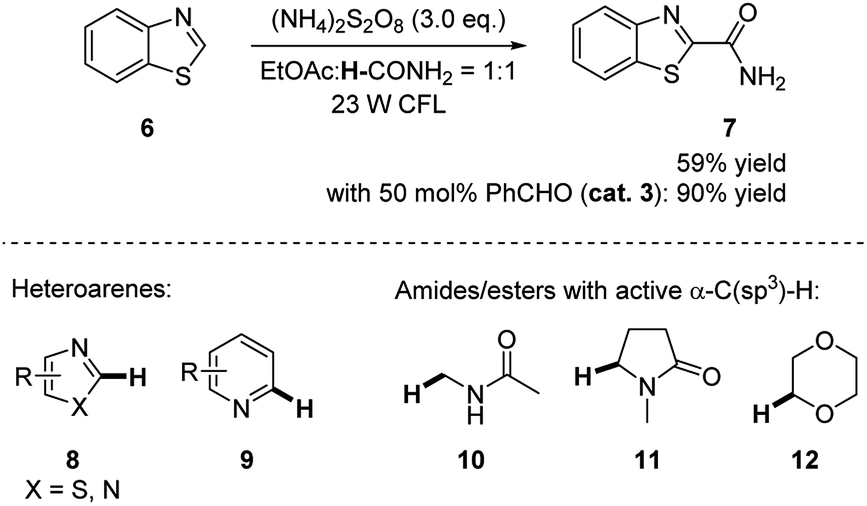 | ||
| Scheme 3 CDC of electron deficient heteroarenes with amides enhanced by benzaldehyde. | ||
Similar to a previously disclosed example utilizing metal-based photoredox catalyst,15 benzaldehyde was shown to be capable of initiating the formation of sulphate radicals in this reaction (Scheme 4). These sulphate radicals in turn oxidize amides to radical intermediate, which subsequently reacts with aromatic substrates by the CDC reaction. The proposed mechanism in full starts with benzaldehyde being illuminated to the photoexited benzaldehyde I and II upon the light irradiation. This promotes the generation of the sulphate radical while forming the possible intermediates III or IV. The sulphate radical then reacts with amide and forms the active amide radical. Subsequently, this amide radical reacts with the protonated heteroarene through nucleophilic addition forming intermediate V, which undergoes a deprotonation and is subsequently oxidized by III or IV to generate the desired product and reforming the aldehyde catalyst to complete the catalytic cycle.
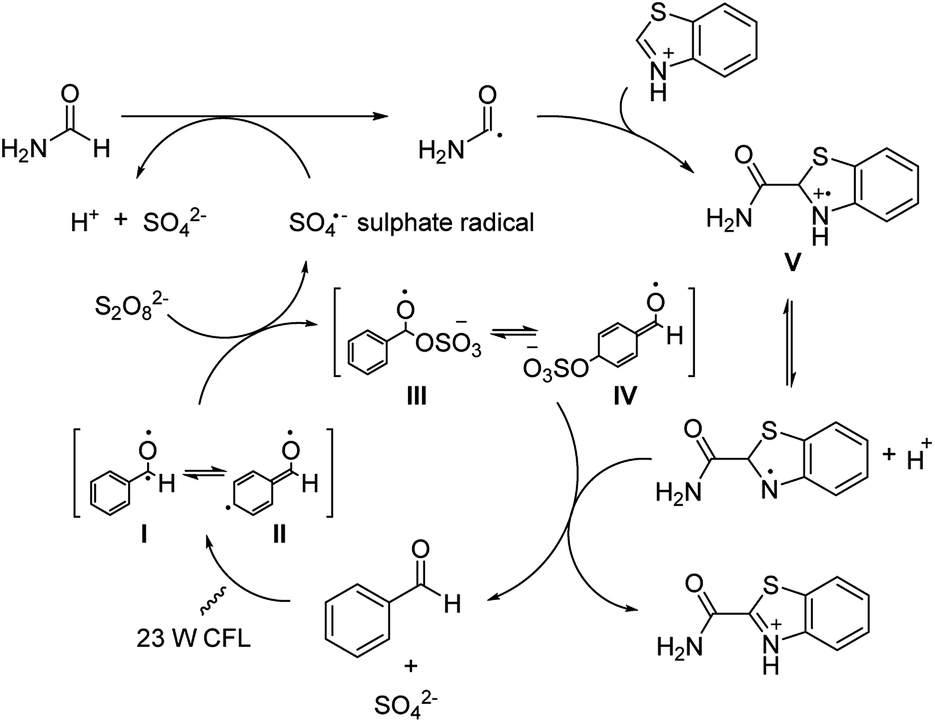 | ||
| Scheme 4 Mechanism of the benzaldehyde enhanced CDC. | ||
In 2019, Hashimi group published examples of benzaldehyde and nickel combined photoredox catalysis for α-C(sp3)–H alkylation/arylation with ethers, amides and thioethers (Scheme 5).16 Following the well-studied aldehyde's triplet exited state activation and direct hydrogen atom transfer (HAT) process, a radical intermediate I was suggested to be generated from the substrate. This radical intermediate thus reacted with aliphatic or aromatic bromide through nickel catalysis forming the coupling products in high yields.
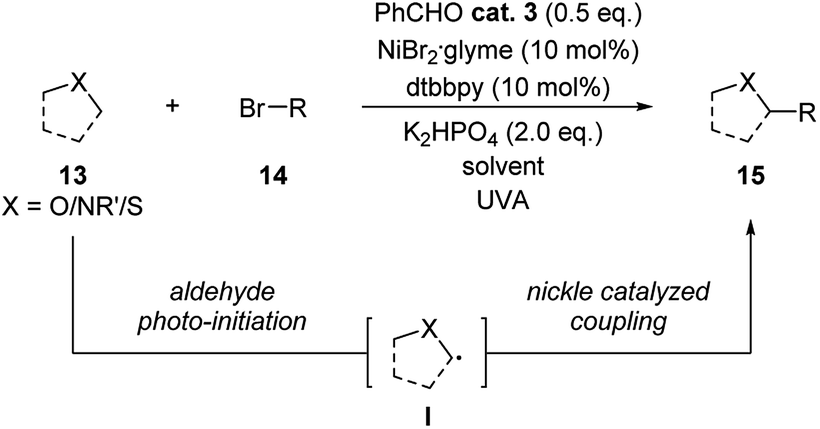 | ||
| Scheme 5 Combination of benzaldehyde and Ni catalysis in the coupling reaction of ethers/amides/thioethers and bromides. | ||
During the same year, with thorough screening of different aromatic aldehydes, Kokotos and coworkers realized the activation of the ortho-hydrogen of aliphatic aldehydes for the hydroacylation with electron-withdrawing alkenes (Scheme 6).17 The use of 10 mol% of p-cyanobenzaldehyde catalyzed the reaction efficiently under the irradiation by CFL lamps.
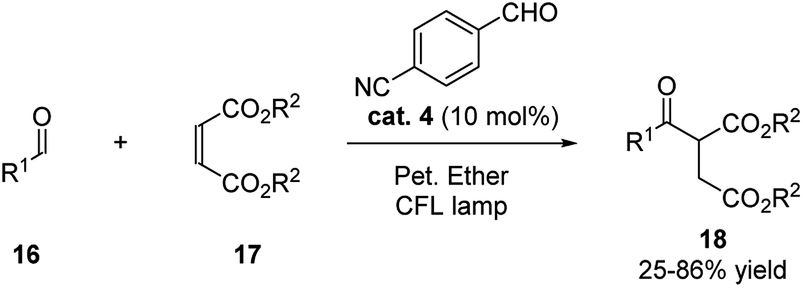 | ||
| Scheme 6 p-Cyanobenzaldehyde catalyzed hydroacylation of electron-withdrawing alkenes. | ||
In 2020, more examples were reported further demonstrating the great potential of aldehydes as privileged alternative catalyst in photo-catalysis. Landais and coworkers used p-anisaldehyde as photosensitizer in the sulfonylcynation of chiral cyclobutenes (Scheme 7).18 The reaction showed high efficiency when compared to the use of other organic dyes. Moreover, excellent diastereoselectivities were observed in these reactions.
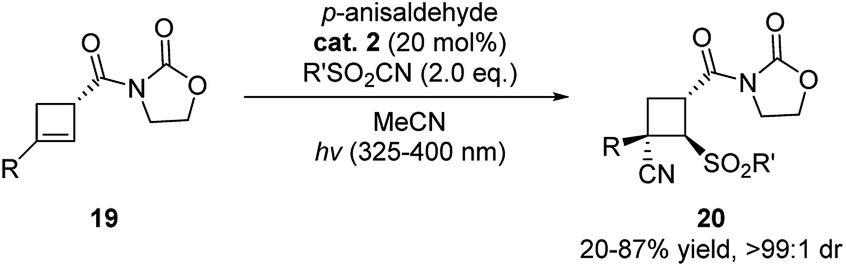 | ||
| Scheme 7 p-Anisaldehyde catalyzed sulfonylcynation of chiral cyclobutenes. | ||
Yajima and coworkers recently reported an example of ATRA of perfluoroalkyl iodides with alkenes or alkynes applying aldehyde catalysis (Scheme 8).19 Diphenylacetaldehyde, pyrrolidine and 23 W CFL were required for promoting this reaction. The iodoperfluoroalkylated products were generated in moderate to excellent yields. For the reaction with alkynes, the yields could be further improved to up to 100% when higher loading of diphenylacetaldehyde and pyrrolidine were used.
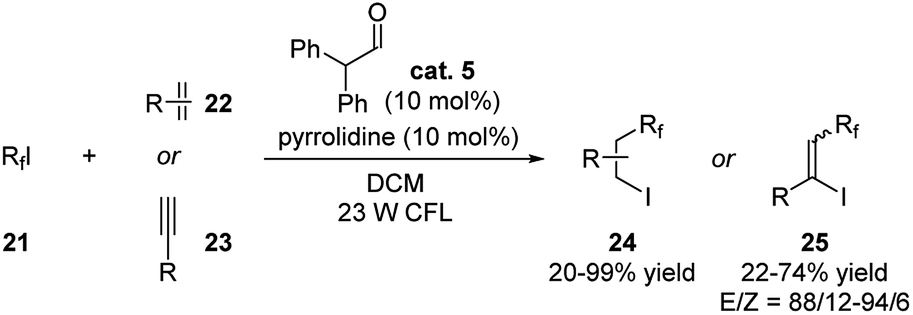 | ||
| Scheme 8 Diphenylacetaldehyde catalyzed ATRA of perfluoroalkyl iodides with alkenes/alkynes. | ||
The reaction was proposed to proceed through a cooperative catalytic pathway with enamine and amine (Scheme 9). The enamine I was formed in situ by the aldehyde and pyrrolidine. Next, an electron-donor–acceptor (EDA) complex II is formed from I and the perfluoroalkyl iodide. Upon the irradiation by 23 W CFL, this complex turns into a radical ion pair III.20 The formed perfluoroalkyl radical (a) then reacts with the alkene/alkyne giving radical IV, and forms the final product through partial iodine transfer from another perfluoroalkyl iodide. At the same time, the iodide ion (c) reduces the enamine radical cation (b) generating an iodine radical V and reforms the enamine intermediate I. This iodine radical V is then captured by free amines in the reaction mixture and also efficiently reacted with radical intermediate IV giving the desired product.
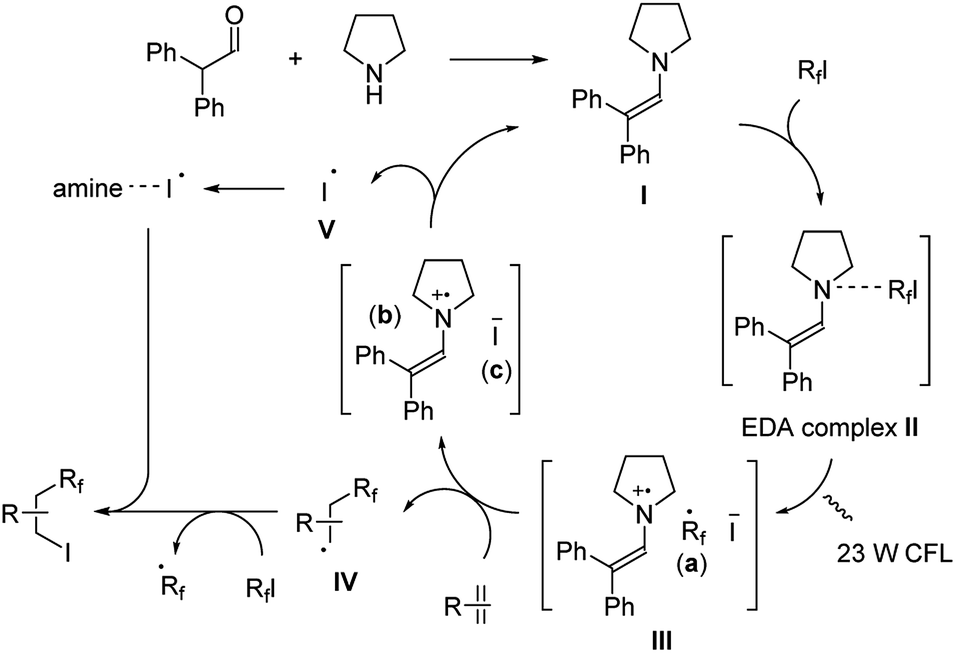 | ||
| Scheme 9 Mechanism of diphenylacetaldehyde-amine catalysis in ATRA of perfluoroalkyl iodides with alkenes/alkynes. | ||
Most recently, Kang and coworkers applied deprotonated salicylaldehyde as photosensitizer in decarboxylative alkylations with isocyanides/thiophenols/disulfanes and N-hydroxyphthalimide esters as reactants (Scheme 10).21 This reaction was carried out under visible light (>400 nm) irradiation owing to the red shift and strong fluorescence the salicylaldehyde displayed upon deprotonation. The mechanism involves the exited state of salicylaldehyde sensitizing N-hydroxyphthalimide through energy transfer pathway generating the reactive radical R, which further reacts with the substrates. These reactions are highly efficient and the catalyst loading could be decreased to as low as 1 mol%.
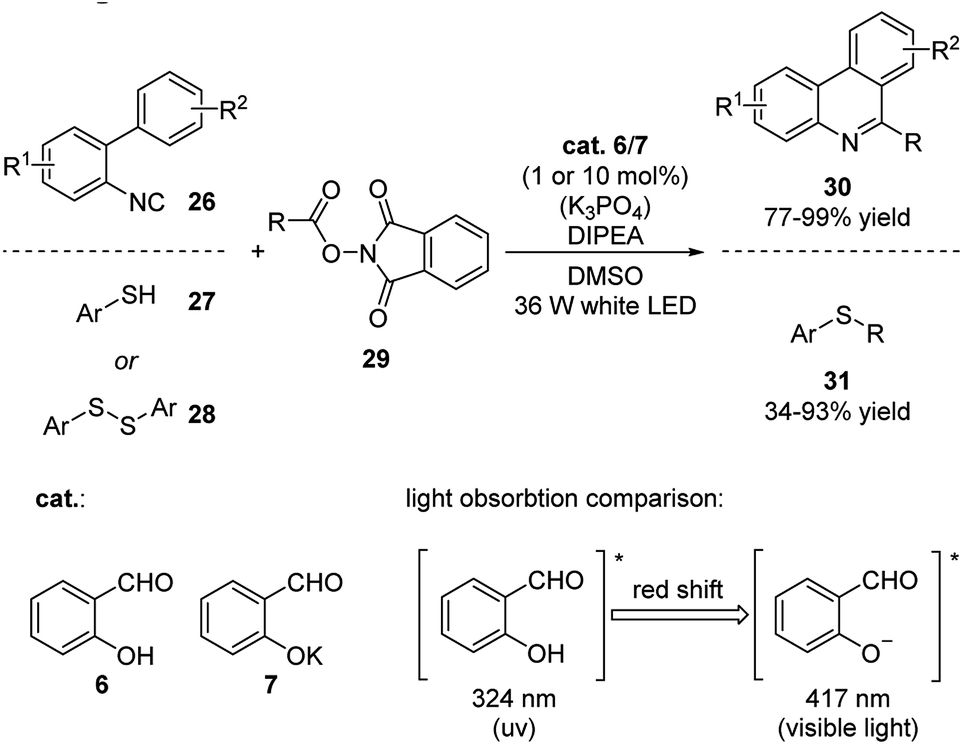 | ||
| Scheme 10 Salicylaldehyde/base catalyzed decarboxylative alkylations. | ||
This strategy was also successfully expanded to the visible light induced aerobic oxidation of N-alkylpyridinium salts (Scheme 11).21 The reaction proceeded smoothly under mild conditions generating isoquinolones in good yields.
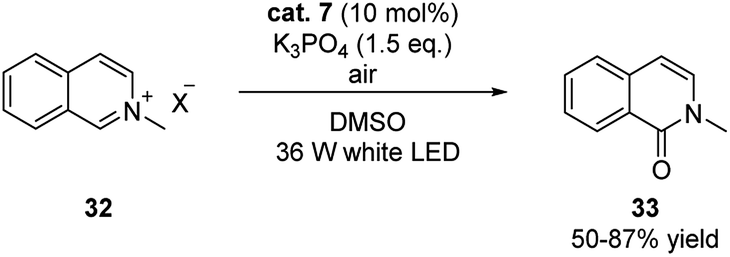 | ||
| Scheme 11 Potassium 2-formylphenolate (cat. 7) catalyzed aerobic oxidation of N-alkylpyridinium salts. | ||
Dehydration
Simple aldehydes (formaldehyde and octanal) were found to be a green alternative for promoting the dehydration of amides.22 These compounds transferred the molecular equivalent of water from amides to the solvent acetonitrile or benzonitrile in the presence of formic acid (Scheme 12). This method enabled the efficient transformation of both aromatic and aliphatic amides to their corresponding nitriles in relatively mild reaction conditions (a). In these reactions, excess amount of aldehyde is generally applied. Even though catalytic amount of aldehyde is less efficient, a good amount of yield was obtained. As shown in equitation (b), 60% yield of 3-phenylpropanenitrile was generated when 20 mol% of octanal was employed to the reaction of 3-phenylpropanamide 36.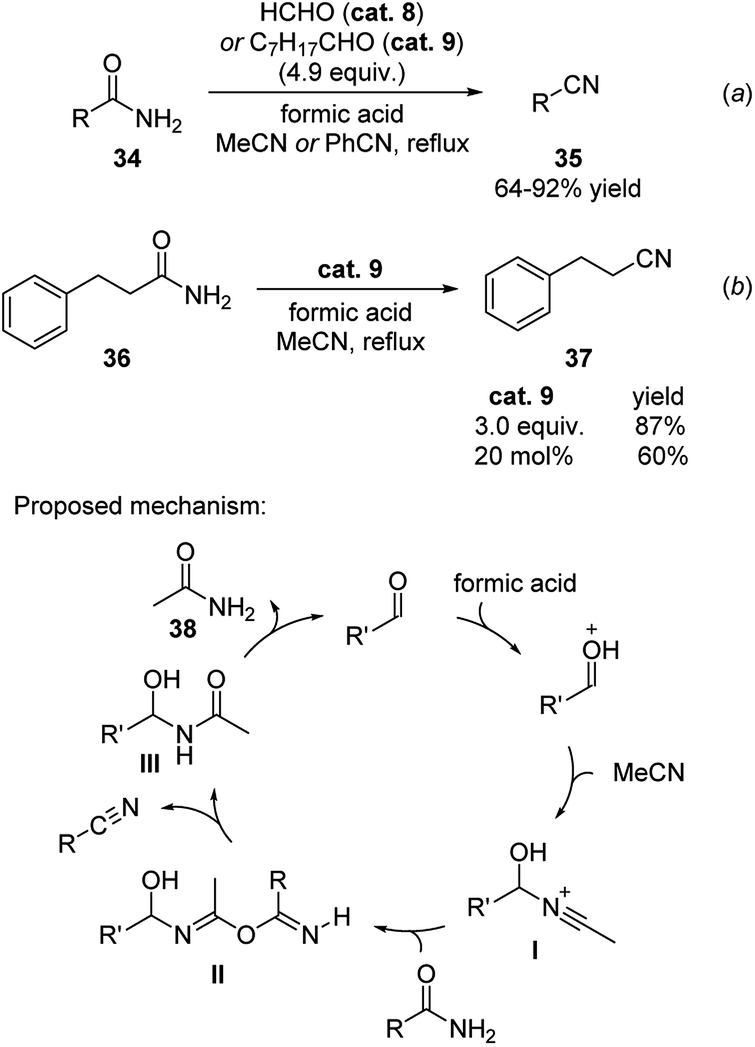 | ||
| Scheme 12 Simple aldehydes promoted dehydration of amides. | ||
The reaction was proposed to undergo via the mechanism shown in Scheme 12. The nitrogen of acetonitrile attacks the aldehyde catalyst, which is activated by the acid additive, forming the cation intermediate I. This intermediate then reacts with the amide substrate to form the new amide intermediate II and the nitrile product. The formed amide intermediate III subsequently releases the amide 38 and reforms the aldehyde catalyst.
Dehydrative transformation
Xu and coworkers developed a series of dehydrative transformation of alcohols with nucleophiles catalyzed by simple aldehydes.23,24,27 These reactions were carried out in a metal- and solvent-free fashion in the presence of base at elevated temperatures. The products were generated in low to excellent yields. Regarding the reaction with primary amines/amides, the catalytic role of aldehyde has been recognized in earlier report.25 However, the author managed to perform this reaction under much milder reaction conditions (Scheme 13).23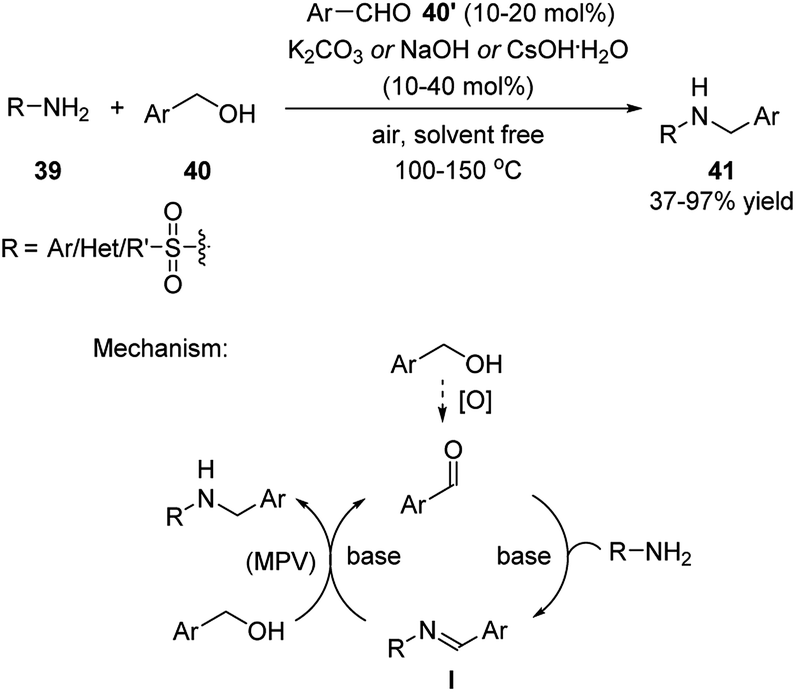 | ||
| Scheme 13 Simple aldehydes catalyzed dehydrative transformation between the corresponding alcohols and primary amines/amides. | ||
The mechanism of the reaction is shown in Scheme 13. The aldehyde catalyst forms an imine intermediate I with the amine substrate. This intermediate then undergoes a metal-free transfer hydrogenative reduction with the primary alcohol and generates the product while reforming the aldehyde catalyst. The later Meerwein–Pondorf–Verley (MPV) type reduction26 is the key to the success of this reaction. The base plays an important role, not only to promote the imine condensation, but also enables the transition metal-free MPV reduction. The use of aromatic aldehydes most probably facilitates the formation of the imine intermediate I with amines.
The dehydrative coupling with 2-substituted alcohols involved multiple MPV redox sequences (Scheme 14).24 Firstly, alcohol 42 is oxidized to the corresponding nucleophilic ketone 42′ through a MPV sequence. Compound 42′ then undergoes an Aldol condensation with the aldehyde catalyst forming an α,β unsaturated ketone intermediate I. This intermediate is sequentially reduced through MPV sequence over two steps by the alcohol substrates 43 forming the final product and recovering the aldehyde catalyst 43′. It is noted that, substrate alcohol 42 also participates in the last two MPV steps to generate intermediate II, the product and side residue ketone 42′.
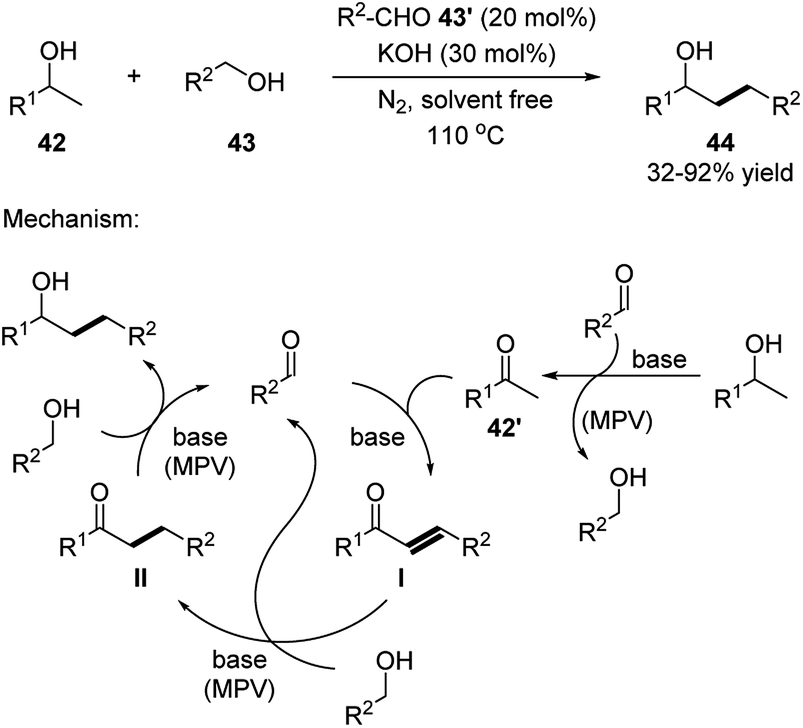 | ||
| Scheme 14 Simple aldehydes catalyzed dehydrative transformation between the corresponding alcohols and 2-substituted alcohols. | ||
In 2017, Xu group continuously expanded this strategy in the dehydrative coupling of primary alcohols with fluorenes (Scheme 15).27 Similar to the previous examples, the aldehyde catalyst forms an unsaturated intermediate (methylenyl fluorene I) with fluorene, which undergoes a hydrogenative reduction with alcohol substrate forming the desired product and regenerates the catalyst.
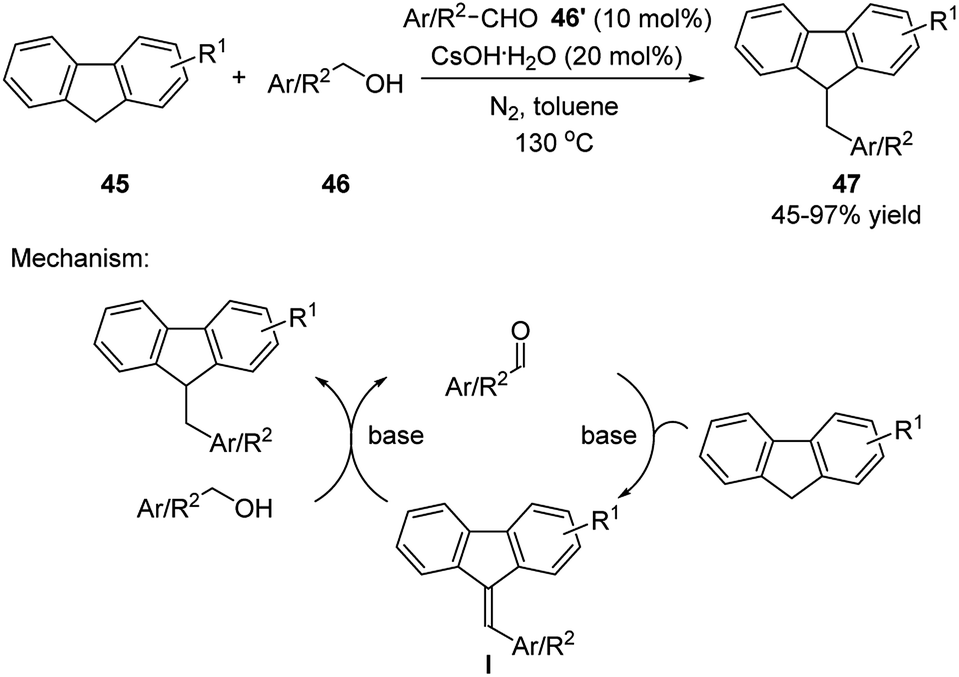 | ||
| Scheme 15 Simple aldehydes catalyzed dehydrative transformation between the corresponding alcohols and fluorenes. | ||
In the above reactions, the aldehydes both act as catalysts and are eventually installed in the final product, when regenerated from the corresponding alcohol substrates upon oxidation. Its utility seems limited since for each catalytic reaction with different hydroxyl group providing substrate, a corresponding aldehyde catalyst is needed. Nevertheless, for simple aldehydes, these reactions in general have broad reaction scopes with good yields.
Hydrolysis/hydrations/hydroaminations
Simple aldehyde-based catalysts are well applied in hydrolysis, hydration or hydroamination reactions of α-heteroatom substituted compounds. As shown in Scheme 16, these reactions mainly follow two mechanisms (A or B). Substrate 48 forms a temporary tether intermediate (I or II) with the aldehyde catalyst. This key intermediate introduces intramolecularity between the substrate and nucleophile 49, facilitating the transformations of the functional group on the substrate.2a,b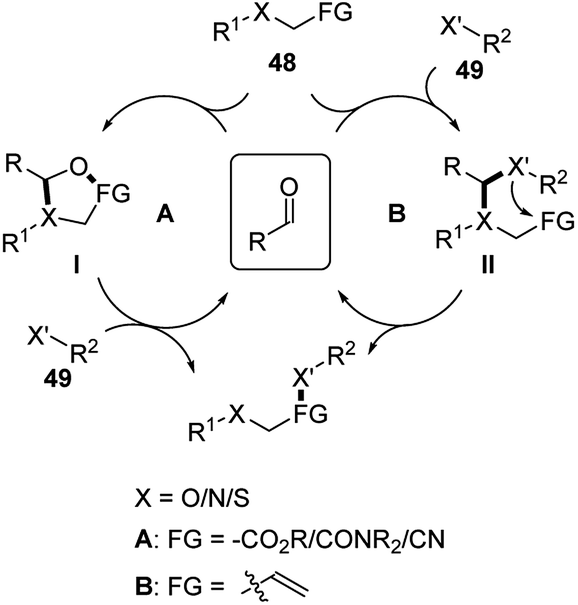 | ||
| Scheme 16 Mechanisms of hydrolysis/hydrations/hydroaminations applying temporary tether strategy. | ||
Early research focused on the hydrolysis of α-hetero atom containing esters or amides following the A mechanistic cycle (Scheme 17, Scheme 16 A).2a Upon the addition of the hetero atom on the substrate 48 to the carbonyl catalyst, an hemiacetal/hemiaminal/hemimercaptal anion intermediate is formed, and the oxygen anion of the immediately attacks the ester or amide forming a five-membered ring intermediate I. This intermediate eventually undergoes a hydrolysis process and generates carboxylic acid product 49. For the hydration of α-amino nitriles, amide product is generated correspondingly in the presence of aldehydes or sugars (Scheme 17).2a,28
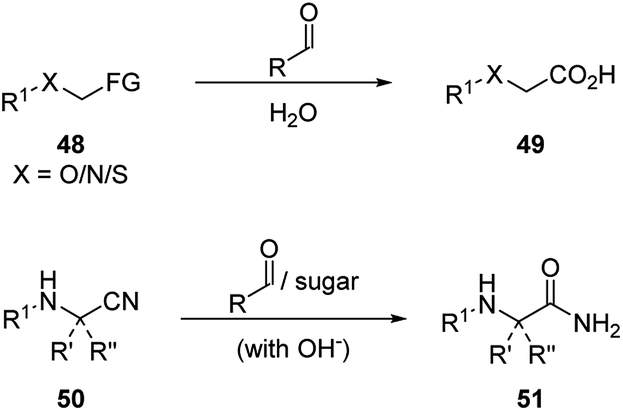 | ||
| Scheme 17 Hydrolysis of 48 and hydration of 50 catalyzed by simple aldehydes or sugars. | ||
Later on, Beauchemin and coworkers continiously applied this temporary tether stragegy to the hydrolysis of phosphinic amides, allowing this reaction to procedes in mild reaction conditions (Scheme 18).29 Compared to the earlier strategy using formaldehyde as catalyst, o-phthalaldehyde displayed superior catalytic activation. It was proposed that a six-membered ring intermediate is generated, which enhanced the electrophilic property of the phosphorous atom. This strategy avoided the use of strong acid/base, Lewis acids, metal, fluorides or exess amount of formaldehyde in this reaction.
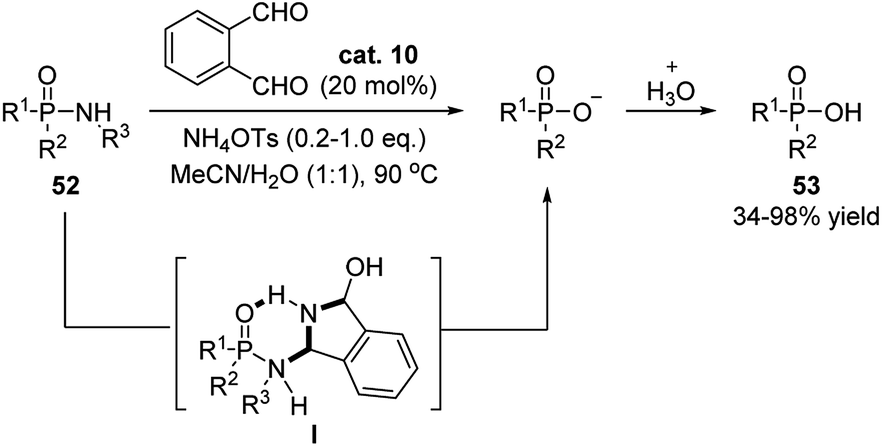 | ||
| Scheme 18 o-Phthalaldehyde catalyzed hydrolysis of phosphinic amides. | ||
Intermolecular hydroamination to strained alkenes is a very powerful, yet challenging, strategy for constructing amines. This is not only due to the high activation energy, but also due to potential selectivity issues. Beauchemin and coworkers overcame the challenges by applying aldehyde catalysis to promote the cope-type hydroamination of a-amino substituted terminal alkenes (allylic amines) with hydroxylamines in mild reaction conditions (Scheme 19).30–32 The reaction mechanism follows the B catalytic cycle shown in Scheme 16.2b Firstly, the aldehyde catalyst forms a nitrone intermediate with the hydroxylamine 55, which generates an aminal intermediate II with the allylic amine. Thus, the aldehyde catalyst forms a temporary tether that holds the two substrates together. Next, this tether intermediate undergoes the hydroamination, which is the rate determine step. Subsequent rapid release of the product completes the catalytic cycle.
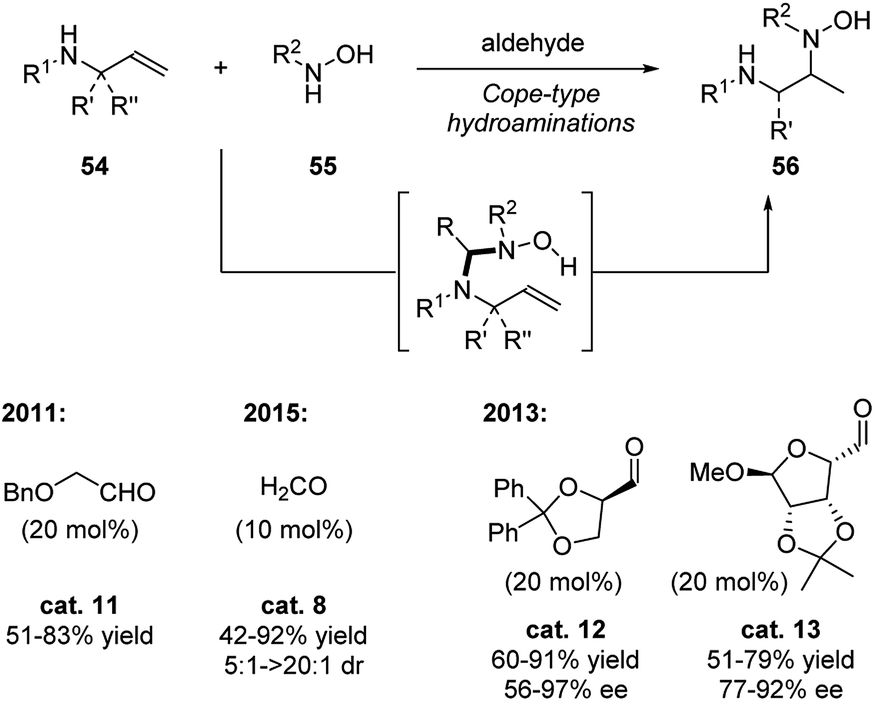 | ||
| Scheme 19 Aldehyde catalyzed hydroaminations. | ||
The choice of the aldehyde catalyst is critical since most aliphatic or aromatic aldehyde forms a stable nitrone with hydroxylamine disfavoring the formation to tether intermediate. Aldehyde with strong electron withdrawing group also disfavors the desired catalytical cycle. Benzyloxyacetaldehyde30 and formaldehyde31 are promising catalysts in this reaction system. Formaldehyde is able to efficiently promote the reaction with chiral substrates in good yields and diastereoselectivities. Moreover, formaldehyde was also found out to be active in highly diluted aqueous solutions, and this catalytic mode might illustrate its important role in prebiotic life. The chiral version of this reaction was initially tested in the earlier work30 and was further developed in 2013.32 By applying chiral catalyst cat. 12 and cat. 13, the α-amino hydroxylamines were obtained in good to high yields and enantiomeric excess.
Coupling reaction
Transition metal mediated C(sp3)–H functionalization is one of the most powerful transformations in synthetic chemistry for the efficient installation of functional groups to the molecules. Normally, proximal directing groups are required in the substrate to guide the transition metal to the position where the reaction occurs. This brings challenges for the substrates without directing groups or with functional groups poorly coordinate to the metal, e.g. primary amines. To circumvent this additional steps are needed for installing the directing group to the substrate. Recently, examples applying aldehyde as the temporary directing functionality have been reported which offers an alternate strategy for the efficient direct functionalization of alkyl primary amines.33The general mechanism is shown in Scheme 20. Normally, the aldehyde catalyst contains an additional coordinating atom, e.g. a heteroatom. Upon imine formation between the aldehyde the amine substrate, the formed imine and the coordinating atom in the catalyst coordinates to the transition metal in a bidentate fashion (I). This leads the metal to the position where C–H activation occurs (II, III). The in situ formed imine function acts as a temporary directing group, which is readily hydrolyzed giving the free amine product and release of the aldehyde catalyst.
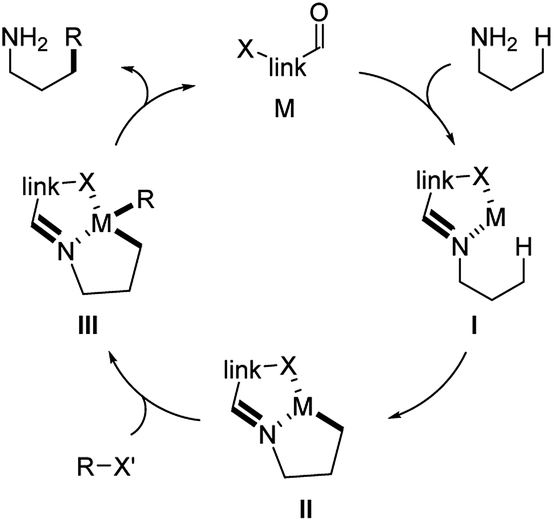 | ||
| Scheme 20 Mechanism of metal catalysis applying aldehydes as transient directing groups. | ||
The first example was developed by Ge and coworkers in 2016 (Scheme 21).34 Catalytic amount of glyoxylic acid was applied, which allowed the γ-arylation of aliphatic amines. The products were prepared as free amine form and no additional protection or deprotection was needed. Shortly after, Yu reported that 2-hydroxynicotinaldehyde showed to be an efficient catalyst in this type of reaction, especially for the γ-arylation on the ring of cyclohexylamines (Scheme 21).35 It is notable that, the loading of the metal catalyst [Pd(OAc)2] and transient aldehyde catalyst can be as low as 3% and 5%, respectively. The products were isolated after a simple Boc-protection of the amine. By applying the same aldehyde as transient directing group, Kamenecka further optimized this reaction for allylic and α-aromatic amino ester substrates and the γ-(sp3)- and γ-(sp2)-arylated products were obtained in decent yields (Scheme 21).36 Instead of using aldehyde directly, Bull realized that simple α-oxylacetal can be applied as the directing aldehyde precursor in the γ-arylation of aliphatic amines (Scheme 21).37
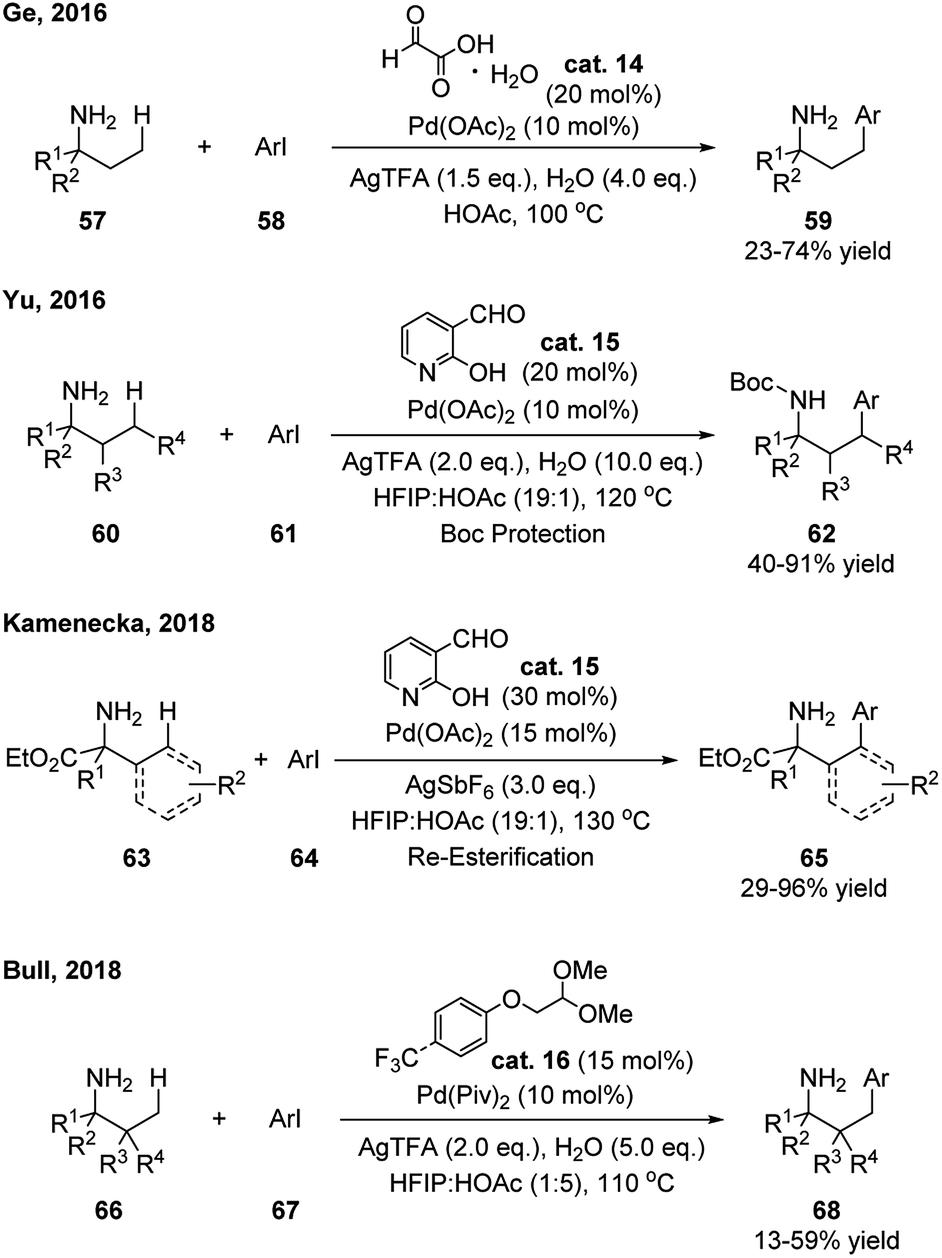 | ||
| Scheme 21 Metal mediated C–H functionalization using catalytic amount of aldehydes as transient directing groups. | ||
Except for above examples where aldehydes (or their precursors) function in catalytic amount under the reaction conditions, stoichiometric amount of aldehyde, such as quinoline-8-carbaldehyde cat. 17 and salicylaldehyde cat. 18, were discovered by Dong and Murakami respectively to promote the γ-(sp3)-C–H activation of primary amines (Scheme 22).38 These reactions expand the substrate scope to o-allylic substituted anilines. The reactivities using catalytic amount of these aldehydes were also tested. They are able to promote the reaction smoothly, albeit in lower yields.
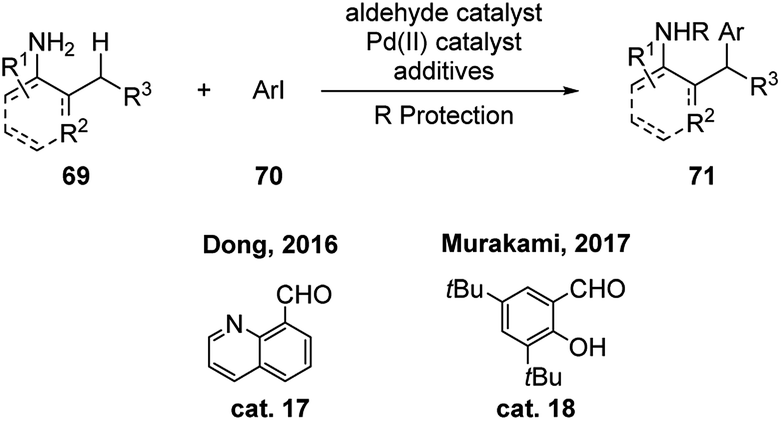 | ||
| Scheme 22 Metal mediated C–H functionalization using stoichiometric amount of aldehydes as transient directing groups. | ||
Yu reported that by combining 2-hydroxybenzaldehyde cat. 19 and pyridin-2-ol the substrate scope of this class of reaction could be extended to include allylic ring substituted primary amines and a wide range of heteroaryl iodides (Scheme 23).39 The previously unreactive heteroaryl bromides also showed high reactivity in this reaction system. In the transition state, pyridin-2-ol transforms to the 2-pyridone form and coordinates to the metal via the nitrogen atom as the additional ligand. The author also realized the δ-C(sp3)–H functionalization of the amino substrate by replacing the ligands to carbonyl compound cat. 20 and pyridin-2-ol.
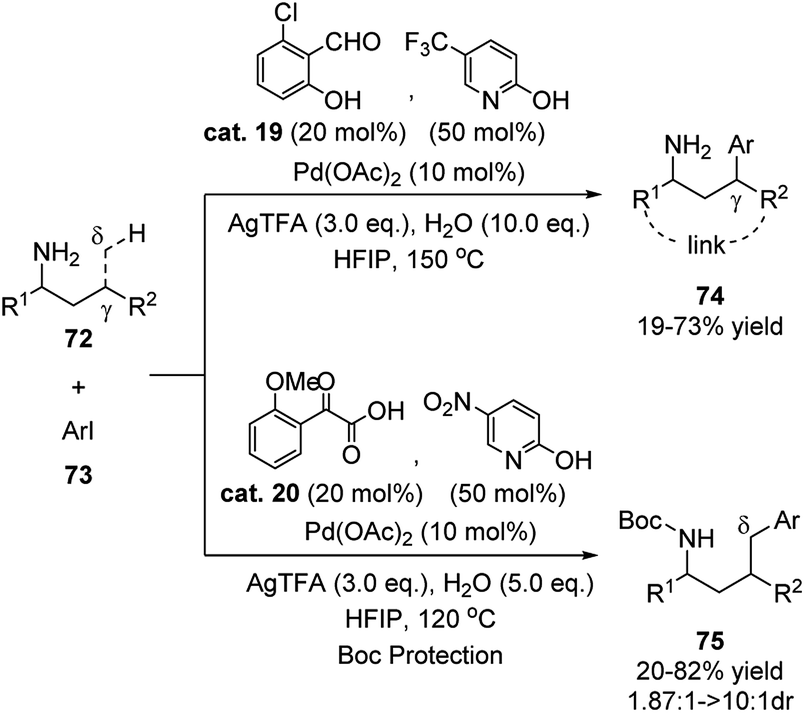 | ||
| Scheme 23 Metal mediated C–H functionalization using 2-hydroxybenzaldehyde cat. 19 and pyridin-2-ol as transient directing groups. | ||
The aldehyde catalysis has also been applied in cooperation with copper catalysis. In 2013, Huang discovered that catalytic amount of aldehyde formed aminal with secondary amines, which underwent an aerobic oxidative C–H amination with benzoxazoles (Scheme 24).40 Although the reaction could be performed without aldehyde catalyst using DCM as solvent (DCM forms aminal with amines), the efficiency and yields were improved when aldehyde was added.
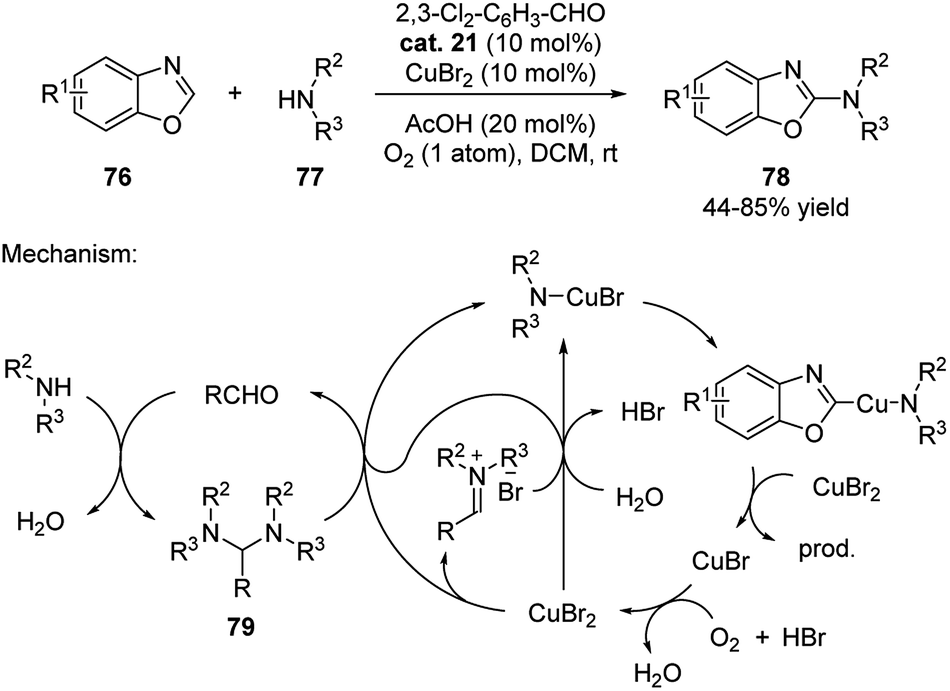 | ||
| Scheme 24 Metal mediated C–H amination of benzoxazoles cooperated with aldehyde cat. 21. | ||
3 Aldehyde catalyst based on PLP skeleton
As the active form in vitamin B6 family, pyridoxal phosphate (PLP) is probably one of the most well studied cofactor which base its mode of action on the principles of aldehyde catalysis (Fig. 2).7 It cooperates with enzymes by binding its functional groups to the specific amino acids of enzymes. The aldehyde group condenses with the lysine residue of the enzyme to form internal aldimines. The nitrogen of the pyridine is protonated by the amino acid residue and exists as a salt form, and the phosphate group interacts with the amino acid residue through hydrogen bonding. The hydroxyl group has proven to be essential for the activity of PLP by forming the hydrogen bond with the nitrogen of the internal aldimine. When amino acids approach the PLP cofactor, the aldehyde group is released from the internal aldimine and forms an external aldimine with the approaching amino acid substrates, enabling various reactions, interconversion or metabolism of the amino acids.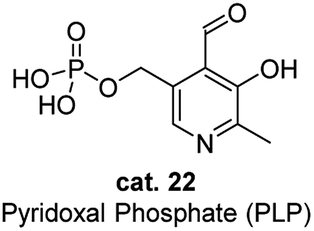 | ||
| Fig. 2 Structure of pyridoxal phosphate (PLP). | ||
Several catalytic pathways and the key intermediates of PLP-dependent enzymes are summarized (Scheme 25). Detailed enzyme types, enzymatic binding/breaking and the catalyst recycling are omitted for the concise description. When amino acid substrate approaches the PLP cofactor, the aldehyde group of the cofactor is released from the internal aldimine and forms the external aldimine 80 with the approaching substrate. This key intermediate 80 may proceed via three main reaction pathways, decarboxylation, elimination and deprotonation. The first two pathways lead to the corresponding amine and glycine (Scheme 25, (a) and (b)). Through the deprotonation, an important carbanionic aldimine intermediate 81 is generated and undergoes a variety of reactions. Reprotonation at C1 of the carbanionic aldimine 2 leads to the racemization of the amino acid substrate (Scheme 25, (c)). Alternatively, reprotonation at C2 leads to a ketimine intermediate that undergoes a transamination process (Scheme 25, (d)). The released pyridoxamine phosphate (PMP) interconverts to PLP in other catalytic cycles. When R group of carbanionic aldimine 81 contains a leaving group at the C3 position, elimination would occur and generate an enamine (Scheme 25, (e)), which continuously undergoes addition reactions. Carbanionic aldimine 81 is nucleophilic and can also react directly with electrophiles through Aldol or Claisen type addition (Scheme 25, (f)).
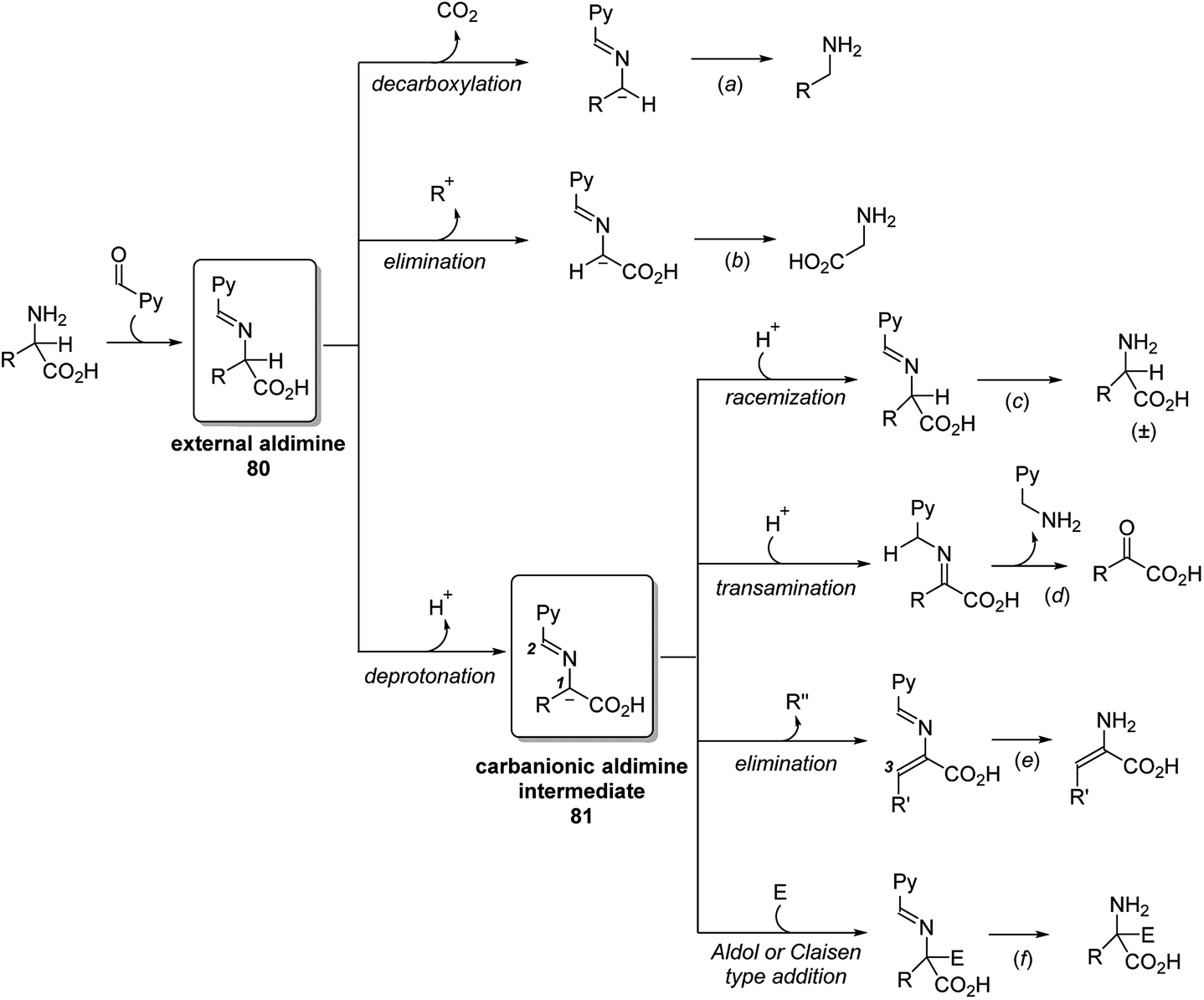 | ||
| Scheme 25 Brief summary of the catalytic routes of PLP-dependent enzymes. | ||
Aside from the diversity of PLP-dependent enzyme catalysis which enables important biological interconversions, metabolisms and transformations of amino acids, non-enzymatic catalytic ability of PLP derivatives has also been long known.41 These sterling properties continuously inspire synthetic chemists developing biomimetic aldehyde catalysis.
Racemization
In 2012, Branda's group developed a photoswitchable carbonyl catalyst which shows the reversible ability of catalyzing the racemization of L-alanine (Scheme 26).42 By mimicking the structure property of PLP, they linked the pyridinium and carbonyl group (see the core structure of PLP) with a dithienylethene (DTE) moiety in between. The generated catalyst combined the advantages of the photoswitchable property of DTE and the catalytic function of the pyridinium aldehyde. When the reaction solution was irradiated with 365 nm light, the catalyst transformed to the pi-conjugated active form, which enables the aldimine intermediate formation with 82, and the exchange of hydrogen to deuterium of was observed. When the irradiation was changed back to visible light, little racemization of L-alanine occurred. The authors were able to obtain up to 95% yield (by NMR) of the racemic alanine from their experiments.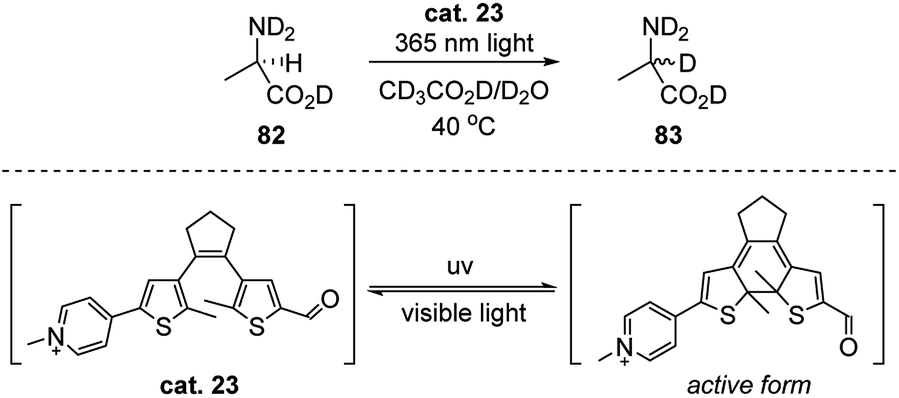 | ||
| Scheme 26 Photoswitchable catalyst promoted racemization of L-alanine. | ||
Transamination
Metal assisted oxidation strategies in nature (e.g. copper amine oxidases) have inspired a set of aerobic catalytic reactions.43 Starting from mid-20th century, researchers have employed PLP derivatives with various metal salts for the oxidative deamination of primary amines (Scheme 27).41 In these reactions, aldehydes including pyridoxal (PL, cat. 24), PLP, 5′-deoxypyridoxal (DPL, cat. 25) and 4-nitrosalicylaldehyde (cat. 26) have shown to be reactive. The metal complex of the Schiff base is shown as intermediate I. PL has also proven to be catalytic reactive without metal cooperation. In 1957, Matsuo successfully applied 50 mol% PL for catalyzing the transamination reaction between alanine and 2-oxobutanoic acid in basic conditions (Scheme 27).41b | ||
| Scheme 27 Non-enzymatic catalyzed transamination using PLP derivatives. | ||
The strategies of enantioselective biomimetic transamination have also been developed for synthesizing chiral amino acids.44 In most cases, stoichiometric amount of pyridoxamine (PM) derived chiral catalysts were applied with the addition of metal salts. PM derived catalyst could be generated from the transamination between the corresponding aldehyde catalyst and α-ketone amino acid substrate in the reaction mixture, thus the PM derived catalyst is considered as the equivalent of the aldehyde catalyst.
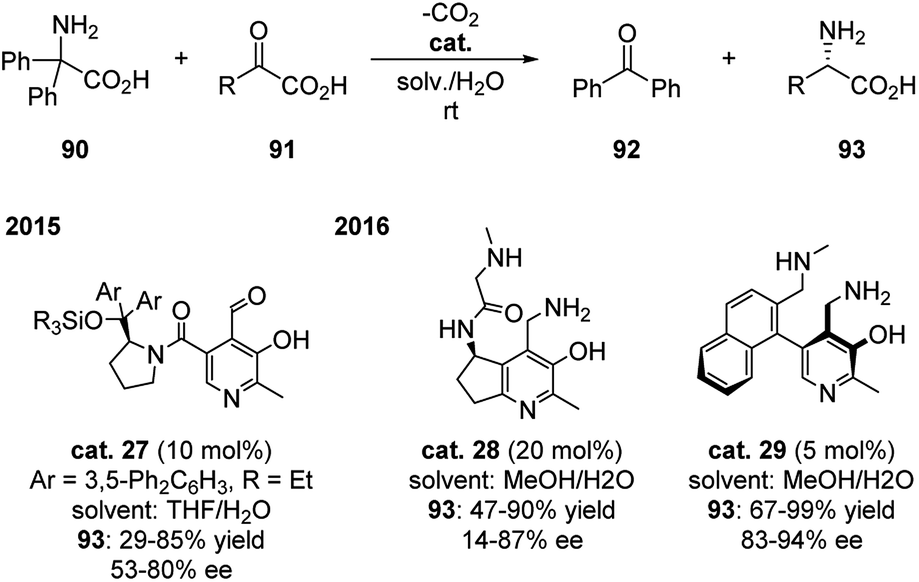
This field did not receive much attention until recently. In 2015, Zhao group developed PL based catalyst bearing a chiral diaryl prolinol sillyl ether moiety (cat. 27), which successfully catalyzed the transamination of 2,2-diphenylglycine with 2-oxobutanoic acid in mild reaction conditions with 10 mol% catalyst. The chiral aminal acids were generated in moderate yields and enantioselectivities (Scheme 28).45 Based on this and the previous work, Zhao continued to develop two different PM based catalysts (cat. 28 and cat. 29) in 2016, which improved the yield up to 99% and enantioselectivity to 94% ee with as low as 5 mol% catalyst (Scheme 28).46
 | ||
| Scheme 28 Asymmetric transamination catalyzed by PL/PM derived chiral catalysts. | ||
Mannich reaction
In 2018, Zhao's group developed an N-quaternized pyridoxal catalyst that facilitated a stereoselective Mannich reaction between glycinate and aryl N-diphenylphosphinyl imines (Scheme 29).47 Pioneering studies showed that chiral pyridoxals, together with metal salts, were able to promote the Aldol addition of glycine to aldehydes (Scheme 29).48 However, stoichiometric amount of pyridoxal catalysts (cat. 30) were needed, and the stereoselectivities of the products were moderate. Based on the enzymatic Aldol reaction, in which the coenzymatic pyridoxal intermediates are protonated, Zhao developed methylated chiral pyridoxal catalyst 31. The more electrophilic aldehyde catalyst alone is able to activate glycinate efficiently while the amide side chain spontaneously activates the imine by hydrogen-bonding. The loading of the bifunctional catalyst can be as low as 0.2 mol%, and with 1.0 mol% catalyst the products were in general obtained in high yields and stereoselectivities.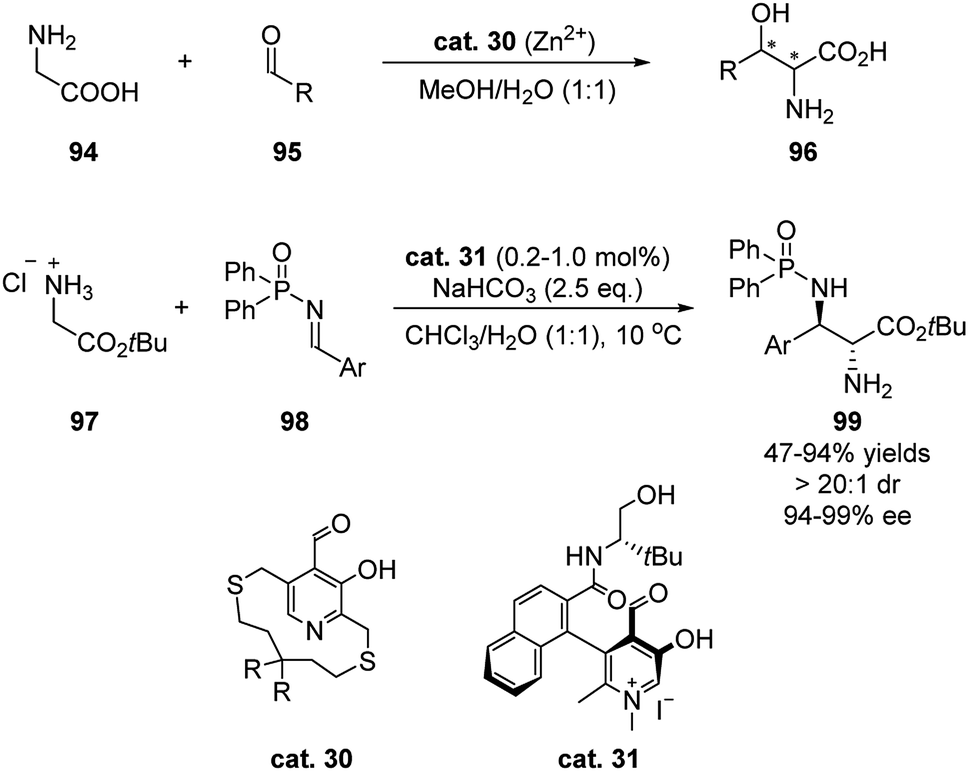 | ||
| Scheme 29 Asymmetric Mannich reaction catalyzed by PL derived chiral catalyst. | ||
4 Aldehyde catalyst based on binaphthyl skeleton
Nucleophilic substitution
Except for the example above, aldehyde catalyst based on another class of chiral backbone has also been shown to be effective in promoting the α-functionalization of amino acid derivatives. Guo and coworkers demonstrated that catalysts based on chiral binaphthyl structure enabled the amino esters to react with electrophiles for the synthesis of tryptophan derivatives and α,α-disubstituted-α-amino acid derivatives in a stereoselective fashion (Scheme 30).49 The catalyst contains the basic binol structure bearing an aldehyde group at 3-position and a substituted group at 3′-position. This catalyst is also proposed to function as bifunctional catalyst that activates both substrates spontaneously. As shown in intermediate I or II, the formed imine coordinates with the hydroxyl group at 2 position (or with ZnCl2), and the hydroxyl group (or deprotonated) at 2′ position activates the electrophile through hydrogen bonding.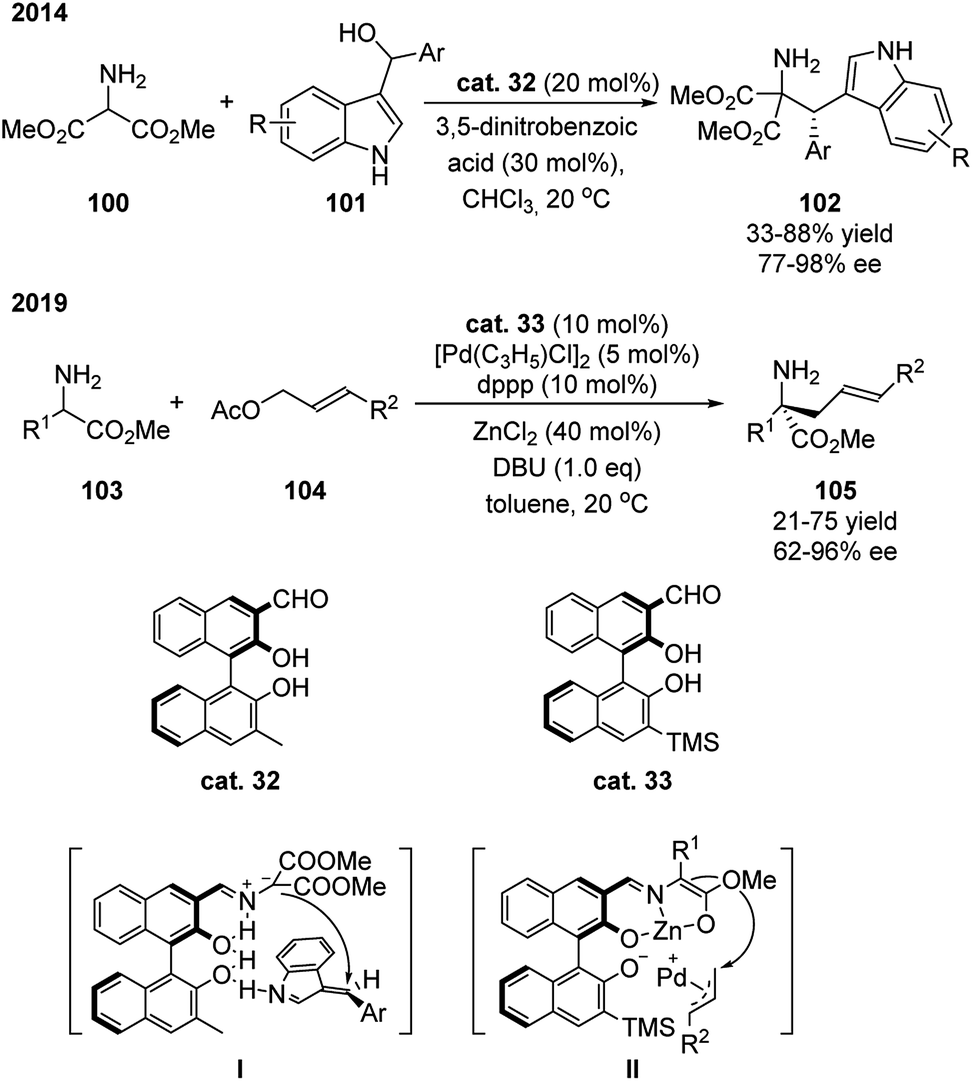 | ||
| Scheme 30 Asymmetric nucleophilic substitution catalyzed by binaphthyl derived catalyst. | ||
Micheal addition/cyclization
This type of chiral binaphthyl based catalyst is also able to promote the Michael addition/cyclization sequence for the synthesis of chiral Δ(1)-pyrroline 108 (Scheme 31).50 In this case, binol based catalysts bearing an aldehyde group at 3-position gave moderate yields and stereoselectivities. Modification of the binaphthyl catalyst overcame these issues in this reaction system. Electron withdrawing substituent were shown to enhance the ability to activate the amine substrate. The changing of the aldehyde position enabled the site of reaction to occur much closer to the chiral axis and hindered substituted aryl group. This catalyst is suitable for a wide scope of substrates such as tert-butyl glycine ester and glycine ester/amide free amine salts and a big range of α,β-unsaturated ketones. Later on, Shi developed a Pd-catalyzed C–H naphthylation for the synthesis of chiral aldehyde catalysts and applied them in the Michael addition/cyclization reaction of glycine derived amides or dipeptides with (E)-chalcone, which in general showed better catalytic activity and stereocontrol compare with the previous examples (Scheme 31).51 | ||
| Scheme 31 Asymmetric Michael addition/cyclization sequence catalyzed by binaphthyl derived catalyst. | ||
5 Artificial enzymes
Elimination/Micheal addition
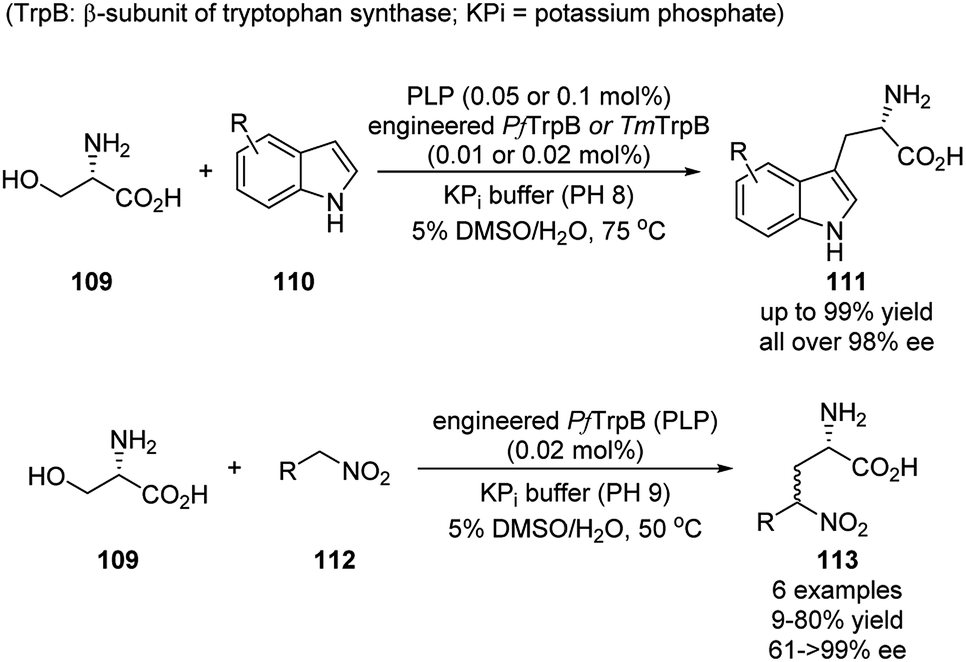
So far, the synthetic applications of asymmetric aldehyde catalysis have been accomplished by mimicking several existing biocatalytic processes for the α-amino acid derivatives. The general strategy is mainly to designing chiral molecules bearing an aldehyde substituent based on either PL-skeleton or binaphthyl-derived-skeleton. Taking a look back to the enzymatic catalysis, the PLP co-enzyme alone is racemic and less/not reactive, and the catalytic activities and stereoselectivities are provided by the cooperative enzymes. Arnold and coworkers focused on the simplification and modification of the cooperative enzyme partner, which provides the alternative strategy for expanding the aldehyde catalysis for more diverse synthesis of noncanonical chiral amino acids.52–55This work was initiated in 2015, when the authors applied directed evolution to the β-subunit of tryptophan synthase from Pyrococcus furiosus (PfTrpB). The obtained PLP-dependent PfTrpB0B2 was proved to be active as a stand-alone catalyst. Later on, a series of engineered PfTrpB and TmTrpB (from Thermotoga maritima) were screened to efficiently catalyze the reaction between L-serine and various substituted indoles. The noncanonical tryptophans were generally obtained in high yields and nearly all in enantiopure form (Scheme 32).52–54 For specific substrate, further directed evolution can be applied to tune the catalytic activity.55 This reaction was also further expanded to nitroalkenes as nucleophiles, with 3 different engineered TrpB variants. A small scope of 6 examples were shown, albeit with less impressive yields and stereoselectivities. Nevertheless, this demonstrated the possibility of applying this biocatalysis for the substrates little similarity to their native case (Scheme 32).56
 | ||
| Scheme 32 Engineered TrpB catalyzed elimination/Micheal addition sequence. | ||
In 2019, Arnold group realized the elimination/addition sequence of L-serine with less nucleophilic 3-subtituted oxindoles. This reaction generated new noncanonical amino acids with quaternary carbon stereocenters (Scheme 33).57 Substrates as lactone and ketone were also compatible with this reaction and formed the quaternary carbon stereocenters. It is also worth noting that by applying optimized engineered TrpB, all products were obtained in enantio-pure fashion and the reaction is highly chemo-selective that no N atom alkylated product was observed for the oxindoles substrates.
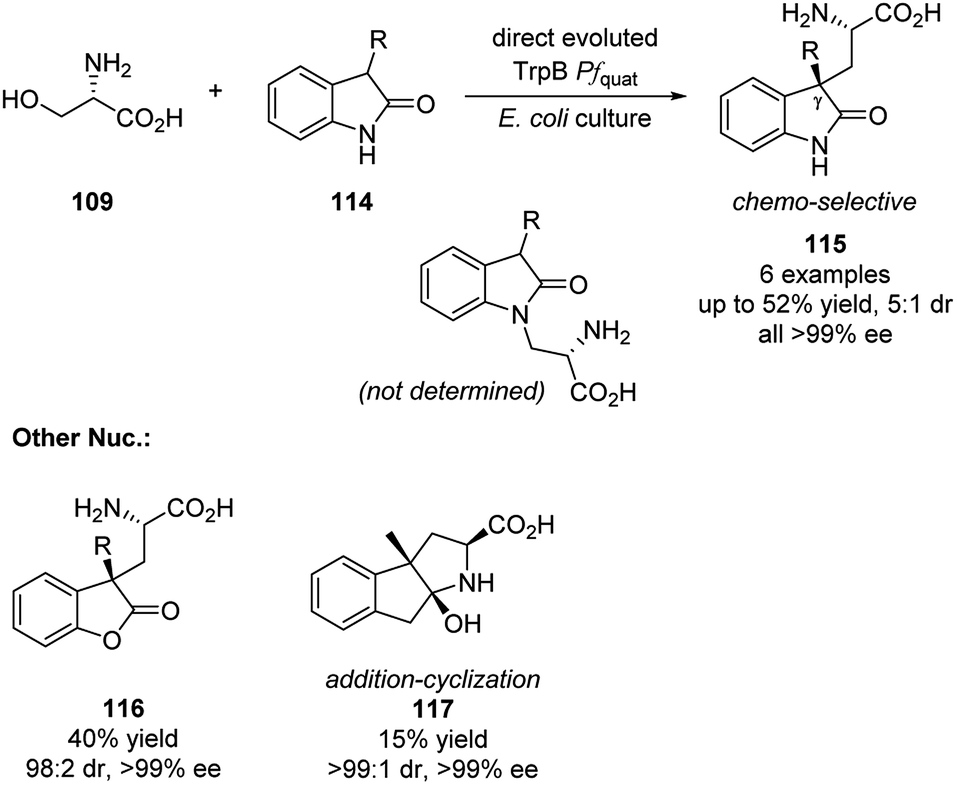 | ||
| Scheme 33 Direct evoluted TrpB Pfquat catalyzed elimination/Micheal addition sequence. | ||
Most recently, Lei and coworkers reported the identification of aspergillomarasmine A (AMA) synthase (PLP-dependent enzyme) in biosynthesis.58 AMA is an inhibitor of New Delhi metallo-β-lactamase that causes β-lactam antibiotic resistance, and its synthesis is of great importance. The authors are able to synthesize a group of AMA analogues not only by using the wild-type enzyme, but also by applying engineered AMA synthase R234A. The results are showing in Scheme 34.
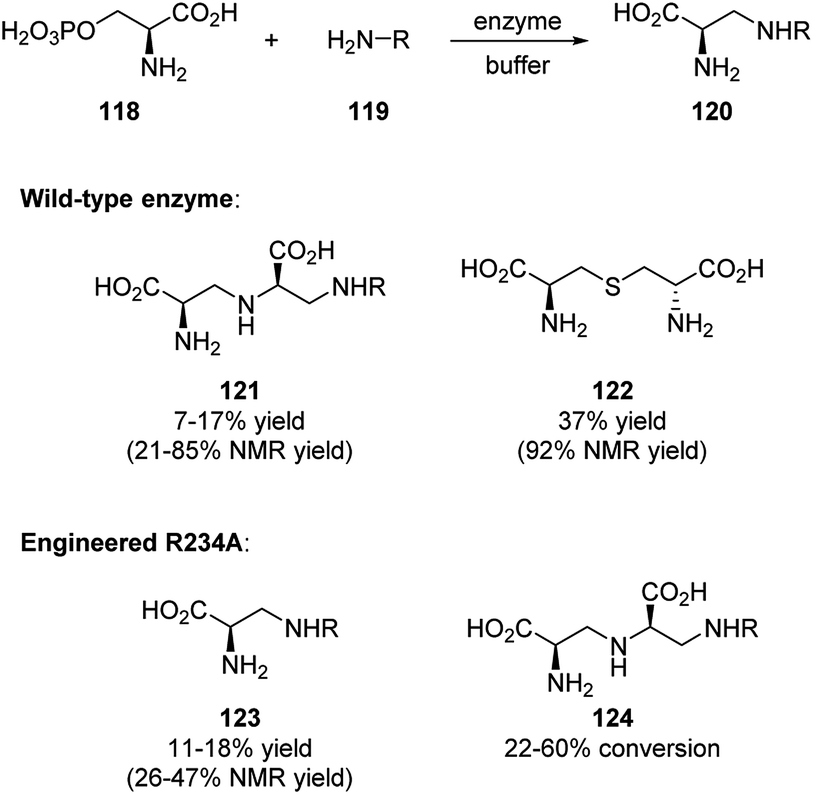 | ||
| Scheme 34 Enzymatic synthesis of AMA and its analogues. | ||
6 Conclusions
Nature is a great treasure house. Through curiosity and passion for exploring, understanding, mimicking and utilizing concepts in nature, chemists have been able to continuously deduce, advance and mature important synthetic strategies and procedures. In the field of aldehyde catalysis, we have witnessed a continuous development, where many nature-inspired reactions are being reported. Different types of aldehyde catalysts have been designed, developed and applied, including simple aldehydes, PLP-derived aldehydes, binaphthyl-derived aldehydes and PLP-dependent engineered enzymes. These catalysts function in different ways on a variety of substrates. As photo-organocatalysts, compounds such as alkenes, aliphatic halides, amides, esters, sulfonyl cyanides and pyridinium salts are activated for radical reactions. As electrophilic catalysts, the aldehyde catalysts can activate amines, alcohols and carbon nucleophiles for dehydrative transformations of primary alcohols. The α-heteroatom substituted functionality can be activated for transformations such as hydrolysis, hydrations and hydroaminations. Amines can be activated by aldehydes in cooperation with transition metals for C–H activations. Moreover, aldehydes have also been demonstrated to activate amino acid derivatives for racemizations, transaminations and nucleophilic reactions. By elaborately design, chiral aldehydes or aldehyde-dependent enzymes have been developed for asymmetric catalysis and high stereoselectivities have been achieved.For further study and future developments, a few areas of interest are worth a special mention. Firstly, except applying aldehydes as directing groups in transition metallic catalysis, the examples of combined catalysis with aldehyde catalysis are still limited. More diverse reactivities might be opened up by combining aldehyde catalysis with other catalytic modes, such as hydrogen-bonding catalysis, iminium-ion activation, phase transfer catalysis and etc. Secondly, the in situ formation of the active catalyst strategy is worthy of more investigation. Less active aldehyde catalyst might be necessary in some reaction systems in order to avoid side reactions, thus an active catalyst form needs to be generated in situ for promoting the desired reaction. Comparably, for less active substrates, aldehyde catalysts with greater catalytic activity are required. Therefore, pre-activation of the aldehyde might be a possible solution. Thirdly, taking advantages of enzymes might open an alternative way for asymmetric synthesis. Designing and synthesizing chiral aldehyde catalysts could be effort and time consuming. Except direct simplify PLP-dependent enzymes, study related to other aldehyde-dependent proteins (e.g. antibodies) remains undeveloped. Fourthly, the catalyst recycling in this field seems to have drawn limited attention. Unlike homogeneous and heterogeneous metal catalyst for which recycling is often important, the reuse of greener and cheaper homogeneous organocatalyst seems less necessary from a sustainability point of view. However, for those well designed delicate chiral aldehyde catalyst and engineered aldehyde-dependent enzymes, their immobilization or efficient recycling are worthy of study to improve cost of goods. Last but not the least, the further fusion of the synthetic and enzymatic aldehyde catalysis is expected. In general, biocatalysis has evolved to be highly efficient and specific for the precise control of the biochemical reactions of significant importance for regulating the chemical activities of life. By borrowing its concept, chemists realized the chemically fabricated catalysts, which often are less efficient or selective, but offer more generality and broader substrate scope. In synthetic catalysis, more complicated aldehydes are being designed and synthesized for the better control the reactivities and stereoselectivities. We have also seen the examples of very detailed fine tuning of the catalysts for achieving higher reactivities and stereocontrol. Meanwhile, effort has also been devoted to enzyme modification to achieve broader reactivities. Finding a better balance of these two types of catalysis continues to be attractive, and we are looking forward to see more development is this field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grants no. 21901177 and 21572218) for financial support.Notes and references
- Selected reviews: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (c) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (d) H. Pellissier, Tetrahedron, 2007, 63, 9267 CrossRef CAS; (e) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS; (f) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8 CrossRef CAS; (g) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (h) B. R. Buckley and M. M. Farah, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2011, 107, 102 RSC; (i) B. R. Buckley, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2013, 109, 189 RSC; (j) V. d. G. Oliveira, M. F. d. C. Cardoso and L. d. S. M. Forezi, Catalysts, 2018, 8, 605 CrossRef; (k) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596 RSC; (l) M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491 CrossRef CAS; (m) G. J. Reyes-Rodríguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221 CrossRef.
- Recent reviews: (a) R. Pascal, Eur. J. Org. Chem., 2003, 2003, 1813 CrossRef; (b) B.-J. Li, C. El-Nachef and A. M. Beauchemin, Chem. Commun., 2017, 53, 13192 RSC; (c) S. Li, X.-Y. Chen and D. Enders, Chem, 2018, 4, 2026 CrossRef CAS; (d) L.-Z. Gong, Sci. China: Chem., 2019, 62, 3 CrossRef CAS; (e) Y. Shimoda, J. Synth. Org. Chem., Jpn., 2019, 77, 369 CrossRef CAS; (f) Q. Wang, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 6818 CrossRef CAS; (g) J. Chen, Y. E. Liu, X. Dong, L. Shi and B. Zhao, Chin. J. Chem., 2019, 37, 103 CAS; (h) M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833 CrossRef CAS.
- Recent examples and reviews: (a) J. Kofoed, J.-L. Reymod and T. Darbre, Org. Biomol. Chem., 2005, 3, 1850 RSC; (b) K. Koch, W. B. Schweizer and A. Eschenmoser, Chem. Biodiversity, 2007, 4, 541 CrossRef CAS; (c) M. Amedjkouh and M. Brandberg, Chem. Commun., 2008, 2008, 3043 RSC; (d) H. J. Cleaves II, Precambrian Res., 2008, 164, 111 CrossRef; (e) S. A. Benner, H.-J. Kim, M.-J. Kim and A. Ricardo, Cold Spring Harbor Perspect. Biol., 2010, 2, a003467 Search PubMed; (f) M. Kajjout, Y. Hebting, P. Albrecht and P. Adam, Chem. Biodiversity, 2012, 9, 714 CrossRef CAS; (g) C. Meinert, I. Myrgorodska, P. de Marcellus, T. Buhse, L. Nahon, S. V. Hoffmann, L. L. S. d'Hendecourt and U. J. Meierhenrich, Sicence, 2016, 352, 208 CrossRef CAS; (h) A. López-Islas, M. Colín-García and A. Negrón-Mendoza, Int. J. Astrobiol., 2019, 18, 420 CrossRef; (i) A. K. Eckhardt, A. Bergantini, S. K. Singh, P. R. Schreiner and R. I. Kaiser, Angew. Chem., Int. Ed., 2019, 58, 5663 CrossRef CAS; (j) H. S. Bernhardt, Life, 2019, 9, 19 CrossRef CAS.
- For a review: A. G. M. Chew and D. A. Bryant, Annu. Rev. Microbiol., 2007, 61, 113 CrossRef CAS.
- For recent reviews: (a) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef; (b) J. Xuan, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Eur. J. Org. Chem., 2013, 2013, 6755 CrossRef CAS; (c) T. Koike and M. Akita, Top. Organomet. Chem., 2014, 48, 371 CrossRef; (d) C. Wang and Z. Lu, Org. Chem. Front., 2015, 2, 179 RSC; (e) A. Aukauloo and W. Leibl, Chapter Three - Bioinspired Photocatalysis, Advances in Botanical Research, ed. B. Robert, Academic Press, 2016, vol. 79, p. 63 Search PubMed; (f) C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596 RSC; (g) K. Zeitler, Metal-free Photo (redox) Catalysis, Visible Light Photocatalysis in Organic Chemistry, ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley-VCH, Weinheim, 2018 Search PubMed; (h) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS.
- Proteolysis: (a) K. Oda, J. Biochem., 2012, 151, 13 CrossRef CAS , keton catalysis mimicking setrine proteases:; (b) M. Ghosh, J. L. Conroy and C. T. Seto, Angew. Chem., Int. Ed., 1999, 38, 514 CrossRef CAS.
- (a) D. E. Metzler, M. Ikawa and E. E. Snell, J. Am. Chem. Soc., 1954, 76, 648 CrossRef CAS; (b) R. A. John, Biochim. Biophys. Acta, 1995, 1248, 81 CrossRef; (c) D. E. Scott, A. Ciulli and C. Abell, Nat. Prod. Rep., 2007, 24, 1009 RSC; (d) R. Percudani and A. Peracchi, BMC Bioinf., 2009, 10, 273 CrossRef; (e) M. D. Toney, Biochim. Biophys. Acta, 2011, 1814, 1407 CrossRef CAS; (f) T. B. Fitzpatrick, Adv. Bot. Res., 2011, 59, 1 CAS; (g) G. T. Wondrak and E. L. Jacobson, Subcell. Biochem., 2012, 56, 291 CAS; (h) R. Casasnovas, J. Frau, J. Ortega-Castro, J. Donoso and F. Muñoz, J. Phys. Chem. B, 2013, 117, 2339 CrossRef CAS; (i) S. Dajnowica, J. M. Parks, X. Hu, R. C. Johnston, A. Y. Kovalevsky and T. C. Mueser, ACS Catal., 2018, 8, 6733 CrossRef; (j) A. J. Borchert, D. C. Ernst and D. M. Downs, Trends Biochem. Sci., 2019, 44, 849 CrossRef CAS; (k) J. Liang, Q. Han, Y. Tan, H. Ding and J. Li, Front. Mol. Biosci., 2019, 6, 1 CrossRef , see also:; (l) N. M. Okeley and W. A. van der Donk, Chem. Biol., 2000, 7, R159 CrossRef CAS; (m) V. L. Davidson, Protein-Derived Cofactors, eLS, John Wiley & Sons, Ltd, 2020 Search PubMed.
- (a) A. R. Fersht, Proc. R. Soc. London, Ser. B, 1974, 187, 397 CrossRef CAS; (b) K. Faber, Biotransformations in Organic Chemistry-A Textbook, Springer-Verlag Berlin Heidelberg, 4th edn, 2000 CrossRef; (c) H. Gröger and Y. Asano, Introduction-Principles and Historical Landmarks of Enzyme Catalysis in Organic Synthesis, Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012 Search PubMed.
- (a) G. S. Hammond, P. A. Leermakers and N. J. Turro, J. Am. Chem. Soc., 1961, 83, 2396 CrossRef CAS; (b) G. S. Hammond, N. J. Turro and P. A. Leermakers, J. Phys. Chem., 1962, 66, 1144 CrossRef CAS.
- (a) N. C. Yang, J. I. Cohen and A. Shani, J. Am. Chem. Soc., 1968, 90, 3264 CrossRef CAS; (b) N. C. Yang, Photochem. Photobiol., 1968, 7, 767 CrossRef CAS.
- For a recent reviews: (a) T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945 CrossRef CAS; (b) T. M. Williams and C. R. J. Stephenson, Atom Transfer Radical Addition using Photoredox Catalysis, Visible Light Photocatalysis in Organic Chemistry, ed. C. Stephenson, T. Yoon and D. W. C. MacMillan, Wiley-VCH, Weinheim, 2018, p. 73 Search PubMed.
- E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064 CrossRef CAS.
- See also: (a) M. Cocivera and A. M. Trozzolo, J. Am. Chem. Soc., 1970, 92, 1772 CrossRef CAS; (b) N. C. Yang, M. Kimura and W. Eisenhardt, J. Am. Chem. Soc., 1973, 95, 5058 CrossRef CAS.
- Y. Zhang, K. B. Teuscher and H. Ji, Chem. Sci., 2016, 7, 2111 RSC.
- J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS.
- (a) L. Zhang, X. Si, Y. Yang, M. Zimmer, S. Witzel, K. Sekine, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 1823 CrossRef CAS; (b) X. Si, L. Zhang and A. S. K. Hashmi, Org. Lett., 2019, 21, 6329 CrossRef CAS.
- I. K. Sideri, E. Voutyritsa and C. G. Kokotos, ChemSusChem, 2019, 12, 4194 CrossRef CAS.
- V. Pirenne, I. Traboulsi, L. Rouvière, J. Lusseau, S. Massip, D. M. Bassani, F. Robert and Y. Landais, Org. Lett., 2020, 22, 575 CrossRef CAS.
- T. Yajima, M. Murase and Y. Ofuji, Eur. J. Org. Chem., 2020, 2020, 3808 CrossRef CAS.
- (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef CAS; (b) H. Matsui, M. Murase and T. Yajima, Org. Biomol. Chem., 2018, 16, 7120 RSC.
- Y.-J. Zhuang, J.-P. Qu and Y.-B. Kang, J. Org. Chem., 2020, 85, 4386 CrossRef CAS.
- M.-P. Heck, A. Wagner and C. Mioskowski, J. Org. Chem., 1996, 61, 6486 CrossRef CAS.
- Q. Xu, Q. Li, X. Zhu and J. Chen, Adv. Synth. Catal., 2013, 355, 73 CrossRef CAS.
- Q. Xu, J. Chen and Q. Liu, Adv. Synth. Catal., 2013, 355, 697 CrossRef CAS.
- For an early example: Y. Sparinzak, J. Am. Chem. Soc., 1956, 78, 3207 CrossRef.
- For a recent review of MPV reaction: A. Porcheddu and G. Chelucci, Chem. Rec., 2019, 19, 1 CrossRef.
- J. Chen, Y. Li, S. Li, J. Liu, F. Zheng, Z. Zhang and Q. Xu, Green Chem., 2017, 19, 623 RSC.
- (a) S. Chitale, J. S. Derasp, B. Hussain, K. Tanveer and A. M. Beauchemin, Chem. Commun., 2016, 52, 13147 RSC; (b) A. J. Wagner, D. Y. Zubarev, A. Aspuru-Guzik and D. G. Blackmond, ACS Cent. Sci., 2017, 3, 322 CrossRef CAS.
- For an early example: W. P. Jencks and M. Gilchrist, J. Am. Chem. Soc., 1964, 86, 1410 CrossRef CAS This work: B.-J. Li, R. D. Simard and A. M. Beauchemin, Chem. Commun., 2017, 53, 8667 RSC.
- M. J. MacDonald, D. J. Schipper, P. J. Ng, J. Moran and A. M. Beauchemin, J. Am. Chem. Soc., 2011, 133, 20100 CrossRef CAS.
- C. R. Hesp, M. J. MacDonald, M. M. Zahedi, D. A. Bilodeau, S.-B. Zhao, M. Pesant and A. M. Beauchemin, Org. Lett., 2015, 17, 5136 CrossRef CAS.
- M. J. MacDonald, C. R. Hesp, D. J. Schipper, M. Pesant and A. M. Beauchemin, Chem.–Eur. J., 2013, 19, 2597 CrossRef CAS.
- For recent reviews: (a) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (b) S. S. John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582 RSC; (c) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955 CrossRef CAS.
- Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26 CrossRef CAS.
- Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554 CrossRef CAS.
- H. Lin, C. Wang, T. D. Bannister and T. M. Kamenecka, Chem.–Eur. J., 2018, 24, 9535 CrossRef CAS.
- S. S. John-Campbell, A. K. Ou and J. A. Bull, Chem.–Eur. J., 2018, 24, 17838 CrossRef.
- (a) Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 CrossRef CAS; (b) A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073 CrossRef CAS.
- Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884 CrossRef CAS.
- Y. Xie, B. Qian, P. Xie and H. Huang, Adv. Synth. Catal., 2013, 355, 1315 CrossRef CAS.
- (a) M. Ikawa and E. E. Snell, J. Am. Chem. Soc., 1954, 76, 4900 CrossRef CAS; (b) Y. Matsuo, J. Am. Chem. Soc., 1957, 79, 2016 CrossRef CAS; (c) K. Tatsumoto, M. Haruta and A. E. Martell, Inorg. Chim. Acta, 1987, 138, 231 CrossRef CAS; (d) V. M. Shanbhag and A. E. Martell, J. Am. Chem. Soc., 1991, 113, 6479 CrossRef CAS; (e) K.-Y. Choi, Biotechnol. Bioprocess Eng., 2015, 20, 988 CrossRef CAS.
- D. Wilson and N. R. Branda, Angew. Chem., Int. Ed., 2012, 51, 5431 CrossRef CAS.
- (a) B. J. Brazeau, B. J. Johnson and C. M. Wilmot, Arch. Biochem. Biophys., 2004, 428, 22 CrossRef CAS; (b) I. Bento, M. A. Carrondo and P. F. Lindley, J. Biol. Inorg Chem., 2006, 11, 539 CrossRef CAS; (c) I. S. MacPherson and M. E. P. Murphy, Cell. Mol. Life Sci., 2007, 64, 2887 CrossRef CAS; (d) A. Mukherjee, V. V. Smirnov, M. P. Lanci, D. E. Brown, E. M. Shepard, D. M. Dooley and J. P. Roth, J. Am. Chem. Soc., 2008, 130, 9459 CrossRef CAS; (e) M. Largerson, Org. Biomol. Chem., 2017, 15, 4722 RSC.
- Y. Xie, H. Pan, M. Liu, X. Xiao and Y. Shi, Chem. Soc. Rev., 2015, 44, 1740 RSC.
- L. Shi, C. Tao, Q. Yang, Y. E. Liu, J. Chen, J. Chen, J. Tian, F. Liu, B. Li, Y. Du and B. Zhao, Org. Lett., 2015, 17, 5784 CrossRef CAS.
- (a) X. Lan, C. Tao, X. Liu, A. Zhang and B. Zhao, Org. Lett., 2016, 18, 3658 CrossRef CAS; (b) Y. E. Liu, Z. Lu, B. Li, J. Tian, F. Liu, J. Zhao, C. Hou, Y. Li, L. Niu and B. Zhao, J. Am. Chem. Soc., 2016, 138, 10730 CrossRef CAS.
- J. Chen, X. Gong, J. Li, Y. Li, J. Ma, C. Hou, G. Zhao, W. Yuan and B. Zhao, Science, 2018, 360, 1438 CrossRef CAS.
- (a) H. Kuzuhara, N. Watanabe and M. Ando, J. Chem. Soc., Chem. Commun., 1987, 1987, 95 RSC; (b) J. T. Koh, L. Delaude and R. Breslow, J. Am. Chem. Soc., 1994, 116, 11234 CrossRef CAS , see also:; (c) L. Liu, M. Rozenman and R. Breslow, Bioorg. Med. Chem., 2002, 10, 3973 CrossRef CAS.
- (a) B. Xu, L.-L. Shi, Y.-Z. Zhang, Z.-J. Wu, L.-N. Fu, C.-Q. Luo, L.-X. Zhang, Y.-G. Peng and Q.-X. Guo, Chem. Sci., 2014, 5, 1988 RSC; (b) L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159 CrossRef CAS.
- W. Wen, L. Chen, M.-J. Luo, Y. Zhang, Y.-C. Chen, Q. Ouyang and Q.-X. Guo, J. Am. Chem. Soc., 2018, 140, 9774 CrossRef CAS.
- G. Liao, H.-M. Chen, Y.-N. Xia, B. Li, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 11464 CrossRef CAS.
- A. R. Buller, S. Brinkmann-Chen, D. K. Romney, M. Herger, J. Murciano-Calles and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14599 CrossRef CAS.
- J. Murciano-Calles, D. K. Romney, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, Angew. Chem., Int. Ed., 2016, 55, 11577 CrossRef CAS.
- D. K. Romney, J. Murciano-Calles, J. E. Wehrmüller and F. H. Arnold, J. Am. Chem. Soc., 2017, 139, 10769 CrossRef CAS.
- C. E. Boville, D. K. Romney, P. J. Almhjell, M. Sieben and F. H. Arnold, J. Org. Chem., 2018, 83, 7447 CrossRef CAS.
- D. K. Romney, N. S. Sarai and F. H. Arnold, ACS Catal., 2019, 9, 8726 CrossRef CAS.
- M. Dick, N. S. Sarai, M. W. Martynowycz, T. Gonen and F. H. Arnold, J. Am. Chem. Soc., 2019, 141, 19817 CrossRef CAS.
- Q. Guo, D. Wu, L. Gao, Y. Bai, Y. Liu, N. Guo, X. Du, J. Yang, X. Wang and X. Lei, ACS Catal., 2020, 10, 6291 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |