
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon monoxide insertion at a heavy p-block element: unprecedented formation of a cationic bismuth carbamoyl†
Jacqueline
Ramler
a,
Jordi
Poater
 bc,
Florian
Hirsch
d,
Benedikt
Ritschel
a,
Ingo
Fischer
d,
F. Matthias
Bickelhaupt
*ef and
Crispin
Lichtenberg
*a
bc,
Florian
Hirsch
d,
Benedikt
Ritschel
a,
Ingo
Fischer
d,
F. Matthias
Bickelhaupt
*ef and
Crispin
Lichtenberg
*a
aInstitute of Inorganic Chemistry, Julius-Maximilians Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: crispin.lichtenberg@uni-wuerzburg.de
bICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain
cUniversitat de Barcelona, Departament de Química Inorgànica i Orgànica & IQTCUB, Martí i Franquès 1-11, 08028 Barcelona, Spain
dInstitute of Physical Chemistry, Julius-Maximilians Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
eVrije Universiteit Amsterdam, Department of Theoretical Chemistry, Amsterdam Center for Multiscale Modeling (ACMM), The Netherlands. E-mail: f.m.bickelhaupt@vu.nl
fRadboud University, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
First published on 28th February 2019
Abstract
Major advances in the chemistry of 5th and 6th row heavy p-block element compounds have recently uncovered intriguing reactivity patterns towards small molecules such as H2, CO2, and ethylene. However, well-defined, homogeneous insertion reactions with carbon monoxide, one of the benchmark substrates in this field, have not been reported to date. We demonstrate here, that a cationic bismuth amide undergoes facile insertion of CO into the Bi–N bond under mild conditions. This approach grants direct access to the first cationic bismuth carbamoyl species. Its characterization by NMR, IR, and UV/vis spectroscopy, elemental analysis, single-crystal X-ray analysis, cyclic voltammetry, and DFT calculations revealed intriguing properties, such as a reversible electron transfer at the bismuth center and an absorption feature at 353 nm ascribed to a transition involving σ- and π-type orbitals of the bismuth-carbamoyl functionality. A combined experimental and theoretical approach provided insight into the mechanism of CO insertion. The substrate scope could be extended to isonitriles.
Introduction
The traditional distinction between transition metal and main-group compounds according to their physical and chemical properties has recently been challenged.1 This is due to the fact that researchers are currently in the process of understanding how to design main group compounds with properties including a well-defined one-electron-redox chemistry, small HOMO–LUMO gaps of <4 eV, and remarkable reactivities in the activation of small molecules such as H2, CO, CO2, and ethylene.Among the main group compounds, complexes of the heavy p-block elements with a principal quantum number of n > 4 show some inherent characteristics with high potential for applications in synthesis and catalysis. These include low homolytic bond dissociation energies,2 reversible homolytic bond cleavage/bond forming reactions,3 and large ionic radii, which allow for the design of cationic species with a considerable soft Lewis acidity. In the case of bismuth, a relatively low toxicity of the element and its complexes should also be noted.4 For compounds based on elements of the sixth row of the periodic table of the elements, relativistic effects come into play and may affect their physical and spectroscopic properties.5
When targeting highly reactive heavy main group species, low-valent compounds, complexes with element–element multiple bonds, and cationic species are promising candidates. However, the synthesis of such compounds may be extremely challenging. This is due to the inherent characteristics of heavy p-block elements, including (i) large ionic and covalent radii, which facilitate (unselective) oligomerization of low-valent species, (ii) relatively low redox potentials, which make over-reduction a serious synthetic problem, (iii) disproportionation reactions of mono-cationic complexes, and (iv) difficulties in accessing extremely Lewis-acidic and Lewis-basic species.
Despite these challenges, considerable efforts in designing heavy main group species that undergo well-defined reactions with small molecules have been crowned with success. The distannyne Sn2Aryl2 has been shown to reversibly add two equivalents of ethylene across its Sn![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Sn bond to give the corresponding distannane (Scheme 1a, Aryl = C6H3-2,6(C6H3-2,6-iPr2)2).6,7 This distannyne and closely related derivatives also react with dihydrogen to give a dinuclear tin species with two bridging hydride ligands, a reaction that has recently been shown to be reversible (Scheme 1b).8 In an alternative strategy without any E–E multiple bonding, the hypervalent bismuth compound Bi(C6H4CH2)2NtBu(OMe) with one polar Bi–O bond has been shown to undergo reversible CO2 fixation under mild reaction conditions (Scheme 1c).9,10
Sn bond to give the corresponding distannane (Scheme 1a, Aryl = C6H3-2,6(C6H3-2,6-iPr2)2).6,7 This distannyne and closely related derivatives also react with dihydrogen to give a dinuclear tin species with two bridging hydride ligands, a reaction that has recently been shown to be reversible (Scheme 1b).8 In an alternative strategy without any E–E multiple bonding, the hypervalent bismuth compound Bi(C6H4CH2)2NtBu(OMe) with one polar Bi–O bond has been shown to undergo reversible CO2 fixation under mild reaction conditions (Scheme 1c).9,10
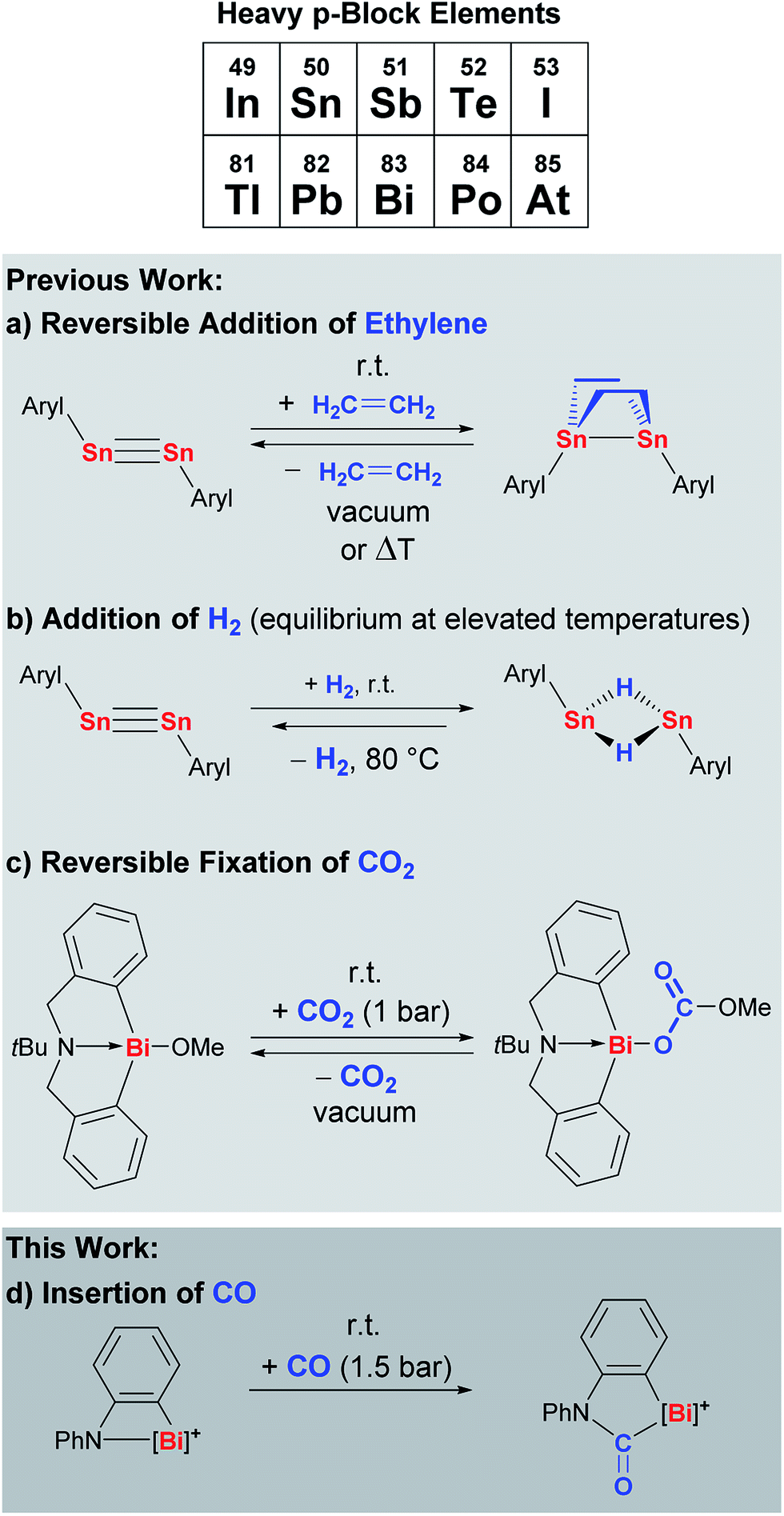 | ||
| Scheme 1 Examples of well-defined compounds of heavy p-block elements that show unusual reactivity patterns towards small molecules. Aryl = C6H3-2,6(C6H3-2,6-iPr2)2. | ||
Apart from H2, CO2, and ethylene, carbon monoxide is an intriguing substrate for insertion chemistry, since it represents an attractive, versatile C1-building block in synthetic chemistry. Traditionally, its utilization for the synthesis of organic molecules such as alcohols, aldehydes, or carboxamides has been realized with transition metal carbonyl species acting as critical intermediates.11 According to the Dewar–Chatt–Duncanson model, the bond between CO and a transition metal constitutes of ligand-to-metal σ-donation and metal-to-ligand π-back-donation. It has been demonstrated that π-back-donation makes an important or even crucial contribution to the overall bonding energy of most synthetically relevant carbonyl compounds.12 This renders carbon monoxide an extremely challenging substrate for compounds based on heavy p-block elements, because synthetically accessible species of this kind usually lack occupied orbitals of sufficient symmetry and energy for π-back-donation. As a result, insights in this field of research are very limited.13,14 Electrochemical sensing of CO at SnO2 or In2O3 surfaces at elevated temperatures has been related to the formation of carbonate and carboxylate species by IR spectroscopy.15 Matrix isolation techniques allowed for the IR spectroscopic characterization of tin and lead carbonyl species such as SnCO, Sn(CO)2, SnnCO, PbnCO, and SnCO− at 7–20 K (n = 1–4).16 These reports have been complemented by additional purely theoretical studies on these and related compounds.17 To the best of our knowledge, however, well-defined insertion reactions of carbon monoxide with compounds based on heavy p-block elements showing a principal quantum number of n > 4 have not been reported to date.
Cationic bismuth amides combine a considerable Lewis acidity with a good accessibility and a high reactivity, making them promising candidates for small molecule activation.18–20 Insertion reactions with carbon monoxide in specific would grant direct access to cationic bismuth carbamoyls, an unprecedented structural motif with high potential for the stabilization of (intermediate) radical species (Scheme 1d).21
Here we demonstrate that a well-defined cationic bismuth amide undergoes highly selective and facile insertion reactions with carbon monoxide and isonitriles under mild conditions, yielding the first examples of cationic bismuth carbamoyls and amidinoyls, which were isolated and fully characterized.
Results and discussion
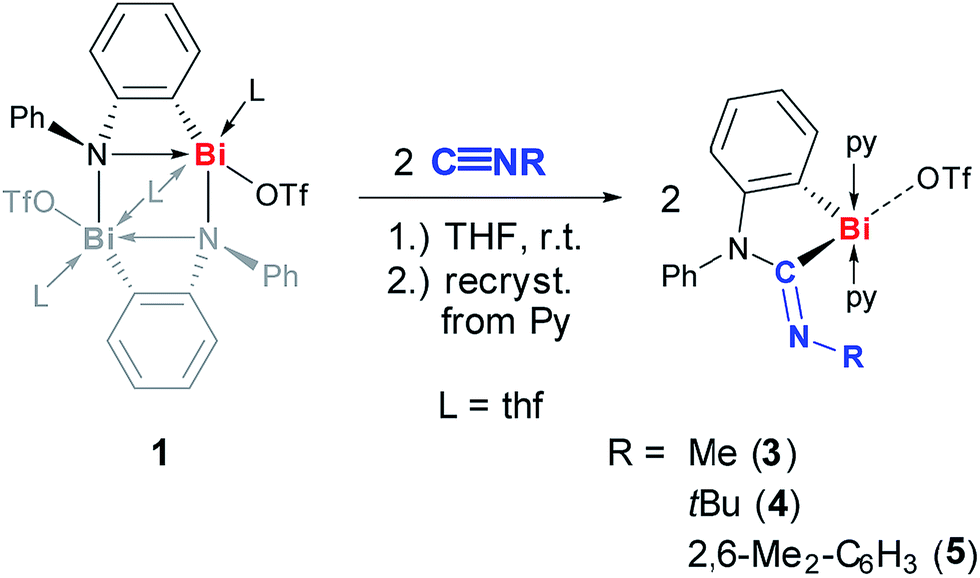
The dinuclear cationic bismuth compound [Bi2(N(C6H4)Ph)2(OTf)2(thf)3] (1) has recently been reported and was synthesized according to the literature (Scheme 2; Tf = SO2CF3).19a,22 The prominent structural feature of compound 1 is a four-membered C2NBi ring. The geometric parameters dictated by the phenylene unit suggest a considerable ring strain and lead to a significantly elongated Bi–N bond as part of the four-membered ring.19 These characteristics seem predetermined for compound 1 to be highly active in small molecule activation reactions. Thus, compound 1 was reacted with carbon monoxide, which represents an extraordinarily challenging substrate in the chemistry of heavier group 15 compounds. When a suspension of yellow 1 in THF was exposed to an atmosphere of 1.5 bar CO the precipitation of a pale yellow solid indicated the consumption of the starting materials. Whereas a quantitative reaction monitoring by IR and NMR spectroscopy was hampered by the moderate to poor solubility of the starting material and the product in THF, these methods still indicated significant conversion of 1 after 1 h. However, extended reaction times of 2 d were necessary to reach full conversion of 1, which was otherwise difficult to separate from the product. Recrystallization of the crude precipitate from pyridine/Et2O/pentane gave the insertion product 2 in 61% yield (Scheme 2).23 Compound 2 is moderately soluble in THF, but adequately soluble in pyridine. In pyridine solution, it is stable at ambient temperature and under ambient lighting for at least five weeks and at 80 °C for at least 2 h. NMR spectroscopy indicated the substitutional lability of the pyridine ligands, which can be removed in vacuo at 80 °C. 1H and 13C and 19F NMR spectroscopic analyses revealed the expected signal patterns for the functional groups present in 2 (a phenylene and a phenyl group, two pyridine ligands, and a triflate). An unusually large 13C NMR chemical shift of 251 ppm was recorded for the CONR2 group, which was ascribed to its interaction with the Lewis acidic cationic bismuth center.24 IR spectroscopic analyses revealed a CO stretching frequency of 1637 cm−1 for the carbamoyl functional group. This is close to those reported for carbamoyl compounds of Sn and Pb, which were prepared by classical salt elimination protocols.25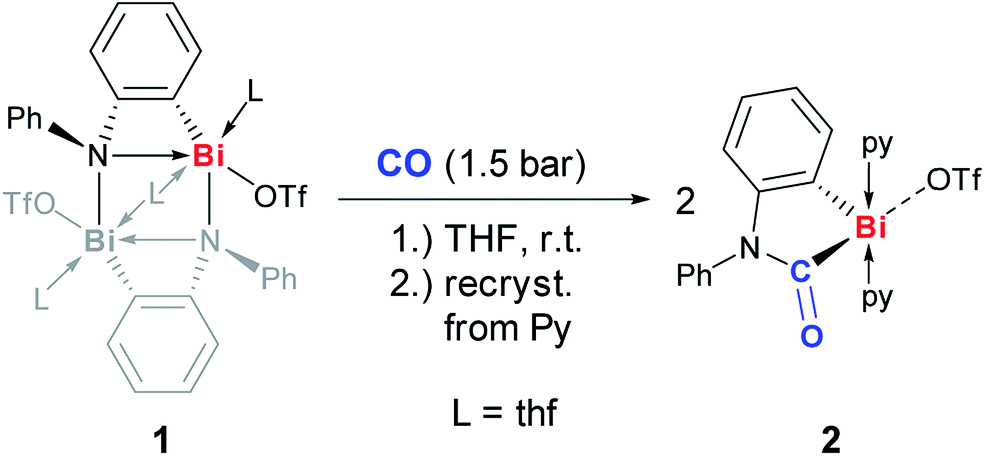 | ||
| Scheme 2 Insertion of CO into Bi–N bond of (masked) cationic bismuth amide 1 to give cationic bismuth carbamoyl compound 2; py = pyridine, Tf = SO2CF3. | ||
The molecular structure of 2 was unambiguously identified by single-crystal X-ray diffraction (monoclinic space group P21/n with Z = 4; Fig. 1). This confirmed the insertion of CO into the former Bi–N bond, resulting in formation of a bismuth carbamoyl moiety, an unprecedented structural motif.26 The Bi1–C13 bond is considerably longer than the Bi1–C2 bond (Δ = +0.11 Å). Despite the cationic nature of 2,22 the Bi–(CONR2) bond length is in the same range as that of electronically stabilized Bi–C bonds in neutral compounds such as Bi(CH(SiMe3)2)3, Bi(η1-2-Me-allyl)3, or Bi(CF2CF3)3.27 Accordingly, the C13–N1 distance of 1.38 Å is in agreement with a partial C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond character. According to an energy decomposition analysis (EDA) of 2 and related species, these differences in Bi–C bond lengths are also reflected by the corresponding interaction energies: in homolytic bond dissociations, the interaction energy of the Bi–Aryl bond is 17.9 kcal mol−1 larger than that of the Bi–CONR2 bond (ESI†). The covalent character of both types of bonds amounts to 44–49% according to EDA. In the solid state, 2 forms a loosely associated contact ion pair. The Bi1⋯O2 distance of 3.16 Å is 12% below the sum of the van-der-Waals radii, but significantly elongated compared to the Bi–OTf bond of 2.37 Å in starting material 1.19a The coordination geometry around the bismuth center in 2 is saturated by two pyridine ligands in trans-position to each other. Overall, this results in a distorted square pyramidal coordination geometry around bismuth with C2 in the apical position (τ5 = 0.17).28
N double bond character. According to an energy decomposition analysis (EDA) of 2 and related species, these differences in Bi–C bond lengths are also reflected by the corresponding interaction energies: in homolytic bond dissociations, the interaction energy of the Bi–Aryl bond is 17.9 kcal mol−1 larger than that of the Bi–CONR2 bond (ESI†). The covalent character of both types of bonds amounts to 44–49% according to EDA. In the solid state, 2 forms a loosely associated contact ion pair. The Bi1⋯O2 distance of 3.16 Å is 12% below the sum of the van-der-Waals radii, but significantly elongated compared to the Bi–OTf bond of 2.37 Å in starting material 1.19a The coordination geometry around the bismuth center in 2 is saturated by two pyridine ligands in trans-position to each other. Overall, this results in a distorted square pyramidal coordination geometry around bismuth with C2 in the apical position (τ5 = 0.17).28
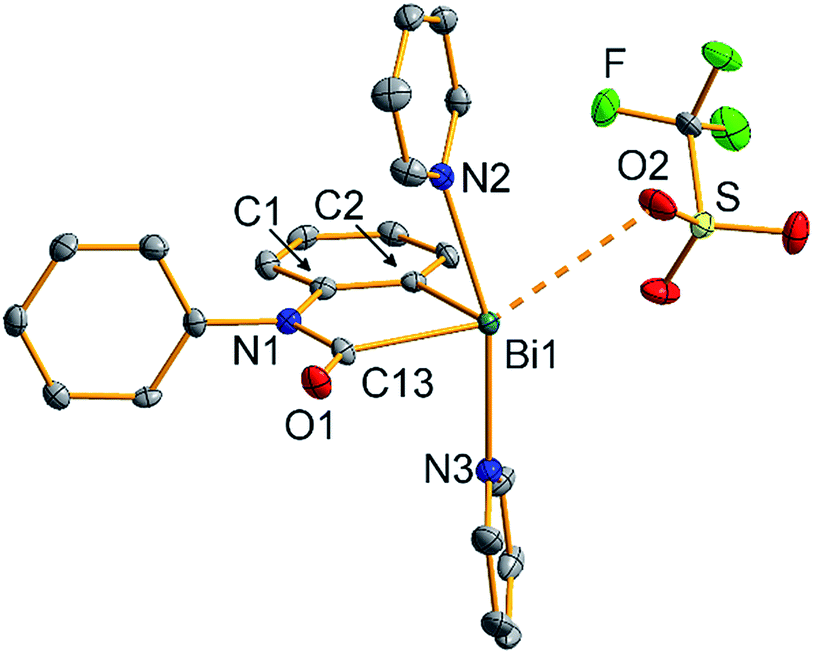 | ||
| Fig. 1 Molecular structure of [Bi(CONPh(C6H4))(NC5H5)2][OTf] (2) in the solid state. Displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Bi1–C2, 2.227(5); Bi1–C13, 2.335(5); Bi1–N2, 2.520(4); Bi1–N3, 2.471(4); Bi1⋯O2, 3.164(4); C13–O1, 1.213(5); C13–N1, 1.382(6); C2–Bi1–C13, 76.60(16); C2–Bi1–N2, 84.21(15); C2–Bi1⋯O2, 103.25(14); N2–Bi1–N3, 163.56(13); C13–Bi1⋯O2, 153.50(14); τ5 = 0.17. | ||
The electrochemical behavior of 2 was investigated by cyclic voltammetry in THF/0.1 M [N(nBu)4][PF6] at 23 °C, which revealed a chemically reversible redox event at an only moderately negative potential of −1.66 V vs. Fc/Fc+ (Fig. 2a and ESI;† Fc = Fe(C5H5)2). According to DFT calculations, the radical generated upon reduction is free of neutral donor ligands, [Bi(CONPh(C6H4))]˙ (2red). 74% of the spin density are located at the bismuth center, occupying a molecular orbital with dominating contributions from a Bi-6p-type atomic orbital (Fig. 2b and ESI†). The remaining spin density is delocalized over the phenylene and the carbamoyl unit of 2red, with spin densities of up to 5% at single C, N, and O atoms. This delocalization of spin density is believed to contribute to the stability of this species under the conditions of the electrochemical experiment. While two examples of isolable BiII radical species and one example of a persistent BiII radical species have recently been reported,3a,29 this is the first example of a quasi-reversible electron transfer at a mononuclear bismuth compound with the bismuth atom being the major spin carrier, demonstrating the potential of the bismuth carbamoyl functional group for the stabilization of (intermediate) radical species.30 The pale yellow compound 2 was further investigated by UV/vis spectroscopy in THF solution, which revealed a broad absorption band at λmax = 353 nm (FWHM = 96 nm; Fig. 3). In dilute solutions, the substitutionally labile pyridine ligands of 2 can presumably be exchanged for thf ligands (ESI†). TD-DFT calculations at the CAMY-B3LYP/TZ2P level of theory on [Bi(CONPh(C6H4))(thf)2]+ indicated a HOMO−1/LUMO singlet–singlet transition at λ = 354 nm, when spin–orbit-coupling was taken into account using perturbation theory (for details see ESI†). This transition is suggested to correspond to the experimentally observed band at λmax = 353 nm. The HOMO−1 shows major contributions by orbitals forming the Bi–C and C–N σ-bonds in the carbamoyl group and by an oxygen-centered p-type orbital. The LUMO consists of a bismuth-centered p-orbital and the π*-orbital of the CO group, both of which are orthogonal to the plane defined by the carbamoyl functionality. Thus, the bismuth carbamoyl functional group is directly involved in the electronic transition that is triggered by light in the UV/vis region (λ ≈ 310–400 nm).
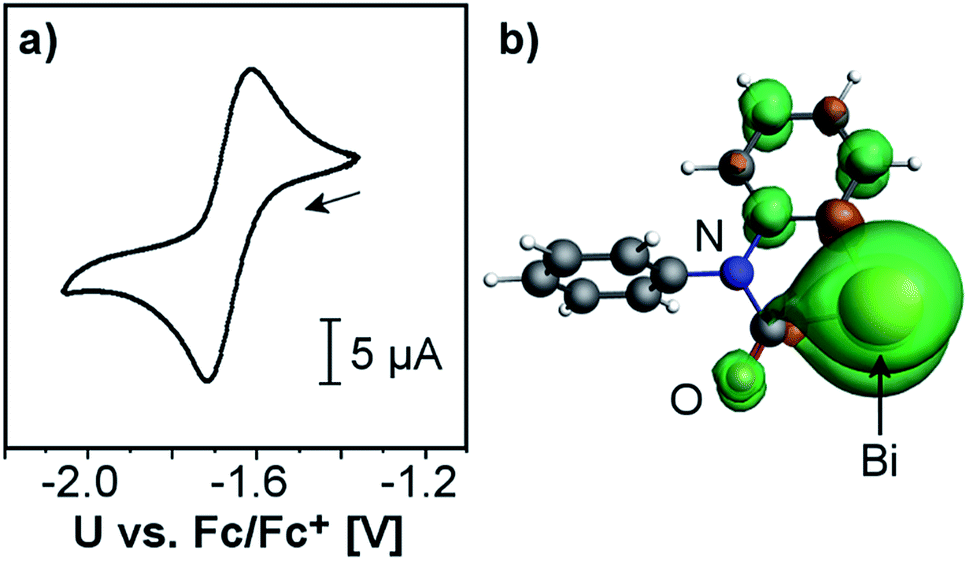 | ||
| Fig. 2 (a) Cyclic voltammogram of 2 in THF/0.1 M [N(nBu)4][PF6] at 23 °C with a scan rate of 500 mV s−1; Fc = Fe(C5H5)2. (b) Optimized geometry and spin density distribution (iso value = 0.001) of reduced species [Bi(CONPh(C6H4))]˙ (2red). | ||
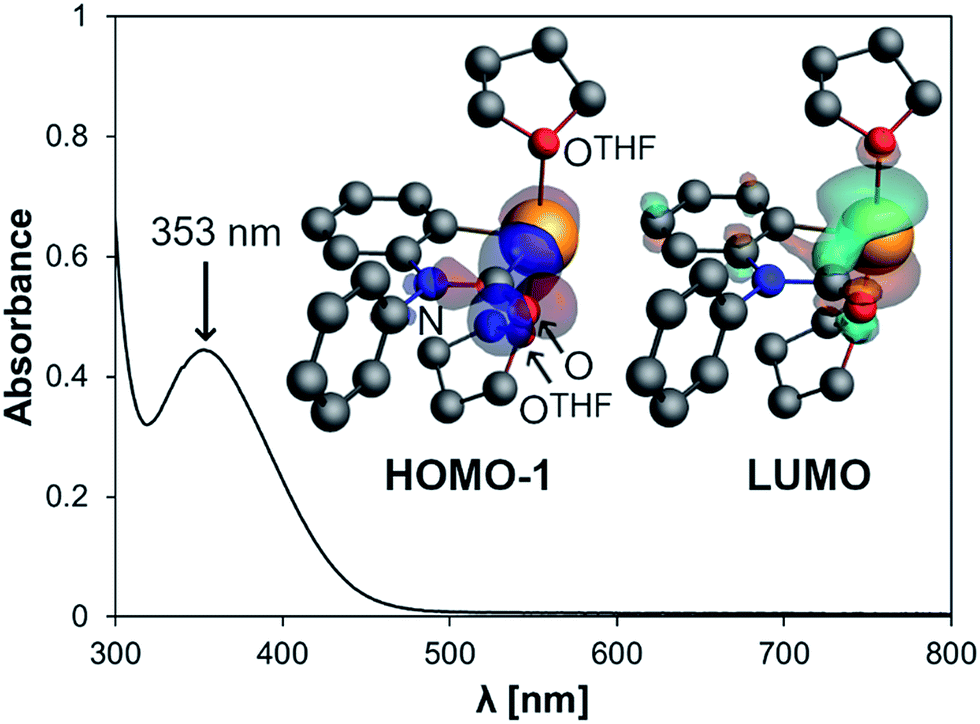 | ||
| Fig. 3 UV/vis spectrum of 2 (0.4 mM solution in THF) and orbitals involved in the transition at 353 nm according to TD-DFT calculations on [Bi(CONPh(C6H4))(thf)2]+. | ||
The mechanism of CO insertion was investigated by DFT calculations (Scheme 3, for details see ESI†).31 All values are given in kcal mol−1. We suggest coordination of CO to a Lewis-acidic bismuth center to be the initiating reaction step leading to intermediate I-1 (ΔH: +9.7; ΔG: +5.9).32 Insertion of the CO molecule proceeds via transition state TS-1, in which the CO unit bridges a Bi and an N atom (barrier: ΔH≠: +8.5; ΔG≠: +10.5), to give intermediate I-2 in an almost thermoneutral reaction (ΔH: +0.2; ΔG: +3.0). A similar sequence of CO-coordination (to give I-3) and formation of a transition state with a Bi⋯(CO)⋯N unit (TS-2, barrier: ΔH≠: +9.3; ΔG≠: +11.6) and insertion leads to dinuclear intermediate I-4. Coordination of THF gives the monomeric compound I-5, which is suggested to be the species that precipitates from the reaction mixture, when 1 is reacted with CO in THF (vide supra).23 Up to this point the overall reaction is exothermic and endergonic according to DFT calculations (ΔH: −6.7; ΔG: +13.0), and the formation of I-5 is presumably aided by its poor solubility in THF. The exchange of THF for pyridine ligands to give isolated product 2 is exothermal and exergonic, reflecting the Lewis acidity of the bismuth atom (ΔH: −14.4; ΔG: −14.2). Monitoring the reaction by IR spectroscopy indicated the accumulation of I-5 in solution until its saturation concentration was reached (characteristic band at 1592 cm−1).23 Whereas intermediates I-1, I-2, and I-3 could not unambiguously be identified due to their low concentrations, signal overlapping, or their short-lived character, bands at 1655 and 1669 cm−1 were tentatively assigned to intermediate I-4 (ESI†).
Putting the unusual reactivity of 1 towards carbon monoxide into the broader context of main group chemistry reveals some remarkable points. Compounds of strongly electropositive elements of Groups 1 and 2, as well as light(er) elements of Group 13 (B–Ga) undergo insertion with CO; examples include species such as nBuLi, LiN(iPr)2, MgEtBr, or Al(tBu)3.33,34 These reactivity patterns may be interpreted as a result of the strong polarization of the M–X bond (X = C, N) and the high Lewis acidity of the main-group compound. In addition, compounds of Group 13 and 14 elements with low-valent character or with E–E (single or multiple) bonds have been reported to undergo intriguing insertion reactions with carbon monoxide (E = Group 13/14 element). Examples include diborynes, diboracumulenes, carbenes, germylenes and a disila-Dewar-benzene.35–37 When it comes to well-defined compounds of the pnictogens P–Bi, selective insertion reactions with CO under mild conditions have only been reported for phosphorous species. Two different strategies have been reported in this context. Frustrated Lewis pairs (FLPs) with a phosphane moiety as the Lewis basic component react with CO under mild conditions to form heterocyclic compounds.38–43 The second strategy exploits the reactivity of a phosphorus biradicaloid, which inserts CO under harsher conditions to give a P2N2C ring motif that maintains the biradicaloid character.44 Overall, compound 1 represents the first molecular heavy p-block compound (n > 4) to undergo a direct and well-defined insertion reaction with carbon monoxide under mild conditions. The Lewis acidity and geometric constraints of 1 play key roles in determining its remarkable reactivity. It may be considered to interpret these driving forces in terms of FLP chemistry with a Lewis basic N and a Lewis acidic Bi site.45c,d To the best of our knowledge, this would be the first example of FLP chemistry of molecular compounds being extended to the heavy main group element bismuth.45–47
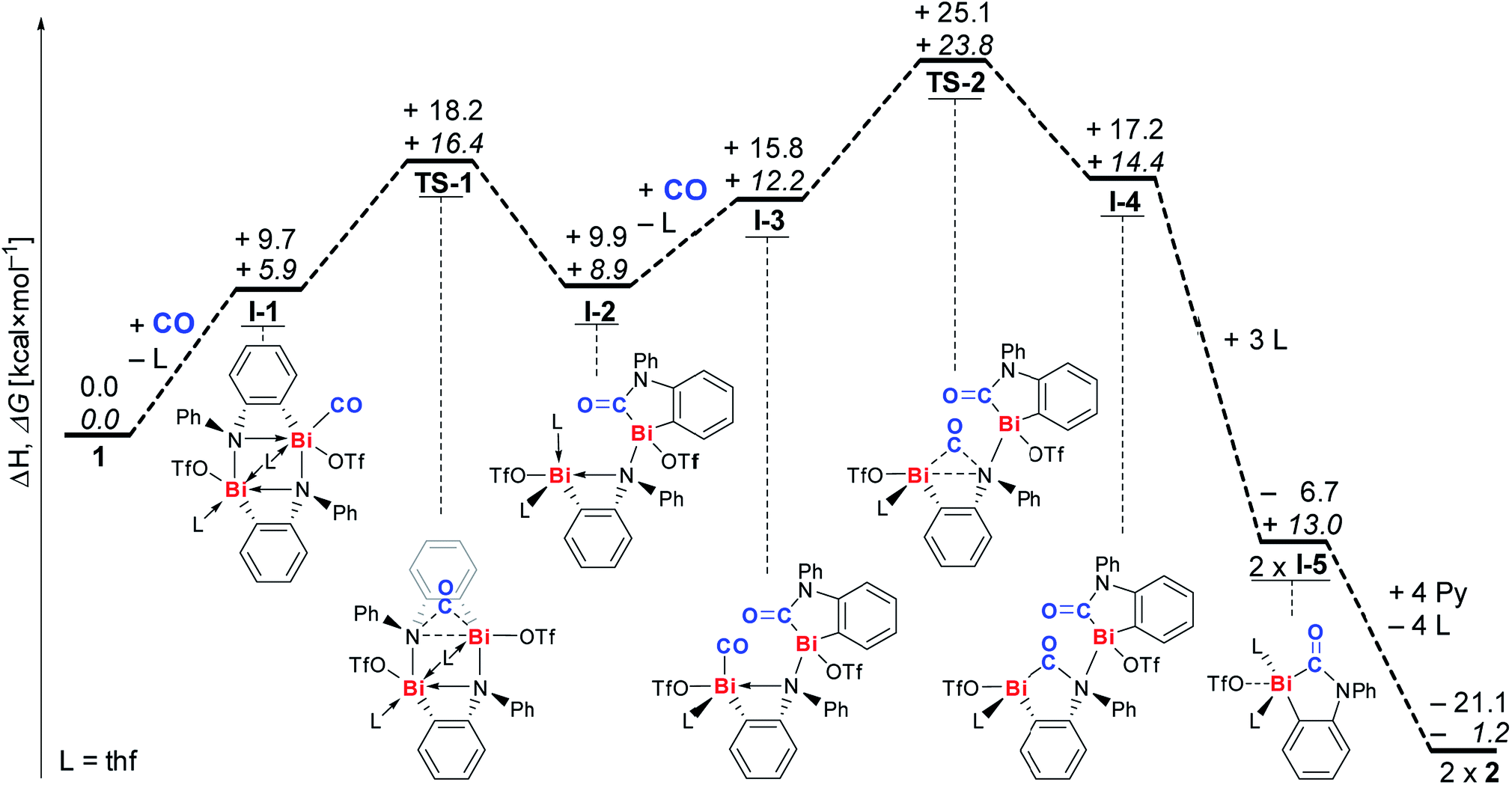 | ||
| Scheme 3 Proposed mechanism for reaction of 1 with CO to give insertion product 2; ΔH values (regular font) and ΔG values (italic) are given in kcal mol−1. L = thf. | ||
In order to extend the scope of the unusual insertion reactions of 1 with unsaturated small molecules, isonitriles were targeted due to their isoelectronic and isolobal relationship with carbon monoxide. Well-defined insertion reactions of pnictogen compounds with isonitriles are rare. Transition metal-stabilized phosphinidenes and arsinidenes [E(C5Me5)(W(CO)5)2] (E = P, As) have been reported to coordinate sterically demanding CNtBu and to undergo insertion reactions with less bulky isonitriles CNR (R = cyclo-hexyl, nBu).48 Only recently, the first example of an insertion reaction between a bismuth compound and an isonitrile has been documented: the strongly nucleophilic nature of the boryl group in [Ph2Bi(BR2)] allowed for isolation of the insertion product [Ph2Bi(C(NPh)BR2)] in 22% yield, when CNPh was used as the substrate (BR2 = 1,2,4,3-triazaborol-3-yl).49 Remarkably, the alkyl isonitrile CN(nBu) was found to undergo fragmentation with formation of [BiPh2(nBu)], implying that isonitrile insertion is not a general reaction pathway for this bismuth boryl compound.
At ambient temperature, compound 1 readily reacted with isonitriles, CNR, that bear alkyl or aryl substituents with a steric profile varying from extremely low (R = Me) to high (R = tBu, 2,6-Me2-C6H3) (Scheme 4). Monitoring the reaction by 1H-NMR spectroscopy in the case of R = tBu revealed full conversion within 10 min and a high spectroscopic yield of >95%. The insertion products 3–5 were isolated in 60–79% yield after re-crystallization from pyridine and fully characterized (Scheme 4, Fig. 4). In all cases the bismuth amidinoyl moiety was exclusively formed in its Z-configuration according to NMR spectroscopy. Compounds 3–5 show 13C NMR chemical shifts between 210 and 223 ppm and IR bands at 1606–1637 cm−1 for the CNRNR2 functionality.
 | ||
| Scheme 4 Insertion of CNR into Bi–N bond of (masked) cationic bismuth amide 1 to give cationic bismuth amidinoyl compounds 3, 4, and 5; py = pyridine, Tf = SO2CF3. | ||
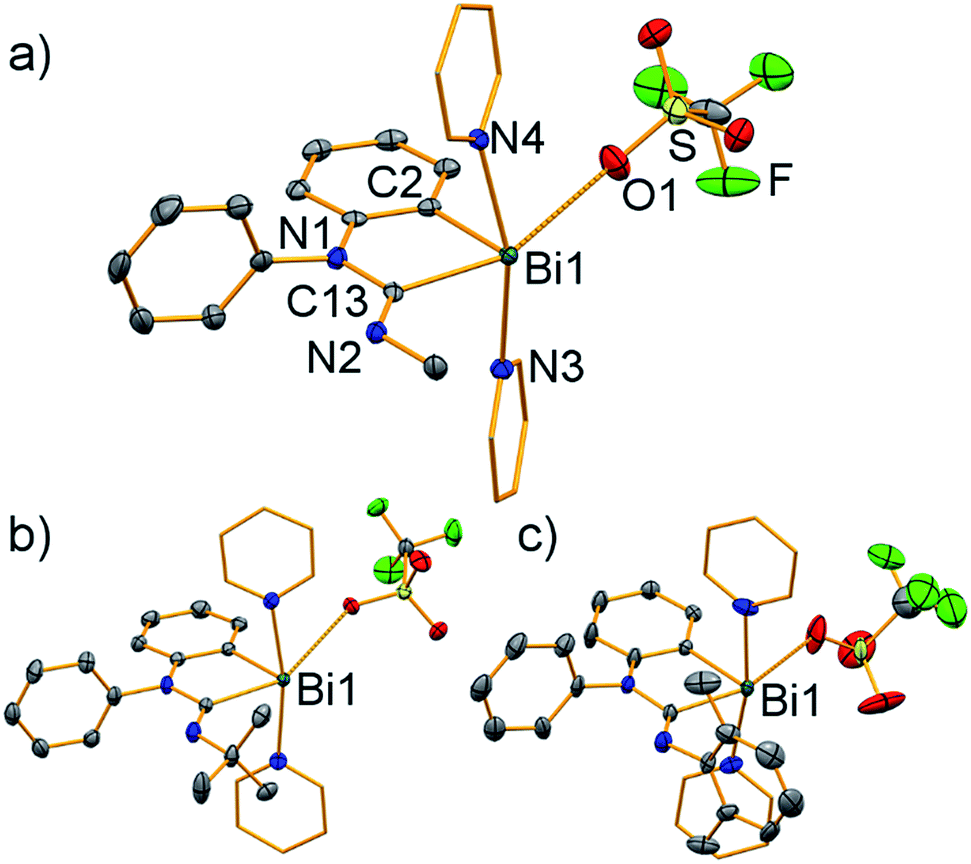 | ||
| Fig. 4 Molecular structures of [Bi(CNRNPh(C6H4))(NC5H5)2][OTf] in the solid state ((a) 3 (R = Me); (b) 4 (R = tBu); (c) 5 (R = 2,6-Me2-C6H3)). Displacement ellipsoids are shown at the 50% probability level; carbon atoms of pyridine ligands are shown as wireframe for clarity. Hydrogen atoms, lattice-bound solvent molecules, and split positions of disordered groups in (c) are omitted for clarity. Atom labelling in (b) and (c) is in analogy with that in (a). Selected bond lengths (Å) and angles (°): (a) Bi1–C2, 2.222(3); Bi1–C13, 2.305(3); Bi1–N3, 2.499(2); Bi1–N4, 2.482(2); Bi1⋯O1, 3.025(2); C13–N1, 1.402(3); C13–N2, 1.263(3); C2–Bi1–C13, 76.84(10); C2–Bi1–N3, 84.48(8); C2–Bi1⋯O1, 79.86(9); N3–Bi1–N4, 164.35(7); C13–Bi1⋯O1, 155.56(8); τ5 = 0.15. (b) Bi1–C2, 2.225(3); Bi1–C13, 2.331(3); Bi1–N3, 2.485(3); Bi1–N4, 2.525(3); Bi1⋯O1, 2.997(2); C13–N1, 1.415(4); C13–N2, 1.261(4); C2–Bi1–C13, 77.35(12); C2–Bi1–N3, 87.63(11); C2–Bi1⋯O1, 80.94(10); N3–Bi1–N4, 169.39(9); C13–Bi1⋯O1, 153.18(9); τ5 = 0.27. (c) Bi1–C2, 2.202(4); Bi1–C13, 2.299(4); Bi1–N3, 2.510(4); Bi1–N4, 2.488(4); Bi1⋯O1, 2.907(15); C13–N1, 1.43(2); C13–N2, 1.269(6); C2–Bi1–C13, 76.89(15); C2–Bi1–N3, 84.96(14); C2–Bi1⋯O1, 82.0(3); N3–Bi1–N4, 169.41(13); C13–Bi1⋯O1, 158.8(3); τ5 = 0.18. | ||
Single-crystal X-ray diffraction analyses revealed a very similar coordination behavior for compounds 3–5 (Fig. 4). In each case, the bismuth atom is found in distorted square pyramidal coordination geometry with C2 in the apical position (τ5 = 0.15–0.27). The Bi–C distances of the amidinoyl unit (Bi1–C13, 2.30–2.33 Å) are significantly larger than those involving the aryl group (Bi1–C2, 2.20–2.23 Å). The N1–C13 bond lengths of 1.40–1.43 Å suggest partial double bond character, albeit they are larger than those in carbamoyl compound 2 (1.38 Å).
Conclusions
In summary, we have reported the facile insertion of carbon monoxide into a bismuth–nitrogen bond, exploiting the Lewis acidity of the cationic bismuth compound and the release of ring strain as driving forces of the reaction. To the best of our knowledge, this is the first example of a molecular compound based on a heavy p-block element with the principal quantum number n > 4 to undergo a well-defined insertion reaction with carbon monoxide under mild conditions. This grants access to a novel cationic bismuth carbamoyl species, which allows for the first time to realize a chemically reversible redox process at the bismuth center of a mononuclear molecular compound. The pale yellow color of this compound was attributed to a singlet-singlet-transition that involves the bismuth carbamoyl functionality and is facilitated by spin–orbit coupling. Isonitriles were shown to analogously undergo these unusual insertion reactions. These results reveal the potential of heavy main group compounds in small-molecule activation, reversible electron–transfer reactions at heavy main group elements, and photochemistry targeting the accessibility of excited states with visible light.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. L. thanks Dr R. D. Dewhurst for helpful discussions and Prof. H. Braunschweig for continuous support. Funding by the Fonds der Chemischen Industrie (Liebig fellowship to C. L.), the DFG (funding to C. L. and GRK 2112), the Netherlands Organization for Scientific Research (NWO), the Catalan Government (2017SGR348), and the Spanish MINECO (CTQ2016-77558-R and MDM-2017-0767) is acknowledged.Notes and references
- (a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (b) P. P. Power, Chem. Rev., 2003, 103, 789–809 CrossRef CAS PubMed; (c) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (d) M.-A. Légaré, C. Pranckevicius, H. Braunschweig, Chem. Rev. DOI:10.1021/acs.chemrev.8b00561; (e) C. Lichtenberg, Angew. Chem., Int. Ed., 2016, 55, 484–486 CrossRef CAS PubMed; (f) C. Weetman, S. Inoue, ChemCatChem, 2019, 10, 4213–4228 Search PubMed.
- E.g.: (a) L. D. Freedman and G. O. Doak, Chem. Rev., 1982, 82, 15–57 CrossRef CAS; (b) G. P. Smith and R. Patrick, Int. J. Chem. Kinet., 1983, 15, 167–185 CrossRef CAS and references therein.
- E.g.: (a) S. Ishida, F. Hirakawa, K. Furukawa, K. Yoza and T. Iwamoto, Angew. Chem., Int. Ed., 2014, 53, 11172–11176 CrossRef CAS PubMed; (b) D. Dakternieks, D. J. Henry and C. H. Schiesser, Organometallics, 1998, 17, 1079–1084 CrossRef CAS; (c) M. Lehnig and K. Dören, J. Organomet. Chem., 1981, 210, 331–341 CrossRef CAS.
- E.g.: J. M. Bothwell, S. W. Krabbe and R. S. Mohan, Chem. Soc. Rev., 2011, 40, 4649–4707 RSC and references therein.
- (a) P. Pyykkö, Chem. Rev., 1988, 88, 563–594 CrossRef; (b) P. Pyykkö, Annu. Rev. Phys. Chem., 2012, 63, 45–64 CrossRef PubMed; (c) R. J. F. Berger, D. Rettenwander, S. Spirk, C. Wolf, M. Patzschke, M. Ertl, U. Monkowius and N. W. Mitzel, Phys. Chem. Chem. Phys., 2012, 14, 15520–15524 RSC; (d) A. Wodyński, O. L. Malkina and M. Pecul, J. Phys. Chem. A, 2016, 120, 5624–5634 CrossRef PubMed.
- Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668–1670 CrossRef CAS PubMed.
- For the insertion of ethylene into the Sn–C bond of a diarylstannylen see: T. Y. Lai, J.-D. Guo, J. C. Fettinger, S. Nagase and P. P. Power, Chem. Commun., 2019, 55, 405–407 RSC.
- (a) Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard and P. P. Power, Chem. Commun., 2008, 6042–6044 RSC; (b) S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590–593 CrossRef CAS PubMed.
- S.-F. Yin, J. Maruyama, T. Yamashita and S. Shimada, Angew. Chem., Int. Ed., 2008, 47, 6590–6593 CrossRef CAS PubMed.
- For reactions of CO2 with bismuth compounds, also see: (a) H. J. Breunig, L. Königsmann, E. Lork, M. Nema, N. Philipp, C. Silvestru, A. Soran, R. A. Varga and R. Wagner, Dalton Trans., 2008, 1831–1842 RSC; (b) K. Wójcik, T. Rüffer, H. Lang, A. A. Auer and M. Mehring, J. Organomet. Chem., 2011, 696, 1647–1651 CrossRef; (c) D. R. Kindra, I. J. Casely, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 7777–7787 CrossRef CAS PubMed.
- (a) H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC; (b) N. Rajesh, N. Basu and B. Sundararaju, Tetrahedron Lett., 2018, 59, 862–868 CrossRef CAS; (c) A. S. Veige, Polyhedron, 2008, 27, 3177–3189 CrossRef CAS.
- (a) K. Nakashima, X. Zhang, M. Xiang, Y. Lin, M. Lin and Y. Mo, J. Theor. Comput. Chem., 2008, 7, 639–654 CrossRef CAS; (b) G. Bistoni, S. Rampino, N. Scafuri, G. Ciancaleoni, D. Zuccaccia, L. Belpassi and F. Tarantelli, Chem. Sci., 2016, 7, 1174–1184 RSC; (c) A. Diefenbach, F. M. Bickelhaupt and G. Frenking, J. Am. Chem. Soc., 2000, 122, 6449–6458 CrossRef CAS.
- Iodine reacts with carbon monoxide and water to give carbon dioxide and hydrogen iodide: W. M. MacNevin and W. N. Carson Jr, J. Am. Chem. Soc., 1950, 72, 42–44 CrossRef CAS.
- Laser photolysis of HI in a carbon monoxide matrix allows for the detection of the formyl radical: J. A. Brivati, N. Keen and M. C. R. Symons, J. Chem. Soc., 1962, 237–243 RSC.
- (a) P. G. Harrison and M. J. Willet, Nature, 1988, 332, 337–339 CrossRef; (b) M.-I. Baraton, L. Merhari, H. Ferkel and J.-F. Castagnet, Mater. Sci. Eng., C, 2002, 19, 315–321 CrossRef.
- (a) A. Bos, J. Chem. Soc., Chem. Commun., 1972, 26–27 RSC; (b) L. N. Zhang, J. Dong and M. F. Zhou, J. Chem. Phys., 2000, 113, 8700–8705 CrossRef CAS; (c) L. Jiang and X. Qiang, Bull. Chem. Soc. Jpn., 2006, 79, 857–863 CrossRef CAS; (d) L. Jiang and Q. Xu, J. Chem. Phys., 2005, 122, 034506 CrossRef PubMed.
- (a) Y. Lu, H. Wang, Y. Xie, H. Liu and H. F. Schaefer, Inorg. Chem., 2014, 53, 6252–6256 CrossRef CAS PubMed; (b) C. Gourlaouen, O. Parisel and J.-P. Piquemal, J. Chem. Phys., 2010, 133, 124310 CrossRef CAS PubMed; (c) A. J. Bridgeman, Inorg. Chim. Acta, 2001, 321, 27–41 CrossRef CAS.
- H. Dengel and C. Lichtenberg, Chem.–Eur. J., 2016, 22, 18465–18475 CrossRef CAS PubMed.
- (a) B. Ritschel, J. Poater, H. Dengel, F. M. Bickelhaupt and C. Lichtenberg, Angew. Chem., Int. Ed., 2018, 57, 3825–3829 CrossRef CAS PubMed; (b) B. Ritschel and C. Lichtenberg, Synlett, 2018, 29, 2213–2217 CrossRef CAS.
- (a) R. J. Schwamm, B. M. Day, M. P. Coles and C. M. Fitchett, Inorg. Chem., 2014, 53, 3778–3787 CrossRef CAS PubMed; (b) R. J. Schwamm, M. P. Coles and C. M. Fitchett, Dalton Trans., 2017, 46, 4066–4074 RSC; (c) C. Hering-Junghans, M. Thomas, A. Villinger and A. Schulz, Chem.–Eur. J., 2015, 21, 6713–6717 CrossRef CAS PubMed; (d) M. Veith, B. Bertsch and V. Huch, Z. Anorg. Allg. Chem., 1988, 559, 73–88 CrossRef CAS.
- For examples of intermediate carbamoyl radical species in synthetic chemistry see: (a) F. Minisci, F. Fontana, F. Coppa and Y. M. Yan, J. Org. Chem., 1995, 60, 5430–5433 CrossRef CAS; (b) Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172–2175 CrossRef CAS PubMed; (c) G. López-Valdez, S. Olguín-Uribe and L. D. Miranda, Tetrahedron Lett., 2007, 48, 8285–8289 CrossRef.
- Compounds 1 and 2 represent contact ion pairs in the solid state; the term “cationic species” is used to describe the weak interactions between the triflate anions and the bismuth atoms in these compounds.
- The solid that precipitated from THF was analyzed by NMR spectroscopy in NC5D5. The spectrum showed all resonances for compound 2 (except for those due to NC5H5) plus resonances for non-coordinate THF. This supports our results from DFT calculations suggesting that the precipitate is compound I-5.
- A 13C NMR chemical shift of 204.5 ppm has been reported for CO groups in the five-membered cyclic complex anion [As[C(O)]2[NDipp]2]−: A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 8536–8541 CrossRef CAS PubMed.
- (a) L. C. Willemsens, J. Organomet. Chem., 1971, 27, 45–51 CrossRef CAS; (b) G.-E. Matsubayashi, M. Hiroshima and T. Tanaka, J. Organomet. Chem., 1973, 59, 207–212 CrossRef CAS.
- The formation of a bismuth carbamoyl species from BiCl3, DMF and amines has been discussed on the basis of conductance measurements and elemental analysis, but no spectroscopic or structural data has been provided: A. K. Mishra and K. N. Tandon, Inorg. Chem., 1971, 10, 1896–1899 CrossRef CAS.
- (a) B. Murray, J. Hvoslef, H. Hope and P. P. Power, Inorg. Chem., 1983, 22, 3421–3424 CrossRef CAS; (b) C. Lichtenberg, F. Pan, T. P. Spaniol, U. Englert and J. Okuda, Angew. Chem., Int. Ed., 2012, 51, 13011–13015 CrossRef CAS PubMed; (c) S. Solyntjes, J. Bader, B. Neumann, H.-G. Stammler, N. Ignat'ev and B. Hoge, Chem.–Eur. J., 2017, 23, 1557–1567 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) R. J. Schwamm, J. R. Harmer, M. Lein, C. M. Fitchett, S. Granville and M. P. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed; (b) C. Ganesamoorthy, C. Helling, C. Wölper, W. Frank, E. Bill, G. E. Cutsail III and S. Schulz, Nat. Commun., 2018, 9, 87 CrossRef PubMed.
- For electrochemical studies on dibismuthenes including a ferrocenyl substituted derivative see: (a) M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Chem.–Asian J., 2013, 8, 690–693 CrossRef CAS PubMed; (b) T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 12582–12588 CrossRef CAS PubMed.
- Relativistic, dispersion-corrected DFT calculations were carried out at the ZORA-BLYP-D3BJ/TZ2P level of theory using COSMO to simulate solvation in THF, as implemented in the ADF program, see: (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS; (b) F. M. Bickelhaupt and E. J. Baerends, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 2000, vol. 15, pp. 1-86 Search PubMed.
- Substitution of the two terminal thf ligands in 1 for two CO molecules was calculated to be endothermal and endergonic (ΔH: +18.0 kcal mol−1; ΔG: +16.2 kcal mol−1) and is therefore suggested not to be involved in the primary reaction pathway (formation of “I-1_2CO”, ESI†).
- (a) M. Ryang and S. Tsutsumi, Bull. Chem. Soc. Jpn., 1962, 35, 1121–1124 CrossRef CAS; (b) H. C. Brown and E. Negishi, J. Am. Chem. Soc., 1967, 89, 5477–5478 CrossRef CAS; (c) N. S. Nudelman and A. A. Vitale, J. Org. Chem., 1981, 46, 4625–4626 CrossRef CAS; (d) D. Seyferth and R. M. Weinstein, J. Am. Chem. Soc., 1982, 104, 5534–5535 CrossRef CAS; (e) W. J. J. M. Sprangers and R. Louw, J. Chem. Soc., Perkin Trans. 2, 1976, 1895–1901 RSC; (f) M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed; (g) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559–13568 CrossRef CAS PubMed; (h) M. R. Mason, B. Song and K. Kirschbaum, J. Am. Chem. Soc., 2004, 126, 11812–11813 CrossRef CAS PubMed; (i) X. Li, C. Ni, H. Song and C. Cui, Chem. Commun., 2006, 1763–1765 RSC; (j) M. R. Mason, B. Song, Y. Han and X. Hu, Inorg. Chim. Acta, 2008, 361, 3332–3337 CrossRef CAS; (k) C. M. Lindsay and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1, 1988, 569–573 RSC.
- An FLP type reactivity towards CO has recently been reported for a phosphorus ylide with a boryl substituent: M. Radius and F. Breher, Chem.–Eur. J., 2018, 24, 15744–15749 CrossRef CAS PubMed.
- (a) X. Wang, Z. Zhu, Y. Peng, H. Lei, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2009, 131, 6912–6913 CrossRef CAS PubMed; (b) Z. D. Brown and P. P. Power, Inorg. Chem., 2013, 52, 6248–6259 CrossRef CAS PubMed.
- (a) H. Braunschweig, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer, I. Krummenacher, J. Mies, A.-K. Phukan and A. Vargas, Nat. Chem., 2013, 5, 1025–1028 CrossRef CAS PubMed; (b) M. Arrowsmith, J. Böhnke, H. Braunschweig and M. A. Celik, Angew. Chem., Int. Ed., 2017, 56, 14287–14292 CrossRef CAS PubMed; (c) N. Nakata, T. Oikawa, T. Matsumoto, Y. Kabe and A. Sekiguchi, Organometallics, 2005, 24, 3368–3370 CrossRef CAS; (d) H. Zhang, Z. Cao, W. Wu and Y. Mo, Angew. Chem., Int. Ed., 2018, 57, 13076–13081 CrossRef CAS PubMed; (e) L. Wallbaum, D. Weismann, D. Löber, C. Bruhn, P. Prochnow, J. E. Bandow and U. Siemeling, Chem.–Eur. J., 2019, 25, 1488–1497 CrossRef CAS PubMed.
- Carbenes and silylenes also form stable and transient (sila-)ketenes with CO, respectively: (a) V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488–3491 CrossRef CAS PubMed; (b) M. Tacke, C. Klein, D. J. Stufkens, A. Oskam, P. Jutzi and E. A. Bunte, Z. Anorg. Allg. Chem., 1993, 619, 865–868 CrossRef CAS; (c) M.-A. Pearsall and R. West, J. Am. Chem. Soc., 1988, 110, 7228–7229 CrossRef CAS; (d) R. Becerra and R. Walsh, J. Am. Chem. Soc., 2000, 122, 3246–3247 CrossRef CAS.
- (a) M. Sajid, A. Lawzer, W. Dong, C. Rosorius, W. Sander, B. Schirmer, S. Grimme, C. G. Daniliuc, G. Kehr and G. Erker, J. Am. Chem. Soc., 2013, 135, 18567–18574 CrossRef CAS PubMed; (b) M. Sajid, L.-M. Elmer, C. Rosorius, C. G. Daniliuc, S. Grimme, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 2243–2246 CrossRef CAS PubMed; (c) L.-M. Elmer, G. Kehr, C. G. Daniliuc, M. Siedow, H. Eckert, M. Tesch, A. Studer, K. Williams, T. H. Warren and G. Erker, Chem.–Eur. J., 2017, 23, 6056–6068 CrossRef CAS PubMed; (d) L. Wang, K. Samigullin, M. Wagner, A. C. McQuilken, T. H. Warren, C. Daniliuc, G. Kehr and G. Erker, Chem.–Eur. J., 2016, 22, 11015–11021 CrossRef CAS PubMed; (e) K.-Y. Ye, G. Kehr, C. G. Daniliuc, L. Liu, S. Grimme and G. Erker, Angew. Chem., Int. Ed., 2016, 55, 9216–9219 CrossRef CAS PubMed.
- Also see reaction of (P(tBu)3 + 2B(C6F5)3) with syngas: R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 4974–4977 CrossRef CAS PubMed.
- For examples of phosphorus compounds reacting with carbonyl ligands see: (a) A. Fuchs, D. Gudat, M. Nieger, O. Schmidt, M. Sebastian, L. Nyulaszi and E. Niecke, Chem.–Eur. J., 2002, 8, 2188–2196 CrossRef CAS PubMed; (b) L. Weber, K. Reizig and R. Boese, Angew. Chem., Int. Ed., 1986, 25, 755–757 CrossRef; (c) A. Marinetti, J. Fischer and F. Mathey, J. Am. Chem. Soc., 1985, 107, 5001–5002 CrossRef CAS; (d) M. Scheer, E. Leiner, P. Kramkowski, M. Schiffer and G. A. Baum, Chem.–Eur. J., 1998, 4, 1917–1934 CrossRef CAS; (e) M. Finze, E. Bernhardt, H. Willner and C. W. Lehmann, Inorg. Chem., 2006, 45, 669–678 CrossRef CAS PubMed.
- For examples of metal complexes with phosphorus ligands reacting with CO, see: (a) P. G. Edwards, M. B. Hursthouse, K. M. A. Malik and J. S. Parry, J. Chem. Soc., Chem. Commun., 1994, 1249–1250 RSC; (b) D. M. Roddick, B. D. Santariero and J. E. Bercaw, J. Am. Chem. Soc., 1985, 107, 4670–4678 CrossRef CAS; (c) R. L. De, D. Wolters and H. Vahrenkamp, Z. Naturforsch., 1986, 41b, 283–291 CAS.
- For an example of Pd-catalyzed synthesis of acylphosphonates using CO as a C1-source see: Y. Masuda, N. Ishida and M. Murakami, Chem.–Asian J., 2015, 10, 321–324 CrossRef CAS PubMed.
- For an example of reversible coordination of CO to a phosphinidene see: M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 2776–2779 CrossRef CAS PubMed.
- (a) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (b) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed; (c) D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC; (d) F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- A 1-phosphane-8-stibonium-naphtalene species with Lewis acidic character has been reported, but no FLP-type reactivity has been described to date: (a) R. A. Ugarte, D. Devarajan, R. M. Mushinski and T. W. Hudnall, Dalton Trans., 2016, 45, 11150–11161 RSC; (b) R. A. Ugarte and T. W. Hudnall, Green Chem., 2017, 19, 1990–1998 RSC.
- In the heterogeneously catalyzed photocatalytic reduction of CO2, the reactivity of frustrated Lewis pairs of In2O3−x(OH)y has been modified by isomorphous substitution of In by Bi: Y. Dong, K. K. Ghuman, R. Popescu, P. N. Duchesne, W. Zhou, J. Y. Y. Loh, A. A. Jelle, J. Jia, D. Wang, X. Mu, C. Kübel, L. Wang, L. He, M. Ghoussoub, Q. Wang, T. E. Wood, L. M. Reyes, P. Zhang, N. P. Kherani, C. V. Singh and G. A. Ozin, Adv. Sci., 2018, 5, 1700743–1700742 Search PubMed.
- M. Seidl, M. Schiffer, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2013, 19, 13783–13791 CrossRef CAS PubMed.
- W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, details of DFT calculations, NMR, IR and UV/vis spectra, cyclic voltammograms, single crystal X-ray analysis data, coordinates of calculated structures. CCDC 1875340–1875343. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00278b |
| This journal is © The Royal Society of Chemistry 2019 |