
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective palladium-catalyzed one-pot, five-fold B–H/C–H cross coupling of monocarboranes with alkenes†
Yunjun
Shen
,
Kang
Zhang
,
Xuewei
Liang
,
Rakesh
Dontha
and
Simon
Duttwyler
 *
*
Department of Chemistry, Zhejiang University, 310027 Hangzhou, Zhejiang, P. R. China. E-mail: duttwyler@zju.edu.cn
First published on 4th March 2019
Abstract
Palladium-catalyzed dehydrogenative B–H/C–H cross coupling of monocarborane anions with alkenes is reported, allowing for the first time the isolation of selectively penta-alkenylated boron clusters. The reaction cascade is regioselective for the cage positions, leading directly to B2–6 functionalization. Under mild and convenient conditions, styrenes, benzylic alkenes and aliphatic alkenes are demonstrated to be viable coupling partners with exclusive vinyl-type B–C bond formation. Multiple subsequent transformations provide access to directing group-free products, chiral derivatives and penta-alkylated cages. The five-fold coupling, combined with the latter reactions, represents a powerful methodology for the straightforward synthesis of new classes of boron clusters.
Introduction
Icosahedral monocarboranes based on the [CB11H12]− anion and dicarbaboranes with the parent skeleton C2B10H12 are polyhedral 12-vertex boranes with one or two BH vertices replaced by CH. They possess unique steric and electronic properties that set them apart from traditional organic or inorganic building blocks.1,2 Their sphere-like distribution of electron density gives rise to remarkable chemical and thermal stability, including low toxicity. Monocarboranes are well known as weakly coordinating anions,3 and various applications of monocarboranes and dicarbaboranes have been reported in areas such as coordination,4 supramolecular5 and medicinal chemistry,6 as well as luminescence7 and materials science.8 In order to exploit the potential of carboranes in these fields, improved methodologies which enable the coupling of cluster vertices to organic substituents are essential.C–C bond forming reactions are at the heart of classical organic synthesis. Likewise, when it comes to the preparation of inorganic–organic hybrid molecules, the construction of E–C bonds (E = main group element other than C) is equally important. For icosahedral carboranes, the modification of boron vertices traditionally relies on direct electrophilic substitution or electrophilic halogenation followed by transformation to a B–X (X = C, N, O, S, P) bond.9,10 Drawbacks of these approaches are harsh conditions, limited control over selectivity of the substituted positions and moderate overall yields. The functionalization of boron vertices by directing group-mediated, metal-catalyzed B–H activation is a highly attractive alternative strategy. Over the past years, the utility of this concept to provide access to selectively modified carborane derivatives has been demonstrated impressively.11,12 Among others, the groups of Xie, Yan, Bregadze, Spokoyny and Peryshkov have established effective methodologies involving neutral dicarbaboranes.13,14 Our group, on the other hand, has focused on catalytic B–H activation of mono- and dianionic boron clusters. These procedures have made use of amide or ureido directing groups for Rh- and Ir-catalyzed reactions with acrylates, styrenes, sulfones, alkynes, azides and N-chlorosuccinimide.15 These transformations allow mono- to tetra-substitution of boron vertices. Reactions with acrylates or styrenes resulted either in one- or two-fold oxidative (vinyl-type) coupling or higher mixed oxidative/reductive coupling.15b,d,f Recently, we have also achieved penta-arylation of the monocarborane-1-carboxylic acid with aryl halides under Pd catalysis (Fig. 1a).15e Herein, we report the first Pd-catalyzed penta-functionalization of [CB11H12]−-based anions by selective B–H/C–H cross coupling with a wide range of alkenes at room temperature under air (Fig. 1b). The substitution is highly regioselective with respect to the cage at the B2–6 positions, and the coupling to the alkene occurs regio- and stereoselectively with regard to the alkene, giving vinyl-type (E) products exclusively. This mild and ligand-free catalytic protocol using a removable directing group represents a powerful method for the construction of highly substituted monocarborane clusters.
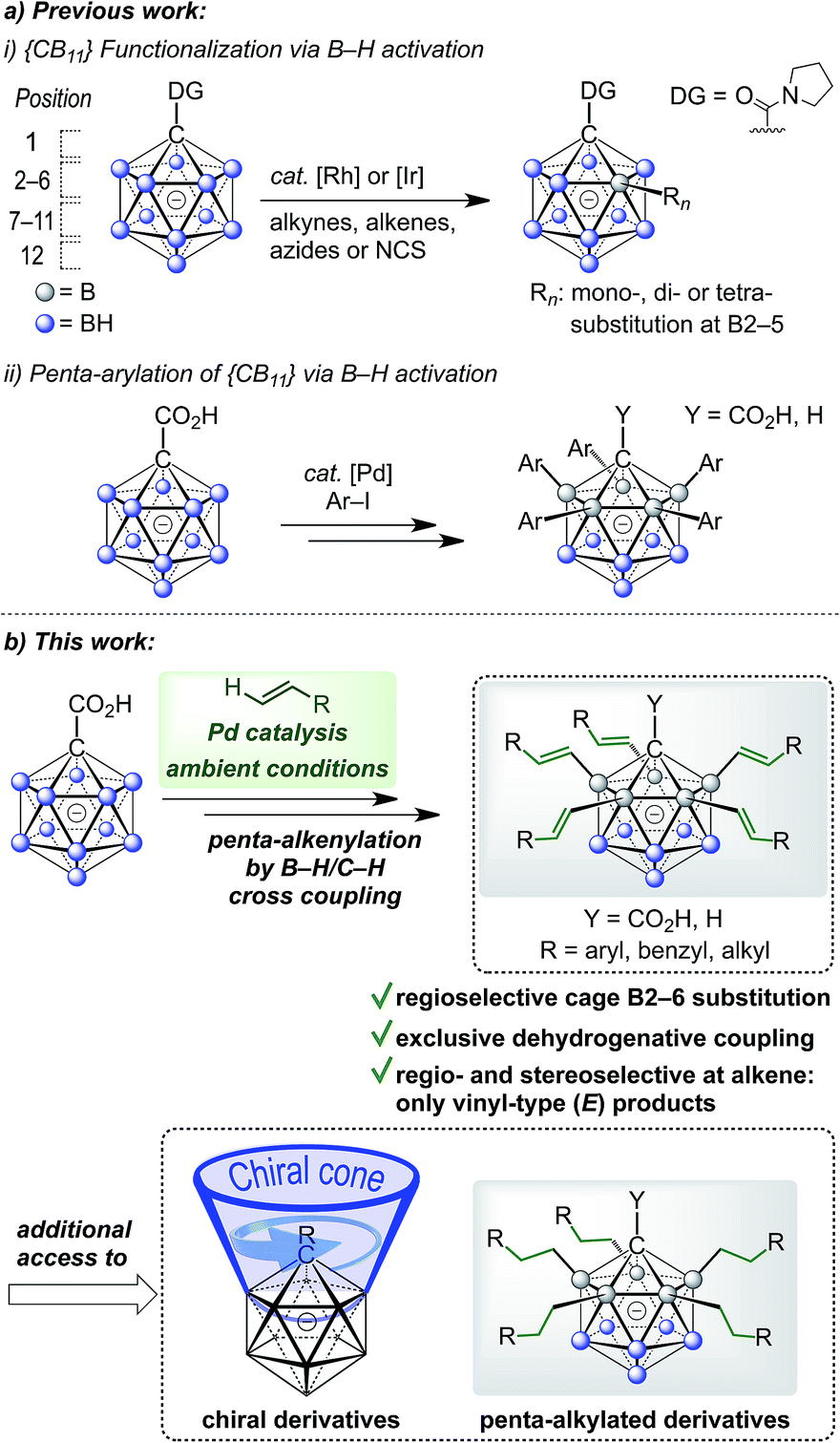 | ||
| Fig. 1 (a) Previously reported metal-catalyzed functionalization of {CB11} cages and (b) regioselective dehydrogenative penta-alkenylation by direct B–H/C–H cross coupling. | ||
Results and discussion
In order to establish conditions for this intermolecular coupling, we studied the model reaction between 1a and 4-fluorostyrene (2a, 5.4 equivalents) to give functionalized product 3a with potential multiple substitution (Table 1). No reaction occurred in the presence of only catalyst Pd(OAc)2 or AgOAc in acetonitrile (entries 1 and 2). On the other hand, a combination of Pd(II) (10 mol%) and Ag(I) (10 equivalents) cleanly afforded 3a in 86% isolated yield, as evidenced by reaction monitoring using ESI-mass spectrometry and NMR spectroscopy of the purified product (entry 3). The penta-substitution occurred at 25 °C within 24 hours; increasing the temperature to 60 °C was associated with a faster completion (ca. 12 hours) and slightly reduced yield (entry 4). The use of other solvents, such as tetrahydrofuran, 1,2-dichloroethane or dimethylacetamide, led to reduced yields (entries 5 and 7). Oxidants such as Cu(OAc)2 or 1,4-benzoquinone were ineffective (entries 8 and 9); interestingly, for Cu(OAc)2 the reaction stopped at the mono- and di-substitution stage (inseparable mixture), and no higher degree of substitution was observed. We also tested the use of smaller amounts of Pd(II) and Ag(I); lowering the catalyst or oxidant loading afforded the desired 3a, but as a mixture with di-, tri- and tetra-substituted products even after 48 hours. All of the above screening reactions were set up in a fumehood under air atmosphere. Applying the conditions from entry 3 under nitrogen in a glovebox gave 3a in 84% yield, indicating that the presence of oxygen does not play a significant role in this transformation.|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | Oxidant | Solv.b | T [°C] | Resultc |
| a 1a (0.15 mmol), Pd(OAc)2 (0.015 mmol), oxidant (1.5 mmol), in 4 mL of solvent. b ACN = acetonitrile, DMA = dimethylacetamide, DCE = 1,2-dichloroethane, THF = tetrahydrofuran. c Isolated yields after purification by silica gel chromatography. | |||||
| 1 | Pd(OAc)2 | None | ACN | 25 | N.R. |
| 2 | None | AgOAc | ACN | 25 | N.R. |
| 3 | Pd(OAc) 2 | AgOAc | ACN | 25 | 86% |
| 4 | Pd(OAc)2 | AgOAc | ACN | 60 | 83% |
| 5 | Pd(OAc)2 | AgOAc | THF | 25 | 32% |
| 6 | Pd(OAc)2 | AgOAc | DCE | 25 | 10% |
| 7 | Pd(OAc)2 | AgOAc | DMA | 25 | 72% |
| 8 | Pd(OAc)2 | Cu(OAc)2 | ACN | 25 | 0 |
| 9 | Pd(OAc)2 | BQ | ACN | 25 | 30% |
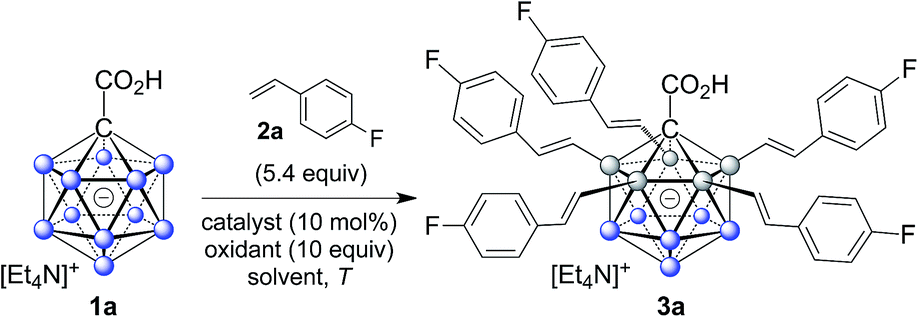
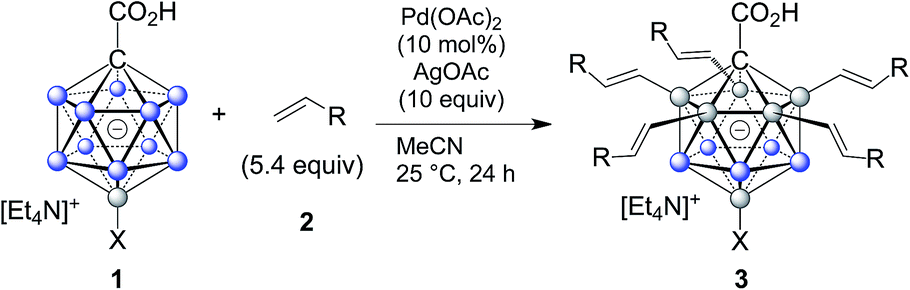
Subsequently, we studied the substrate scope of the penta-alkenylation under the aforementioned conditions of Table 1, entry 3. All reactions were conveniently set up and run under air. The ortho B–H alkenylation of acid carborane 1 with an array of terminal alkenes proceeded smoothly to give the desired products in moderate to high yields (Table 2). Coupling with electron-neutral and electron-deficient styrenes bearing 4-fluoro, 4-hydrogen, 4-trifluoromethyl, and 4-cyano groups gave the desired products 3a–e in yields of 53–86%. 4-Methoxystyrene and 4-methylstyrene as an electron-rich alkene were also tested; however, these substrates afforded only small amounts of product, and attempted chromatographic separation of the reaction mixtures showed that compounds with a lower degree of substitution were also present. Benzylic alkenes with CH2-phenyl and CH2-pentafluorophenyl substituents gave 3f and 3g in yields of 72% and 59%. The reaction was equally successful with aliphatic substrates of different steric requirements, such as hex-1-ene, 4-methylpent-1-ene, 4-methyl-hex-1-ene, 4,4-dimethylpent-1-ene and 4-phenylbut-1-ene, with consistently good yields of 79–84% (3h–3l). Notably, functionalized oxygen-containing alkenes were found to be compatible applying this protocol to give ester 3m and ether 3n in yields of 66% and 74%, respectively. Even though for alkenes 2 with R = benzyl or alkyl, isomeric allyl-type products could form, only vinyl-type coupling was observed (vide infra, discussion of the mechanism). We then chose styrene and 4-fluorostyrene as coupling partners to explore the penta-alkenylation of multiple acid carboranes 1 bearing different substituents at the B12 position. The five-fold selective ortho B–H alkenylation occurred with B12–R (R = Cl, Br, Me, Ph, CN) in yields of 62–82% (3o–t). Furthermore, the method is also reproducible on a gram scale; starting from 1.0 g of 1a, product 3a was obtained in 80% isolated yield (2.3 g). Clusters 3 were fully characterized by 1H, 1H{11B}, 13C{1H}, 11B and 11B{1H} NMR spectroscopy as well as high-resolution mass spectrometry. Upon substitution of the B2–6 positions, a characteristic change in the 11B NMR spectra was observed. Comparing the starting material 1a and 3a as a representative product, the B2–6 resonances appeared at −14.1 and −6.1 ppm, while almost no change was observed for positions B7–11 (−13.3/–12.9 ppm) and B12 (−6.5/−6.1 ppm). Deshielding of the substituted positions by ca. 8 ppm occurred in all cases, while the chemical shift differences for the other vertices were very small. For 3b, the molecular structures was in addition elucidated by X-ray crystallography; Fig. 2a shows an ORTEP representation of this product.16
|
|
||||
|---|---|---|---|---|
| Carborane | Alkene | Product | ||
| a 1 (0.15 mmol), 2 (0.81 mmol), Pd(OAc)2 (0.015 mmol), AgOAc (1.5 mmol), in acetonitrile (4 mL); for further details, see the ESI. b Yields = isolated yields after purification by silica gel chromatography. | ||||
| 1a | X = H | R = aryl | R = C6H4-4-F | 3a 86% |
| X = H | R = C6H5 | 3b 75% | ||
| X = H | R = C6H4-4-CF3 | 3c 70% | ||
| X = H | R = C6H4-4-CN | 3d 63% | ||
| X = H | R = C6F5 | 3e 53% | ||
| X = H | R = benzyl | R = CH2-C6H5 | 3f 72% | |
| X = H | R = CH2-C6F5 | 3g 59% | ||
| X = H | R = alkyl |

|
3h 81% | |
| X = H |

|
3i 84% | ||
| X = H |
|
3j 82% | ||
| X = H |

|
3k 79% | ||
| X = H |

|
3l 76% | ||
| X = H |

|
3m 66% | ||
| X = H |

|
3n 74% | ||
| 1b | X = Cl | R = aryl | R = C6H5 | 3o 71% |
| 1c | X = Br | R = C6H4-4-F | 3p 78% | |
| 1d | X = Me | R = C6H4-4-F | 3q 82% | |
| 1e | X = Ph | R = C6H5 | 3r 62% | |
| 1e | X = Ph | R = C6H4-4-F | 3s 69% | |
| 1f | X = CN | R = C6H4-4-F | 3t 72% | |
 | ||
| Fig. 2 X-ray crystal structures of (a) 3b and (b) 4b (cations, solvent molecules and styryl H atoms omitted for clarity, 30% displacement ellipsoids). | ||
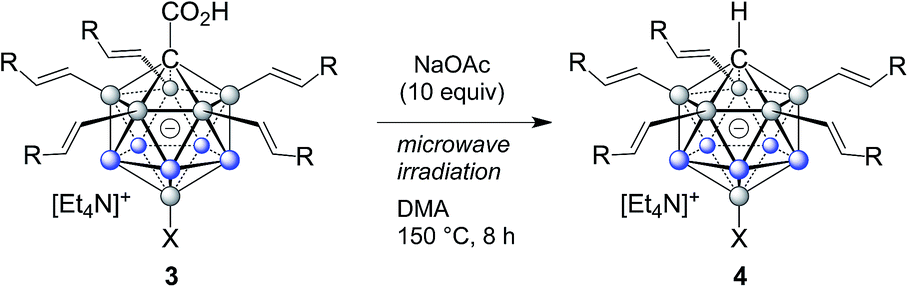
The carboxylic acid moiety at the C1 cluster position is a highly versatile directing group. On the one hand, it can be transformed to the acid chloride, which in turn is a very useful functional group handle for the preparation of, e.g., esters and amides.17 Product 3c was chosen as a representative example, and details about its clean conversion to the acid chloride 3c–Cl are provided in the ESI (p. S12).† Moreover, we recently demonstrated that penta-arylated monocarborane C1-carboxylic acids can be decarboxylated in dimethylformamide at 100 °C to give the C1–H product.15e For products 3, these conditions did not lead to complete directing group removal even after prolonged periods of heating. However, we found that decarboxylation takes place cleanly under microwave irradiation under slightly basic conditions. Specifically, at 150 °C in dimethylacetamide solvent and with the addition of 10 equivalents of NaOAc, formation of the C1–H products 4 occurred within 8 hours (Table 3). This transformation was applied to 3a, 3b, 3q, 3r, and 3t, affording 4a, 4b, 4q, 4r and 4t in isolated yields of 76–86% after purification by silica gel chromatography. This method is particularly attractive because it is transition metal-free and can be set up and run under air. Notably, under these microwave conditions, starting material 1a remains mostly unchanged (ca. 10% decarboxylation), which indicated that substitution at the B2–6 positions facilitates the process, likely by stabilizing the intermediate formal C1 exo-carbanion.15e The molecular structure of 4b was determined by X-ray crystallography, and an ORTEP representation is displayed in Fig. 2b.
|
|
|||
|---|---|---|---|
| a 3 (0.1 mmol), NaOAc (1 mmol), in dimethylacetamide (4 mL) in a sealed microwave vial; for further details, see the ESI. b Yields = isolated yields after purification by silica gel chromatography. | |||
| 3a | X = H | R = C6H4-4-F | 4a 86% |
| 3b | X = H | R = C6H5 | 4b 79% |
| 3q | X = Me | R = C6H4-4-F | 4q 87% |
| 3r | X = Ph | R = C6H5 | 4r 76% |
| 3t | X = CN | R = C6H4-4-F | 4t 80% |
The five groups at B2–6 are oriented in the direction of the B12–C1 axis and thus define a large C5-symmetrical cone. If five chiral substituents with identical absolute stereochemistry are attached, a chiral “unidirectional” cone results (Fig. 1). Such compounds have the potential to serve as ligands for enantiodiscriminative analysis, the build-up of chiral supramolecular structures and enantioselective transformations within the chiral space. To demonstrate the feasibility of the formation of optically active products, an optically pure chiral ether was chosen as the coupling partner, namely, the benzyl ether of pent-4-en-2-ol (Scheme 1). The enantiopure ethers 2u and 2v were prepared starting from commercially available (R)- and (S)-pent-4-en-2-ol. By applying our methodology for penta-alkenylation, desired products 3u and 3v were obtained in yields of 73% and 71%. Subsequent removal of the directing group afforded decarboxylated 4u and 4v in 84% and 80% isolated yields. The two enantiomers 3u and 3v were characterized by circular dichroism (CD) spectroscopy. The mirror-imaged CD spectra featured maxima at 215 nm, and no strong bands were observed above 225 nm. Given the negative charge of 3 and 4 and the ability for hydrogen bonding of the carboxylic acid unit of 3, these novel chiral clusters are promising candidates for the above-mentioned applications, especially for enantioselective recognition and reactions involving cationic guests.
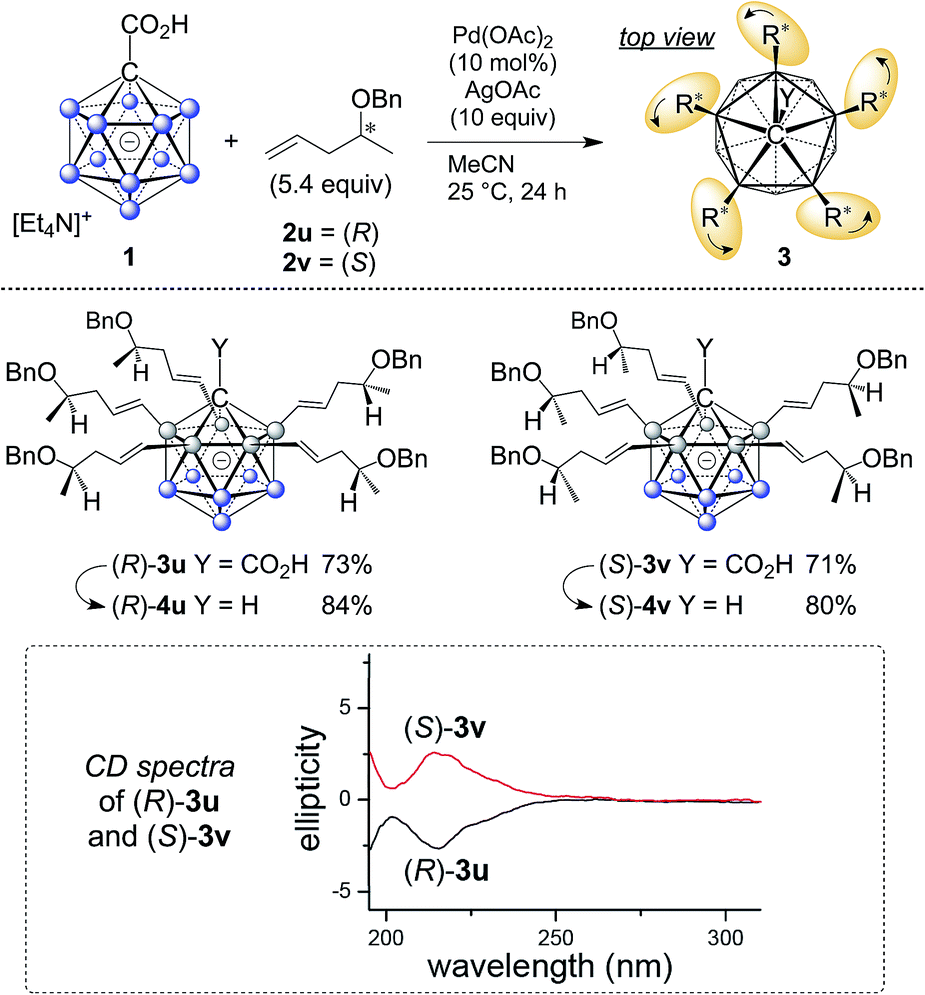 | ||
| Scheme 1 Synthesis of chiral 3u/v and 4u/v; curved arrows around R* indicate identical sense of absolute configuration. | ||
The efficient and selective introduction of C(sp3) substituents to {CB11} clusters has been an long-standing synthetic challenge.9 To further demonstrate the synthetic utility of our B–H functionalization methodology, we examined the hydrogenation of alkenylated products. Treatment of 3a and 3k under hydrogen atmosphere with Pd/C as the catalyst afforded the corresponding products 5a and 5k, which were conveniently purified by column chromatography and isolated in 90% and 87% yields. Remarkably, complete reduction of all five double bonds occured within two hours at room temperature without the need for high pressure (hydrogen balloon). On the one hand, access to carboranes bearing multiple fully reduced substituents is important from a fundamental perspective because it rounds off the synthetic palette; on the other hand, it becomes relevant in the context of modulating physical properties, such as solubility in solvents of low polarity, redox behavior and melting point as well as applications associated with them (Scheme 2).
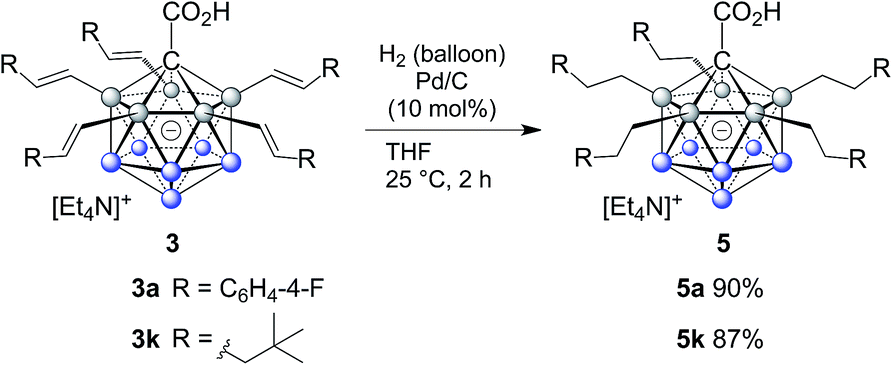 | ||
| Scheme 2 Reduction of the double bond. | ||
In a stoichiometric experiment between 1c and Pd(OAc)2 in acetonitrile, cyclometaled complex 1c–Pd formed cleanly and was isolated in 82% yield (see the ESI for details†). Its 11B NMR spectrum contained diagnostic signals at −21.9 and −3.5 ppm for the B2–Pd and B12–Br positions, respectively (Fig. 2a).4h,15e,18 All other boron vertices overlapped and resonated in the range of −10.5 to −17.5 ppm. Furthermore, the solid-state structure of 1c–Pd was elucidated by X-ray crystallography (Fig. 3b). The Pd–B and Pd–O distances are 2.010(14) and 2.059(8) Å, and the structural features are comparable to those of recently reported monocarborane–palladium complexes.4h,15e,18 Treatment of 1c–Pd with 6.0 equivalents of 4-fluorostyrene and 10 equivalents of AgOAc in acetonitrile cleanly afforded penta-substituted 3p. Furthermore, a control experiment without the addition of AgOAc was carried out. The reaction of 1c–Pd with 6.0 equivalents of 4-fluorostyrene in acetonitrile-d3 cleanly afforded mono-substituted product 3p-mono within 1 hour at 25 °C, as evidenced by NMR spectroscopy and mass spectrometry (see the ESI for details†). The spectra remained unchanged after 3, 5 and 12 hours of total reaction time, i.e., no products with higher degree of substitution were formed. These results suggest that the palladium complex 1c–Pd is an intermediate relevant to the catalytic cycle and that AgOAc is necessary for catalytic turnover.
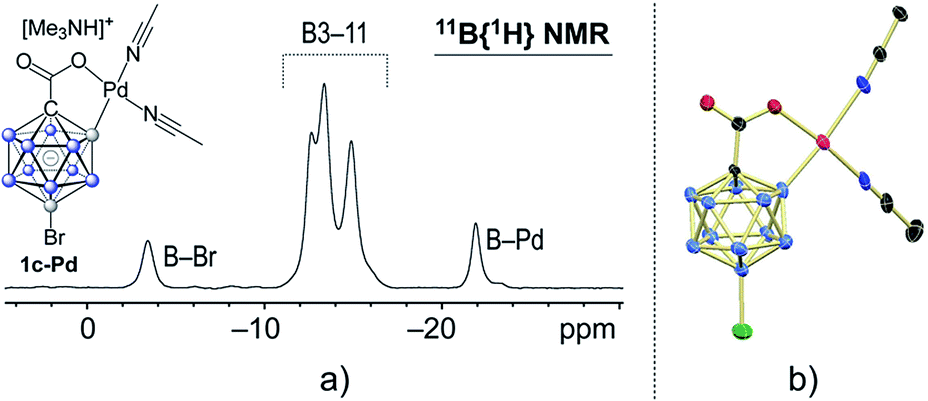 | ||
| Fig. 3 (a) 11B{1H} NMR spectrum and (b) X-ray crystal structure of palladium complex 1c–Pd (cation and H atoms omitted for clarity; 30% displacement ellipsoids). | ||
A plausible mechanistic cycle involving five sequential B–H bond activation/B–C coupling events is displayed in Scheme S1.† Carborane acid 1 can bind to Pd(II) via its carboxylate group, affording the initial intermediate IM-a. Cyclometalation–deprotonation then gives palladacycle 1–Pd with a direct B–Pd bond. Alkene coordination/insertion (intermediates IM-b/IM-c) is followed by β-hydride elimination, furnishing Pd–H complex IM-d. Subsequent reaction with AgOAc leads to IM-a′, which contains one alkenyl substituent. From this point on, further B–H activation/alkenylation steps occur in a similar manner until the final product 3 and Pd(II) are liberated. The use of benzyl and alkyl substrates 2 in principle allows for the occurrence of isomeric products with a B–CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH bonding pattern (Scheme 3). It is remarkable that there is no indication of the formation of such isomers (analysis of crude NMR spectra). The regioselective β-hydride elimination may be explained by stabilization of IM-dvia πalkene → Pd coordination, which we believe would be weaker in the case of the alternative pathway.
CH bonding pattern (Scheme 3). It is remarkable that there is no indication of the formation of such isomers (analysis of crude NMR spectra). The regioselective β-hydride elimination may be explained by stabilization of IM-dvia πalkene → Pd coordination, which we believe would be weaker in the case of the alternative pathway.
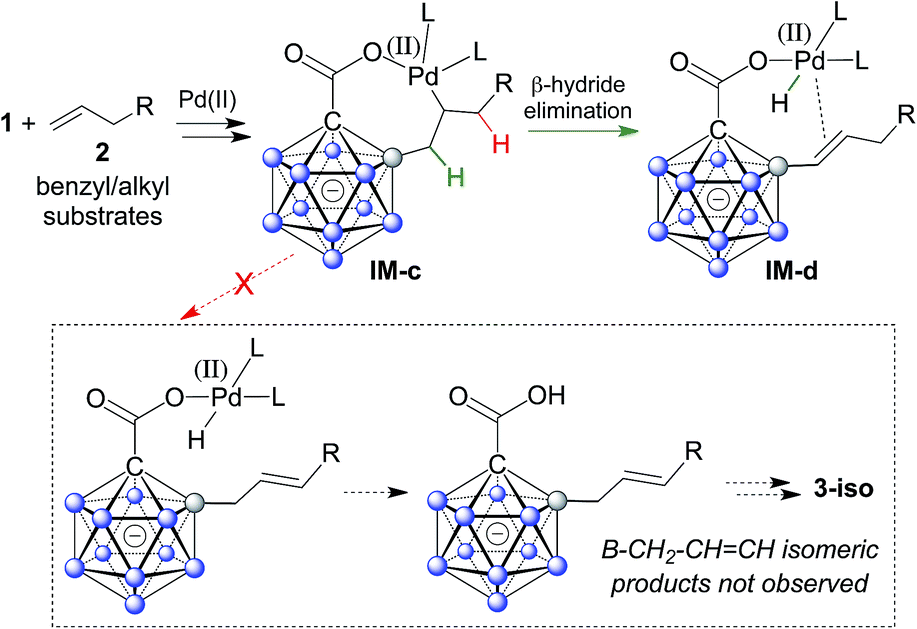 | ||
| Scheme 3 Putative selective β-hydride elimination to explain the regioselectivity of double bond formation with benzyl and alkyl substrates. | ||
Conclusions
In conclusion, we have developed a mild and versatile methodology for the regioselective B2–6 cage alkenylation of monocarborane anions through a palladium-catalyzed B–H/C–H cross coupling cascade. Styrenes and benzylic as well as aliphatic alkenes serve as efficient coupling partners, providing access to a variety of penta-substituted products in moderate to high yields. The carboxylic acid directing group can be readily removed without the need for transition metals. Furthermore, the introduction of homochiral substituents and the possibility to reduce the double bond allows for the preparation of novel chiral and selectively alkylated monocarboranes. Therefore, we believe this methodology can be of broad utility for the construction of new classes of carborane-based compounds, which will be beneficial for synthetic chemists and also researchers in multiple areas of applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Natural Science Foundation of China (Grant No. 21472166), the National Basic Research Program of China (973 project 2015CB856500) and the Special Funds for Basic Scientific Research of Zhejiang University. We thank Prof. Dr Bernhard Spingler from the University of Zurich for checking the X-ray crystal structures.Notes and references
- (a) N. S. Hosmane, Boron Science: New Technologies and Applications, Taylor & Francis Books/CRC, Boca Raton, FL, 2011 CrossRef; (b) R. N. Grimes, Carboranes, Elsevier, Amsterdam, 3rd edn, 2016 Search PubMed.
- (a) C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179–PR233 CrossRef PubMed; (b) R. N. Grimes, Dalton Trans., 2015, 44, 5939–5956 RSC; (c) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed; (d) R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC; (e) S. P. Fischer, A. W. Tomich, J. Gui and V. Lavallo, Chem. Commun., 2019, 55, 1684–1701 RSC.
- For reviews and selected examples, see: (a) K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Mueller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825–827 CrossRef CAS PubMed; (b) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed; (c) T. Kato and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 2908–2911 CrossRef CAS PubMed; (d) C. Douvris and O. V. Ozerov, Science, 2008, 31, 1188–1190 CrossRef PubMed; (e) C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed; (f) O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574–577 CrossRef CAS PubMed; (g) V. Volkis, C. Douvris and J. Michl, J. Am. Chem. Soc., 2011, 133, 7801–7809 CrossRef CAS PubMed; (h) C. Knapp, Weakly Coordinating Anions: Halogenated Borates and Dodecaborates, in Comprehensive Inorganic Chemistry II, Elsevier, Amsterdam, 2013, vol. 1, pp. 651–679 Search PubMed; (i) N. Kordts, C. Borner, R. Panisch, W. Saak and T. Müller, Organometallics, 2014, 33, 1492–1498 CrossRef CAS; (j) Y. Shoji, N. Tanaka, K. Mikami, M. Uchiyama and T. Fukushima, Nat. Chem., 2014, 6, 498–503 CrossRef CAS PubMed; (k) K. M. Osman, D. R. Powell and R. J. Wehmschulte, Inorg. Chem., 2015, 54, 9195–9200 CrossRef CAS PubMed; (l) Y. Kitazawa, R. Takita, K. Yoshida, A. Muranaka, S. Matsubara and M. Uchiyama, J. Org. Chem., 2017, 82, 1931–1935 CrossRef CAS PubMed; (m) C. J. Pell, Y. Zhu, R. Huacuja, D. E. Herbert, R. P. Hughes and O. V. Ozerov, Chem. Sci., 2017, 8, 3178–3186 RSC; (n) B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 403–1407 CrossRef PubMed; (o) T. Müller, K. Natalie, K. Sandra, S. Rathjen, T. Sieling, H. Großekappenberg and M. Schmidtmann, Chem.–Eur. J., 2017, 23, 10068–10079 CrossRef PubMed; (p) L. Omann, B. Pudasaini, E. Irran, H. F. T. Klare, M.-H. Baik and M. Oestreich, Chem. Sci., 2018, 9, 5600–5607 RSC; (q) Q. Wu, Z.-W. Qu, L. Omann, E. Irran, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 9176–9179 CrossRef CAS PubMed; (r) I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- For selected publications, see: (a) M. Finze, J. A. P. Sprenger and B. B. Schaack, Dalton Trans., 2010, 39, 2708–2716 RSC; (b) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed; (c) M. E. El-Zaria, H. Arii and H. Nakamura, Inorg. Chem., 2011, 50, 4149–4161 CrossRef CAS PubMed; (d) Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef CAS; (e) A. El-Hellani and V. Lavallo, Angew. Chem., Int. Ed., 2014, 53, 4489–4493 CrossRef CAS PubMed; (f) L. E. Riley, A. P. Y. Chan, J. Taylor, W. Y. Man, D. Ellis, G. M. Rosair, A. J. Welch and I. B. Sivaev, Dalton Trans., 2016, 45, 1127–1137 RSC; (g) J. Holmes, C. M. Pask, M. A. Fox and C. E. Willans, Chem. Commun., 2016, 52, 6443–6446 RSC; (h) J. Estrada, C. A. Lugo, S. G. McArthur and V. Lavallo, Chem. Commun., 2016, 52, 1824–1826 RSC; (i) F. Šembera, J. Plutnar, A. Higelin, Z. Janoušek, I. Císařová and J. Michl, Inorg. Chem., 2016, 55, 3797–3806 CrossRef PubMed; (j) Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef CAS PubMed; (k) P. Coburger, J. Schulz, J. Klose, B. Schwarze, M. B. Sárosi and E. Hey-Hawkins, Inorg. Chem., 2017, 56, 292–304 CrossRef CAS PubMed; (l) C. Selg, W. Neumann, P. Lönnecke, E. Hey-Hawkins and K. Zeitler, Chem.–Eur. J., 2017, 23, 7932–7937 CrossRef CAS PubMed; (m) A. Ilie, O. Crespo, M. C. Gimeno, M. C. Holthausen, A. Laguna, M. Diefenbach and C. Silvestru, Eur. J. Inorg. Chem., 2017, 2643–2652 CrossRef CAS; (n) J. Estrada and V. Lavallo, Angew. Chem., Int. Ed., 2017, 56, 9906–9909 CrossRef CAS PubMed; (o) P.-F. Cui, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2017, 46, 15535–15540 RSC; (p) B. J. Eleazer, M. D. Smith, A. A. Popov and D. V. Peryshkov, Chem. Sci., 2018, 9, 2601–2608 RSC; (q) T. Rädisch, N. Harmgarth, P. Liebing, M. J. Beltrán-Leiva, D. Páez-Hernández, R. Arratia-Pérez, F. Engelhardt, L. Hilfert, F. Oehler, S. Bussea and F. T. Edelmann, Dalton Trans., 2018, 47, 6666–6671 RSC; (r) Y. Zhang, Z.-X. Zhang, Y. Wang, H. Li, F.-Q. Bai and H.-X. Zhang, Inorg. Chem. Front., 2018, 5, 1016–1025 RSC; (s) E. Oleshkevich, I. Romero, F. Teixidor and C. Viñas, Dalton Trans., 2018, 47, 14785–14798 RSC.
- (a) Y.-F. Han and G.-X. Jin, Acc. Chem. Res., 2014, 47, 3571–3579 CrossRef CAS PubMed; (b) C. E. Housecroft, J. Organomet. Chem., 2015, 798, 218–228 CrossRef CAS; (c) J. Estrada, S. E. Lee, S. G. McArthur, A. El-Hellani, F. S. Tham and V. Lavallo, J. Organomet. Chem., 2015, 798, 214–217 CrossRef CAS; (d) D. J. Clingerman, W. Morris, J. E. Mondloch, R. D. Kennedy, A. A. Sarjeant, C. Stern, J. T. Hupp, O. K. Farhaab and C. A. Mirkin, Chem. Commun., 2015, 51, 6521–6523 RSC; (e) V. Ďordovič, Z. Tošner, M. Uchman, A. Zhigunov, M. Reza, J. Ruokolainen, G. Pramanik, P. Cígler, K. Kalíková, M. Gradzielski and P. Matějíček, Langmuir, 2016, 32, 6713–6722 CrossRef PubMed; (f) S. Rodríguez-Hermida, M. Y. Tsang, C. Vignatti, K. C. Stylianou, V. Guillerm, J. Pérez-Carvajal, F. Teixidor, C. Viñas, D. Choquesillo-Lazarte, C. Verdugo-Escamilla, I. Peral, J. Juanhuix, A. Verdaguer, I. Imaz, D. Maspoch and J. G. Planas, Angew. Chem., Int. Ed., 2016, 55, 16049–16053 CrossRef PubMed; (g) E. Oleshkevich, C. Viñas, I. Romero, D. Choquesillo-Lazarte, M. Haukka and F. Teixidor, Inorg. Chem., 2017, 56, 5502–5505 CrossRef CAS PubMed; (h) H. Xiong, D. Zhou, X. Zheng, Y. Zheng, Y. Qi, Y. Wang, X. Jing and Y. Huang, Chem. Commun., 2017, 53, 3422–3425 RSC; (i) K. Zhang, Y. Shen, J. Liu, B. Spingler and S. Duttwyler, Chem. Commun., 2018, 54, 1698–1701 RSC.
- For selected reviews, see: (a) A. F. Armstrong and J. F. Valliant, Dalton Trans., 2007, 4240–4251 RSC; (b) I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 1433–1450 CrossRef CAS; (c) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed; (d) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (e) H. S. Ban and H. Nakamura, Chem. Rec., 2015, 15, 616–635 CrossRef CAS PubMed; (f) D. Gabel, Pure Appl. Chem., 2015, 87, 173–179 CAS; (g) Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758 CrossRef PubMed; (h) J. Wang, L. Chen, J. Ye, Z. Li, H. Jiang, H. Yan, M. Y. Stogniy, I. B. Sivaev, V. I. Bregadze and X. Wang, Biomacromolecules, 2017, 18, 1466–1472 CrossRef CAS PubMed; (i) G. Calabrese, A. Daou, E. Barbu and J. Tsibouklis, Drug Discovery Today, 2017, 23, 63–75 CrossRef PubMed.
- For selected publications, see: (a) J. C. Axtell, K. O. Kirlikovali, P. I. Djurovich, D. Jung, V. T. Nguyen, B. Munekiyo, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 15758–15765 CrossRef CAS PubMed; (b) Z. Wang, P. Jiang, T. Wang, G. J. Moxey, M. P. Cifuentes, C. Zhang and M. G. Humphrey, Phys. Chem. Chem. Phys., 2016, 18, 15719–15726 RSC; (c) X. Li, H. Yan and Q. Zhao, Chem.–Eur. J., 2016, 22, 1888–1898 CrossRef CAS PubMed; (d) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC; (e) M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Inorg. Chem., 2017, 56, 13274–13285 CrossRef CAS PubMed; (f) Y. Yin, X. Li, S. Yan, H. Yan and C. Lu, Chem.–Asian J., 2017, 12, 2207–2210 CrossRef PubMed; (g) M. R. Son, Y.-J. Cho, S.-Y. Kim, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 24485–24492 RSC; (h) D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS PubMed; (i) H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed; (j) X. Li, X. Tong, Y. Yin, H. Yan, C. Lu, W. Huang and Q. Zhao, Chem. Sci., 2017, 8, 5930–5940 RSC; (k) N. V. Nghia, J. Oh, J. Jung and M. H. Lee, Organometallics, 2017, 36, 2573–2580 CrossRef CAS; (l) N. Shin, S. Yu, J. H. Lee, H. Hwang and K. M. Lee, Organometallics, 2017, 36, 1522–1529 CrossRef CAS; (m) J. Cabrera-González, A. Ferrer-Ugalde, S. Bhattacharyya, M. Chaari, F. Teixidor, J. Gierschner and R. Núñez, J. Mater. Chem. C, 2017, 5, 10211–10219 RSC; (n) M. Hailmann, N. Wolf, R. Renner, B. Hupp, A. Steffen and M. Finze, Chem.–Eur. J., 2017, 23, 11684–11693 CrossRef CAS PubMed; (o) X. Wu, J. Guo, Y. Quan, W. Jia, D. Jia, Y. Chen and Z. Xie, J. Mater. Chem. C, 2018, 6, 4140–4149 RSC; (p) J. Li, C. Yang, X. Peng, Y. Chen, Q. Qi, X. Luo, W.-Y. Lai and W. Huang, J. Mater. Chem. C, 2018, 6, 19–28 RSC; (q) M. Z. Shafikov, A. F. Suleymanova, A. Schinabeck and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 702–709 CrossRef CAS PubMed; (r) S.-Y. Kim, Y.-J. Cho, H.-J. Son, D. W. Cho and S. O. Kang, J. Phys. Chem. A, 2018, 122, 3391–3397 CrossRef CAS PubMed; (s) H. Mori, K. Nishino, K. Wada, Y. Morisaki, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2018, 2, 573–579 RSC; (t) A. V. Marsh, N. J. Cheetham, M. Little, M. Dyson, A. J. P. White, P. Beavis, C. N. Warriner, A. C. Swain, P. N. Stavrinou and M. Heeney, Angew. Chem., Int. Ed., 2018, 57, 10640–10645 CrossRef CAS PubMed; (u) K. Nishino, K. Uemura, K. Tanaka and Y. Chujo, New J. Chem., 2018, 42, 4210–4214 RSC.
- (a) J. Pecyna, P. Kaszyński, B. Ringstrand, D. Pociecha, S. Pakhomov, A. G. Douglass and V. G. Young Jr, Inorg. Chem., 2016, 55, 4016–4025 CrossRef CAS PubMed; (b) R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC; (c) R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed; (d) S. G. McArthur, R. Jay, L. Geng, J. Guo and V. Lavallo, Chem. Commun., 2017, 53, 4453–4456 RSC; (e) M. Y. Tsang, S. Rodríguez-Hermida, K. C. Stylianou, F. Tan, D. Negi, F. Teixidor, C. Viñas, D. Choquesillo-Lazarte, C. Verdugo-Escamilla, M. Guerrero, J. Sort, J. Juanhuix, D. Maspoch and J. G. Planas, Cryst. Growth Des., 2017, 17, 846–857 CrossRef CAS; (f) B. Dong, A. Oyelade and J. A. Kelber, Phys. Chem. Chem. Phys., 2017, 19, 10986–10997 RSC; (g) Y. Zhu and N. S. Hosmane, Eur. J. Inorg. Chem., 2017, 4369–4377 CrossRef CAS; (h) M. P. Grzelczak, S. P. Danks, R. C. Klipp, D. Belic, A. Zaulet, C. Kunstmann-Olsen, D. F. Bradley, T. Tsukuda, C. Viñas, F. Teixidor, J. J. Abramson and M. Brust, ACS Nano, 2017, 11, 12492–12499 CrossRef CAS PubMed; (i) E. Oleshkevich, F. Teixidor, A. Rosell and C. Viñas, Inorg. Chem., 2018, 57, 462–470 CrossRef CAS PubMed; (j) Z.-Y. Wang, M.-Q. Wang, Y.-L. Li, P. Luo, T.-T. Jia, R.-W. Huang, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2018, 140, 1069–1076 CrossRef CAS PubMed; (k) R. M. Dziedzic, M. A. Waddington, S. E. Lee, J. Kleinsasser, J. B. Plumley, W. C. Ewing, B. D. Bosley, V. Lavallo, T. L. Peng and A. M. Spokoyny, ACS Appl. Mater. Interfaces, 2018, 10, 6825–6830 CrossRef CAS PubMed; (l) J. C. Thomas, D. P. Goronzy, A. C. Serino, H. S. Auluck, O. R. Irving, E. Jimenez-Izal, J. M. Deirmenjian, J. Macháček, P. Sautet, A. N. Alexandrova, T. Baše and P. S. Weiss, ACS Nano, 2018, 12, 2211–2221 CrossRef CAS PubMed; (m) X. Wu, J. Guo, Y. Quan, W. Jia, D. Jia, Y. Chen and Z. Xie, J. Mater. Chem. C, 2018, 6, 4140–4149 RSC; (n) M. Kar, O. Tutusaus, D. R. MacFarlane and R. Mohtadi, Energy Environ. Sci., 2018, 12, 566–571 RSC.
- For selected examples of B–C bond formation by electrophilic reactions or halogenation/cross coupling, see: (a) B. T. King, Z. Janousek, B. Gruner, M. Trammell, B. C. Noll and J. Michl, J. Am. Chem. Soc., 1996, 118, 3313–3314 CrossRef CAS; (b) B. Gruner, Z. Janousek, B. T. King, J. N. Woodford, C. H. Wang, V. Vsetecka and J. Michl, J. Am. Chem. Soc., 1999, 121, 3122–3126 CrossRef; (c) C. W. Tsang, Q. C. Yang, E. T. P. Sze, T. C. W. Mak, D. T. W. Chan and Z. W. Xie, Inorg. Chem., 2000, 39, 3582–3589 CrossRef CAS PubMed; (d) C. W. Tsang and Z. W. Xie, Chem. Commun., 2000, 1839–1840 RSC; (e) A. Franken, C. A. Kilner, M. Thornton-Pett and J. D. Kennedy, Collect. Czech. Chem. Commun., 2002, 67, 869–912 CrossRef CAS; (f) K. Vyakaranam, Z. Janousek, L. Eriksson and J. Michl, Heteroat. Chem., 2006, 17, 217–223 CrossRef CAS; (g) B. T. King, S. Korbe, P. J. Schreiber, J. Clayton, A. Nemcova, Z. Havlas, K. Vyakaranam, M. G. Fete, I. Zharov, J. Ceremuga and J. Michl, J. Am. Chem. Soc., 2007, 129, 12960–12980 CrossRef CAS PubMed; (h) M. Finze, Inorg. Chem., 2008, 47, 11857–11867 CrossRef CAS PubMed; (i) M. Valášek, J. Štursa, R. Pohl and J. Michl, Inorg. Chem., 2010, 49, 10247–10254 CrossRef PubMed; (j) A. Himmelspach and M. Finze, J. Organomet. Chem., 2010, 695, 1337–1345 CrossRef CAS; (k) A. Himmelspach, G. J. Reiss and M. Finze, Inorg. Chem., 2012, 51, 2679–2688 CrossRef CAS PubMed; (l) J. Kaleta, J. Tarabek, A. Akdag, R. Pohl and J. Michl, Inorg. Chem., 2012, 51, 10819–10824 CrossRef CAS PubMed; (m) D. Olid, R. Nunez, C. Vinas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC.
- For a nucleophilic substitution approach, see: (a) C. Tang, J. Zhang and Z. Xie, Angew. Chem., Int. Ed., 2017, 56, 8642–8646 CrossRef CAS PubMed; (b) C. Tang, J. Zhang, J. Zhang and Z. Xie, J. Am. Chem. Soc., 2018, 140, 16423–16427 CrossRef CAS PubMed.
- For recent reviews, see: (a) W.-B. Yu, P.-F. Cui, W.-X. Gao and G.-X. Jin, Coord. Chem. Rev., 2017, 350, 300–319 CrossRef CAS; (b) X. Zhang and H. Yan, Coord. Chem. Rev., 2019, 378, 466–482 CrossRef CAS; (c) S. Duttwyler, Pure Appl. Chem., 2018, 90, 733–744 CAS; (d) Y. Quan, Z. Qiu and Z. Xie, Chem.–Eur. J., 2018, 24, 2795–2805 CrossRef CAS PubMed.
- For selected historically important examples of B–H activation of closo-carboranes, see: (a) E. L. Hoel and M. F. Hawthorne, J. Am. Chem. Soc., 1975, 79, 6388–6395 CrossRef; (b) E. Molinos, G. Kociok-Köhn and A. S. Weller, Chem. Commun., 2005, 3609–3611 RSC; (c) E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Organometallics, 2007, 26, 2370–2382 CrossRef CAS.
- For selected publications, see: (a) Y. Quan and Z. Xie, J. Am. Chem. Soc., 2014, 136, 15513–15516 CrossRef CAS PubMed; (b) Y. Quan, Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2014, 136, 7599–7602 CrossRef CAS PubMed; (c) H. Lyu, Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 10623–10626 CrossRef CAS PubMed; (d) K. Cao, Y. Huang, J. Yang and J. Wu, Chem. Commun., 2015, 51, 7257–7260 RSC; (e) Y. Quan and Z. Xie, J. Am. Chem. Soc., 2015, 137, 3502–3505 CrossRef CAS PubMed; (f) Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 1295–1298 CrossRef CAS PubMed; (g) Y. Quan, C. Tang and Z. Xie, Chem. Sci., 2016, 7, 5838–5845 RSC; (h) H. Lyu, Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 11840–11844 CrossRef CAS PubMed; (i) H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2016, 138, 12727–12730 CrossRef CAS PubMed; (j) H. Li, F. Bai, H. Yan, C. Lu and V. I. Bregadze, Eur. J. Org. Chem., 2017, 1343–1352 CrossRef; (k) Y. Quan, H. Lyu and Z. Xie, Chem. Commun., 2017, 53, 4818–4821 RSC; (l) X. Zhang, H. Zheng, J. Li, F. Xu, J. Zhao and H. Yan, J. Am. Chem. Soc., 2017, 139, 14511–14517 CrossRef CAS PubMed; (m) C.-X. Li, H.-Y. Zhang, T.-Y. Wong, H.-J. Cao, H. Yan and C.-S. Lu, Org. Lett., 2017, 19, 5178–5181 CrossRef CAS PubMed; (n) R. Cheng, Z. Qiu and Z. Xie, Nat. Commun., 2017, 8, 14827–14833 CrossRef CAS PubMed; (o) R. Cheng, B. Li, J. Wu, J. Zhang, Z. Qiu, W. Tang, S. You, Y. Tang and Z. Xie, J. Am. Chem. Soc., 2018, 140, 4508–4511 CrossRef CAS PubMed; (p) X. Zhang and Y. Hong, Chem. Sci., 2018, 9, 3964–3969 RSC; (q) T.-T. Xu, K. Cao, J. Wu, C.-Y. Zhang and J. Yang, Inorg. Chem., 2018, 57, 2925–2932 CrossRef CAS PubMed.
- For recent reports on B–H activation in cage walking processes, see: (a) R. M. Dziedzic, J. L. Martin, J. C. Axtell, L. M. A. Saleh, T.-C. Ong, Y.-F. Yang, M. S. Messina, A. L. Rheingold, K. N. Houk and A. M. Spokoyny, J. Am. Chem. Soc., 2017, 139, 7729–7732 CrossRef CAS PubMed; (b) B. J. Eleazer, M. D. Smith, A. A. Popov and D. V. Peryshkov, Chem. Sci., 2017, 8, 5399–5407 RSC.
- (a) Y. Zhang, Y. Sun, F. Lin, J. Liu and S. Duttwyler, Angew. Chem., Int. Ed., 2016, 55, 15609–15614 CrossRef CAS PubMed; (b) Y. Shen, Y. Pan, K. Zhang, X. Liang, J. Liu, B. Spingler and S. Duttwyler, Dalton Trans., 2017, 46, 3135–3140 RSC; (c) F. Lin, Y. Shen, Y. Zhang, Y. Sun, J. Liu and S. Duttwyler, Chem.–Eur. J., 2018, 24, 551–555 CrossRef CAS PubMed; (d) Y. Zhang, T. Wang, L. Wang, Y. Sun, F. Lin, J. Liu and S. Duttwyler, Chem.–Eur. J., 2018, 24, 15812–15817 CrossRef CAS PubMed; (e) F. Lin, J.-L. Yu, Y. Shen, S.-Q. Zhang, B. Spingler and S. Duttwyler, J. Am. Chem. Soc., 2018, 140, 13798–13807 CrossRef CAS PubMed; (f) X. Liang, Y. Shen, K. Zhang, J. Liu and S. Duttwyler, Chem. Commun., 2018, 54, 12451–12454 RSC.
- CCDC 1886699–1886701 contain the supplementary crystallographic data for this paper.†.
- Y. Shen, K. Zheng, R. Dontha, Y. Pan, J. Liu and S. Duttwyler, RSC Adv., 2018, 8, 22447–22451 RSC.
- Y. Shen, J. Liu, T. Sattasatchuchana, K. K. Baldridge and S. Duttwyler, Eur. J. Inorg. Chem., 2017, 4420 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization and X-ray crystallographic data in CIF format. CCDC 1886699–1886701. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00078j |
| This journal is © The Royal Society of Chemistry 2019 |