
Surface functionalized 3D carbon fiber boosts the lithium storage behaviour of transition metal oxide nanowires via strong electronic interaction and tunable adsorption energy†
Lei
Hu‡
b,
Yingxia
Gao‡
a,
Tuzhi
Xiong
a,
David
Adekoya
c,
Weitao
Qiu
b,
Hao
Yang
 b,
M.-Sadeeq (Jie Tang)
Balogun
*a,
Shanqing
Zhang
c,
Anlian
Pan
a,
Yuping
Li
*a and
Yexiang
Tong
*b
b,
M.-Sadeeq (Jie Tang)
Balogun
*a,
Shanqing
Zhang
c,
Anlian
Pan
a,
Yuping
Li
*a and
Yexiang
Tong
*b
aCollege of Materials Science and Engineering, Hunan University, Changsha 410082, P. R. China. E-mail: balogun@hnu.edu.cn; liypli@hnu.edu.cn
bMOE of the Key Laboratory of Bioinorganic and Synthetic Chemistry, The Key Lab of Low-carbon Chemistry & Energy Conservation of Guangdong Province, KLGHEI of Environment and Energy Chemistry, School of Materials Science and Engineering, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: chedhx@mail.sysu.edu.cn
cCentre for Clean Environment and Energy, Griffith School of Environment, Gold Coast Campus, Griffith University, QLD 4222, Australia
First published on 4th July 2019
Abstract
The Li-ion storage properties of transition metal oxide (TMOs) electrodes such as Li-ion intercalation-based electrodes are usually enhanced by hybridizing with 3D carbon scaffolds. However, understanding of the large variation in performance enhancement is rarely reported. As a proof of concept, intercalation reaction-based TMO (V2O5 and TiO2) nanowires were hybridized with two types of 3D carbon scaffolds (namely pristine carbon fiber cloth, CFC and porous N-doped CFC, PNCFC). Theoretical calculation predicts that the PNCFC@TMO hybrids displayed reasonably lower adsorption energy towards easier Li-ion intercalation than those of CFC@TMOs. Electrochemical properties further disclosed that PNCFC-based hybrids exhibit the best lithium storage performance. Furthermore, in situ Raman, XPS and charge redistribution studies not only decipher that strong electronic interaction exists between PNCFC and TMOs but also consistently affirm that such interaction is ascribed to the shift of the p-adsorption energy, facilitating rapid kinetics and leading to improved Li storage properties.
New conceptsPoor kinetics has rendered Li-ion intercalation reaction-based transition metal oxides to be hybridized with highly three-dimensional (3D) carbon-based current collectors. It has been found that the Li-storage performance of these transition metal oxides usually increases with the support of the 3D carbon-based current collectors. The most common reasons for the performance enhancement are habitually ascribed to “all-in-one” integrated advantages arising from the 3D conductive network and high surface area. Less efforts have been devoted to the search for possible mechanisms that attract such enhanced Li-storage performance. In this study, both theoretical and experimental analyses were utilized to address the horizons of enhanced Li-storage properties in 3D carbon scaffold and intercalation reaction-based transition metal oxide hybrids. Li-storage test, in situ Raman, and XPS analyses and density functional theory (DFT) calculations consistently reveal that the enhanced performance is attributed to the alteration in the p-adsorption energy, which helps to facilitate rapid Li-ion diffusion. This present study is beneficial for understanding the Li-ion intercalation reaction storage mechanism towards designing other electrodes for LIBs and beyond. |
Introduction
Li-ion intercalation reaction (LiIR)-based electrodes are one of the most promising electrode materials for lithium ion batteries (LIBs) because they offer the advantages of low volume expansion, stable solid electrolyte interface layer formation, little irreversible capacity loss and no electrolyte decomposition.1–5 The intercalation reaction of TMOs is shown as follows:6| MO + xLi+ + xe− ⇌ LixMO | (1) |
Transition metal oxides (TMOs)2,7–9 such as Ti oxide-based10,11 electrodes especially TiO2 and V oxide-based12–15 electrodes and especially V2O5 usually exhibit such LiIR mechanism. Meanwhile, these TMOs exhibit either poor electrochemical stability (V2O5) or poor electrical conductivity (V2O5 and TiO2) leading to loss in capacity and low rate capability, respectively.16,17 In order to improve the dual dilemma of both V2O5 and TiO2, one of the common strategies employed is to design self-supporting electrode materials by the direct growth of the active materials on 3D carbon scaffolds (e.g. carbon cloth, graphene, carbon nanotubes and so on).18–22 Such strategy could automatically prevent the intercalation of Li-ions into the carbon scaffold (Li-ion usually intercalates in carbon-based material at a working potential below 1.0 V) and make it easy to determine the capacity contribution of the active V2O5 and TiO2 materials. Previous literature has shown that the performance enhancement of the carbon scaffolds and TMO hybrids can be attributed to the conductive 3D network and high surface area that allows rapid ion/electron transfer, enhanced conductivity and improved mechanical flexibility.19,23 However, despite the enhanced storage properties,24 the understanding of the intrinsic properties of the carbon scaffold and active electrodes is rarely reported.25,26 In addition, less or no attention is given to the theoretical insight into the effect of such hybridization towards enhanced electrochemical activity.27
In this work, density functional theory (DFT) calculations were first studied to probe the intercalation of Li-ions into V2O5 on pristine carbon fiber cloth (CFC) and porous N-doped carbon fiber cloth (PNCFC). The PNCFC-based sample requires lower adsorption energy to complete the intercalation process than the CFC counterparts. CFC was selected among the commonly reported carbon scaffolds owing to its good mechanical strength, stability, and wide application in energy storage research.24,27–29 The electrochemical performance of PNCFC@V2O5 presents superior Li storage properties over CFC@V2O5. In situ Raman spectra and XPS consistently reveal that there exist strong electronic interactions between PNCFC and V2O5, which is valuable towards improving Li storage kinetics. Such strong electronic interaction allows the downshift of the p-band adsorption center, which is favorable for enhancing the Li storage performance. More interestingly, based on the understanding of PNCFC@V2O5, theoretical and experimental analyses further verify that a hybrid of TiO2 and PNCFC can also display a similar phenomenon by modulating the p-orbital adsorption energy.
Results
Theoretical predictions
Previous literature has reported that series of TMOs directly synthesized on 3D carbon scaffolds usually relate the enhanced performance to the 3D network.19,29,30 However, theoretical studies on the intercalation process of carbon compared to the active materials in their powdery forms are rarely studied. DFT calculations were first employed to investigate the optimum Gibb's free energy change (ΔGH*) required for Li-ion diffusion through pristine V2O5 and CFC@V2O5.31,32 The diffusion barrier during a diffusion process of a Li-ion is very essential for understanding the electrode behaviour and the reasons for enhanced Li-ion storage performance.33,34 The optimized cluster structures of V2O5 and CFC@V2O5 for the DFT calculations can be found in the ESI,† Fig. S1 and S2 and the free energy change diagrams of V2O5 and CFC@V2O5 are shown in Fig. 1a and b, respectively. The free energy change diagrams for these three electrodes are derived based on theoretical intercalation of Li-ions (i.e. lithiated state) into these electrodes. For example, Fig. S3 (ESI†) shows the lithiated state of V2O5, while Fig. S4 and S7 (ESI†) also show the lithiated state of both CFC@V2O5 and PNCFC@V2O5, respectively. Li-ions at the initial state (Fig. S3(I) and S4(I), ESI†) account for ΔGH* of 0.0 eV in both electrodes (Fig. 1a(i) and b(iii)). At the V2O5 surface, CFC@V2O5 requires a lower diffusion barrier of 0.14 eV (Fig. S4(II), ESI† and Fig. 1b-ts-3), whereas the ΔGH* is much higher in V2O5 (0.19 eV, Fig. S3(II), ESI† and Fig. 1a-ts-1). Upon complete Li-ion intercalation into the lattice of the electrodes forming LixV2O5 (Fig. S3(III), ESI†), V2O5 will still have to overcome an energy barrier of 1.81 eV (Fig. 1a(ts-2)). The lower diffusion barrier (ΔGH*) of 0.95 eV (Fig. 1b(ts-4) and Fig. S4(III), ESI†) for CFC@V2O5 compared to that of pristine V2O5 indicates that rapid Li-ion transportation and enhanced kinetics throughout the intercalation process could result in superior performance of CFC@V2O5 over pristine V2O5. Finally, the energy barrier of V2O5 and CFC@V2O5 returned to 0.0 eV (Fig. 1a(ii) and b(iv)) after the entire intercalation process (Fig. S3(IV) and S4(IV), ESI†), respectively.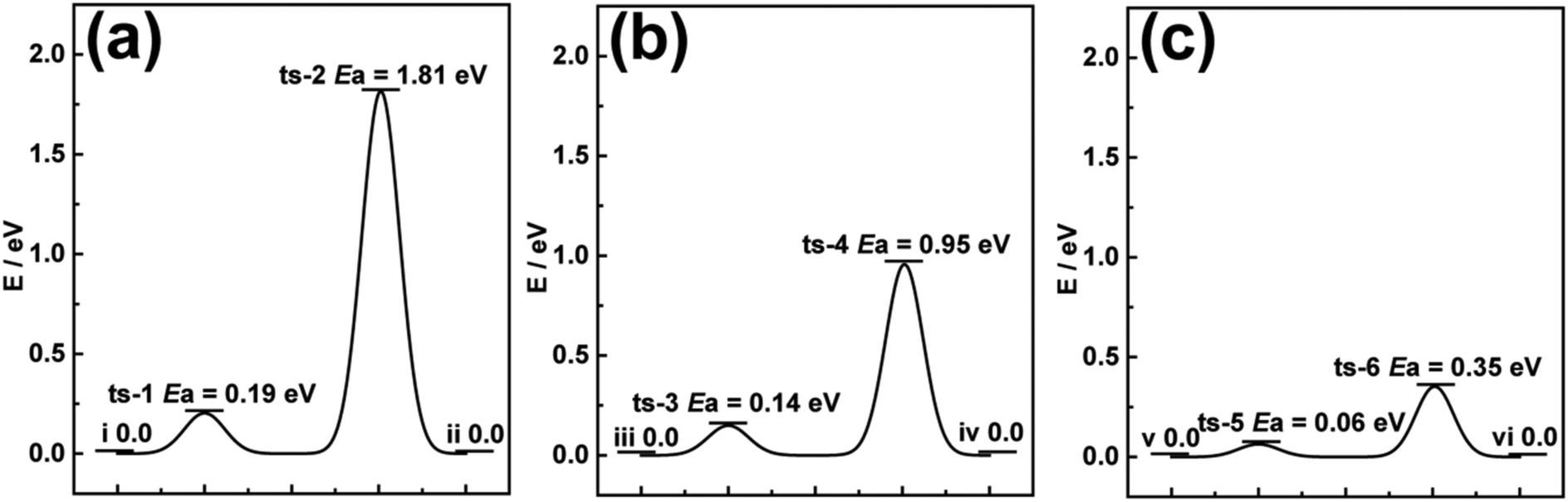 | ||
| Fig. 1 Calculated migration ΔGH* to Li-ion diffusion through (a) V2O5, (b) CFC@V2O5 and (c) PNCFC@V2O5 from DFT calculations. | ||
Previously, we have shown that the surface of CFC can be functionalized with nitrogen (N) atoms, high porosity, and better conductivity, which leads to high surface area and also serves as a promising anode material for LIBs.27,35 When the porous N-doped functionalized CFC (denoted as PNCFC) was used as a current collector for the growth of conversion reaction-based TMOs such as NiCo2O4, experimental and theoretical analyses confirmed that there exists an electronic interaction between PNCFC and NiCo2O4, which is responsible for achieving high capacity of the hybrid.27 Additionally, V2O5 nanobelt36 and V2O5 nanosheet37 cathodes that were synthesized on graphene foam and carbon nanotubes (CNTs) displayed enhanced performance due to the 3D network and harmonious integration.36,37 Motivated by this concept, we utilized the PNCFC as a current collector for the direct synthesis of V2O5 nanowires (denoted as PNCFC@V2O5 NWs) and compare its performance with those of CFC@V2O5 NWs.
Theoretical predictions on a PNCFC@V2O5 hybrid was conducted based on the optimized structure model for PNCFC@V2O5 (Fig. S5, ESI†). The enlarged snapshot of covalent bonds between C (a functional group of PNCFC) and O (from V2O5), and between O (functional group of PNCFC) and V (from V2O5) are depicted in Fig. S6a and b (ESI†), respectively. This bonding shows that the functional groups revealed a close relationship between individual elements in the electrode. Compared to CFC@V2O5, the optimum Gibb's free energy change (ΔGH*) required for Li-ion transportation in PNCFC@V2O5 is shown in Fig. 1c. During Li-ion intercalation in PNCFC@V2O5 (Fig. S7, ESI†), the ΔGH* at ts-5 and ts-6 are 0.06 and 0.35 eV, also indicating the complete lithiated state of V2O5, and significantly lower than those of CFC@V2O5 and pristine V2O5.
The result further suggests that the support of a functionalized 3D carbon scaffold with TMOs could not only hasten the Li-ion diffusion pathway and enhance the kinetics but also lower the adsorption energy towards achieving high performance LIBs.
Synthesis and morphological characterization
The scanning electron microscopy (SEM) images of the two current collectors, CFC and PNCFC (highly networked self-supported current collectors) are shown in Fig. S8a and b (ESI†), respectively. The direct growth of V2O5 NWs on the CFC and PNCFC self-supported materials was accomplished through hydrothermal and air-annealing treatments as previously reported.19 Schematic illustrations of the V2O5 NW fabrication process can be seen in Scheme S1 (ESI†) (details about the synthetic method can be found in the Experimental section). Firstly, a 24 cm2 piece of CFC was converted to PNCFC hydrothermal reaction, thermal reduction and etching treatments (Scheme S1(I), ESI†).38 The SEM image clearly shows the porous and rough nature of the PNCFC surface as seen in Fig. 2a. Transmission electron microscopy (TEM) analysis further reveals an exfoliated and porous surface on the PNCFC (Fig. 2a inset). For the CFC current collector, the approximate thickness of a single fiber is 9.6 μm (Fig. S9a, ESI†), which reduces to 7.7 μm after modification to PNCFC (Fig. S9b, ESI†). | ||
| Fig. 2 Morphological characterization. (a) SEM image of PNCFC showing the exfoliated porous surface. Inset is the corresponding TEM image. (b) SEM image of PNCFC@V2O5 showing V2O5 NWs. (c) SEM image of CFC@V2O5 showing the contact between CFC and V2O5. (d) SEM image of PNCFC@V2O5 showing V2O5 NWs strictly adhered to the PNCFC surface. (e) TEM image of PNCFC@V2O5 NWs showing the core-double shell-like architecture. Elemental mapping of PNCFC@V2O5 showing distribution of the elements from (f) HAADF, (g) C, (h) V, (i) O, and (j) N. (k) TEM image of PNCFC@V2O5 NWs and (l) HRTEM image of PNCFC@V2O5 NWs displaying the lattice fringes. | ||
Previous reports have demonstrated that lightweight substrates or current collectors with porous morphology could serve as favorable scaffolds for the growth of active materials, while reducing the weight of the entire cell24,36,39 and also contributed to the flexible properties of the electrodes.24,35,40,41 Thus, VOx NWs (Fig. S10, ESI†) were synthesized on both CFC and PNCFC current collectors and annealed at 400 °C in the air to obtain CFC@V2O5 (Scheme S1(II), ESI†) and PNCFC@V2O5 NWs (Scheme S1(III), ESI†), respectively.
According to the XRD pattern, both samples were characterized with V2O5 peaks (JCPDF Card #65-0131) but there's no significant difference in the phases of the two samples (Fig. S11, ESI†). Both the SEM images for CFC@V2O5 and PNCFC@V2O5 consist of interwoven NWs with diameters in the range of 300–800 nm (Fig. S12, ESI† and Fig. 2b), respectively. Fig. 2c clearly depicts that V2O5 NWs were successfully coated on the smooth surface of CFC, while Fig. 2d affirms the growth of the NWs firmly adhered to the PNCFC current collector. Further information regarding the morphology of the adhesiveness of V2O5 NWs and the PNCFC current collector interface was accomplished by TEM analyses. Interestingly, the TEM images revealed three-layer (core–double shell) architecture of the sample (Fig. 2e and Fig. S13, ESI†), consisting of an inner-core CFC layer, an exfoliated carbon middle-shell layer and a V2O5 NW outer-shell layer. Energy dispersive X-ray spectroscopy (EDS) elemental mapping collected from Fig. 2e with its corresponding HAADF (Fig. 2f) displayed the distribution of C (Fig. 2g-red), V (Fig. 2h-yellow) and O (Fig. 2i-blue) and N (Fig. 2j-orange). Fig. 2g discloses that the intensity of C (red colour) decreases from bulk (top side) to the surface (down side) and the presence of V and O at the outermost shell layer (Fig. 2h) also confirms the formation of V2O5. The results obtained from the EDS analyses further certified that the V2O5 NWs were successfully coated on a PNCFC current collector demonstrating a core–double shell design. Additionally, the TEM image collected from the PNCFC@V2O5 NW surface (Fig. 2k) showed the single crystalline nature (Fig. S14, ESI†) and lattice spacing of 0.34 nm (Fig. 2l), which corresponds to the (110) plane of V2O5 and also affirms the successful formation of V2O5 on a PNCFC substrate.
Electronic structure characterization
The electronic structure and valence states of both CFC@V2O5 and PNCFC@V2O5 and more information on the relationship between V2O5 and the two types of current collectors were studied using X-ray photoelectron spectroscopy (XPS). The XPS survey spectra of CFC@V2O5 and PNCFC@V2O5 can be found in Fig. S15a (ESI†). C 1s of the PNCFC@V2O5 exhibited a broad signal for the C–C bond and an additional broad peak compared to the CFC@V2O5 sample, which suggests some extent of modifications in the modified CFC coated samples (Fig. 3a).35,42 Moreover, a peak at ∼400 eV in the N 1s spectra of PNCFC@V2O5 is totally absent in the CFC@V2O5 sample (Fig. 3b). This confirmed that the PNCFC@V2O5 sample is N-doped. The N-doping can be obtained during thermal reduction of the Ni(OH)2·0.75H2O precursor in a N2 atmosphere.27,38 Compared to the V 2p XPS spectra of CFC@V2O5, the V 2p XPS spectra of both samples can be deconvoluted into two synthetic peaks (516.3 eV for V4+ and 517.6 eV for V5+) (Fig. 3c(I)–(II)) confirming that the V2O5 samples are mixed-valence in nature.43–47 Interestingly, the V5+ V 2p XPS spectra of CFC@V2O5, shifted to a lower binding energy (ca. 0.36 eV) in the PNCFC-based sample, whereas, the V4+ V 2p XPS spectra shifted to a lower binding energy (ca. 0.36 eV) for the PNCFC-based sample.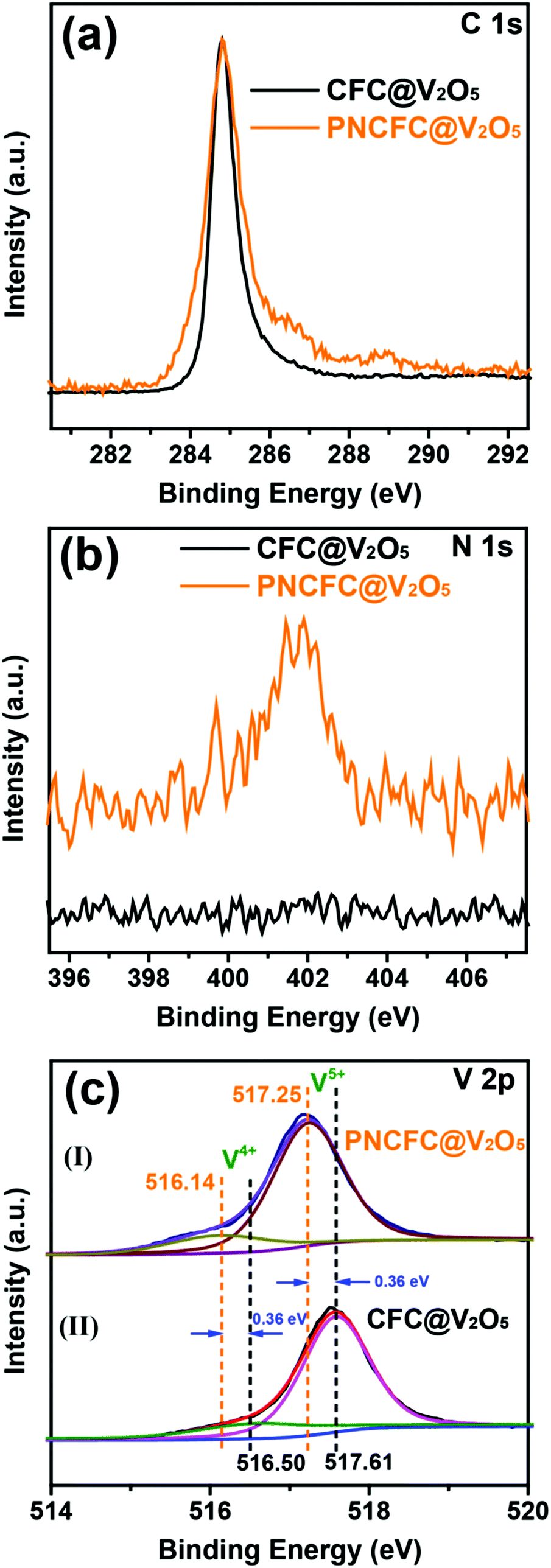 | ||
| Fig. 3 (a) C 1s, (b) N 1s and (c) V 2p3/2 XPS spectra of CFC@V2O5 and PNCFC@V2O5. | ||
This phenomenon indicates a change in the electronic bonding environment state, justifies strong electronic interaction between PNCFC and V2O5 and suggests rapid transfer of electrons in the PNCFC@V2O5 sample.27,48 Moreover, the higher shift in the binding energy of the V4+ suggests stronger electronic interaction on the low valence state of PNCFC@V2O5, which could be beneficial for enhancing the sample conductivity and kinetics.49 Additionally, the percentage ratio of the V4+/V5+ in the samples increased from 38% in CFC@V2O5 to 47% in the PNCFC@V2O5 sample, meaning that a higher composition of V4+ improved the electronic interactions between V2O5 and PNCFC, which could also be an avenue for improving the electrochemical performance of PNCFC@V2O5.43
It has been previously proposed that the valence of vanadium oxide composites can be tailored, and could be a promising way to achieve high capacity, good conductivity and good cycling stability.44,45 For instance, self-doping of V4+ into V2O5 nanoflakes exhibits high structural stability, and long-term and fast reversible Li storage capability resulting from the presence of V4+.43 Likewise V2O5 xerogels showed improved lithium storage performance benefiting from the multi-vanadium oxidation states and electrical conductivity.50 Thus, we proposed that the high composition of V4+ in PNCFC@V2O5 arises from the electronic interaction between V2O5 and PNCFC, and this could eventually lead to improving the electrochemical performance of PNCFC@V2O5 over the CFC@V2O5 sample. According to Fig. S15b (ESI†), the O 1s XPS spectra of PNCFC@V2O5 are slightly different from that of CFC@V2O5, also confirming that characteristic functional groups from the PNCFC current collector and such functional groups are beneficial in improving electrode kinetic properties.27,51 Based on the ex situ Raman spectroscopy analyses, the entire Raman peak below 1000 cm−1 corresponds with V2O5 Raman peaks reported in other literature (Fig. S16a, ESI†).52 More importantly, the Raman spectra present a D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G peak intensity ratio of 1.48 for PNCFC@V2O5 and also larger than that of the CFC@V2O5 (1.32) (Fig. S16b, ESI†). In addition, the G-band of the PNCFC@V2O5 sample becomes broader further affirming modification of the treated CFC coated sample. This can be attributed to the porous exfoliated surface of the PNCFC substrate that consists of some surface disorder and the presence of functional groups induced by etching, which is in agreement with the O 1s XPS spectra.35,53
:G peak intensity ratio of 1.48 for PNCFC@V2O5 and also larger than that of the CFC@V2O5 (1.32) (Fig. S16b, ESI†). In addition, the G-band of the PNCFC@V2O5 sample becomes broader further affirming modification of the treated CFC coated sample. This can be attributed to the porous exfoliated surface of the PNCFC substrate that consists of some surface disorder and the presence of functional groups induced by etching, which is in agreement with the O 1s XPS spectra.35,53
Li-storage properties
Li storage behaviour and performance for both CFC@V2O5 and PNCFC@V2O5 positive electrodes was studied in a coin cell. The Li-ion insertion–deinsertion reactions in V2O5 can be seen from the 5th cyclic voltammetry (CV) curves for both electrodes (Fig. S17, ESI†) and the corresponding reaction mechanism can be seen as V2O5 + xLi+ + xe− ⇌ LixV2O5.36 Regular peaks of V2O5 can be found in both electrodes.54 The PNCFC@V2O5 electrode shows stronger current density peaks than the CFC@V2O5 electrode and also shifts to the higher voltage region, which implies that the PNCFC@V2O5 electrode has a higher electrochemical reactivity and enhanced conductivity compared to the CFC@V2O5.36 Electrochemical impedance spectroscopy (EIS) analysis was carried out to gain information on the kinetics of both CFC@V2O5 and PNCFC@V2O5 electrodes.The Nyquist plot is characterized with a semi-circle and diffusion slope representing the charge transfer resistance (Rct) and Warburg impedance, respectively.19 The smaller the Rct, the more the increase there is in the electron transfer rate of V2O5 in the PNCFC@V2O5 cell. As seen in Fig. 4a, PNCFC@V2O5 displays a smaller Rct value (86 Ω) than that of CFC@V2O5 (122 Ω), confirming the superior conductivity of the PNCFC@V2O5 electrode over CFC@V2O5. As shown in Fig. 4b, the discharge capacity of PNCFC@V2O5 is 268 mA h g−1 after the 10th electrochemical cycle, while that of CFC@V2O5 is lower (233 mA h g−1). It should be noted that energy efficiency (%) is a vital parameter for the practical development of new electrode materials. The energy efficiency of a battery usually depends on the overpotentials of charging and discharging profiles and can be determined according to the formula below;55
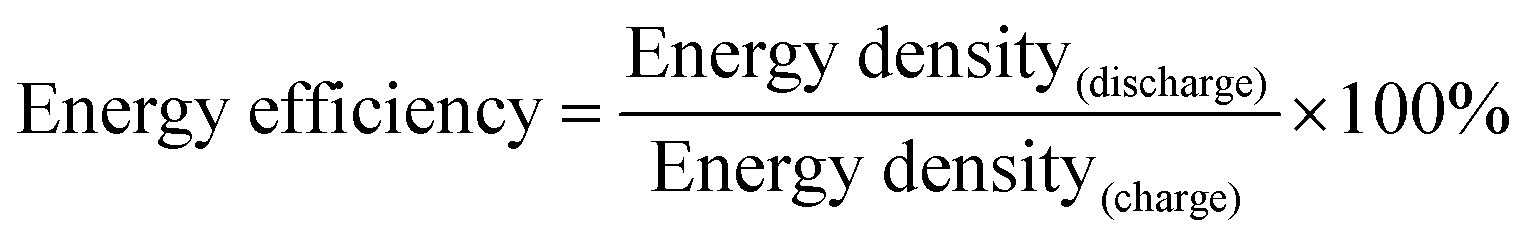 | (2) |
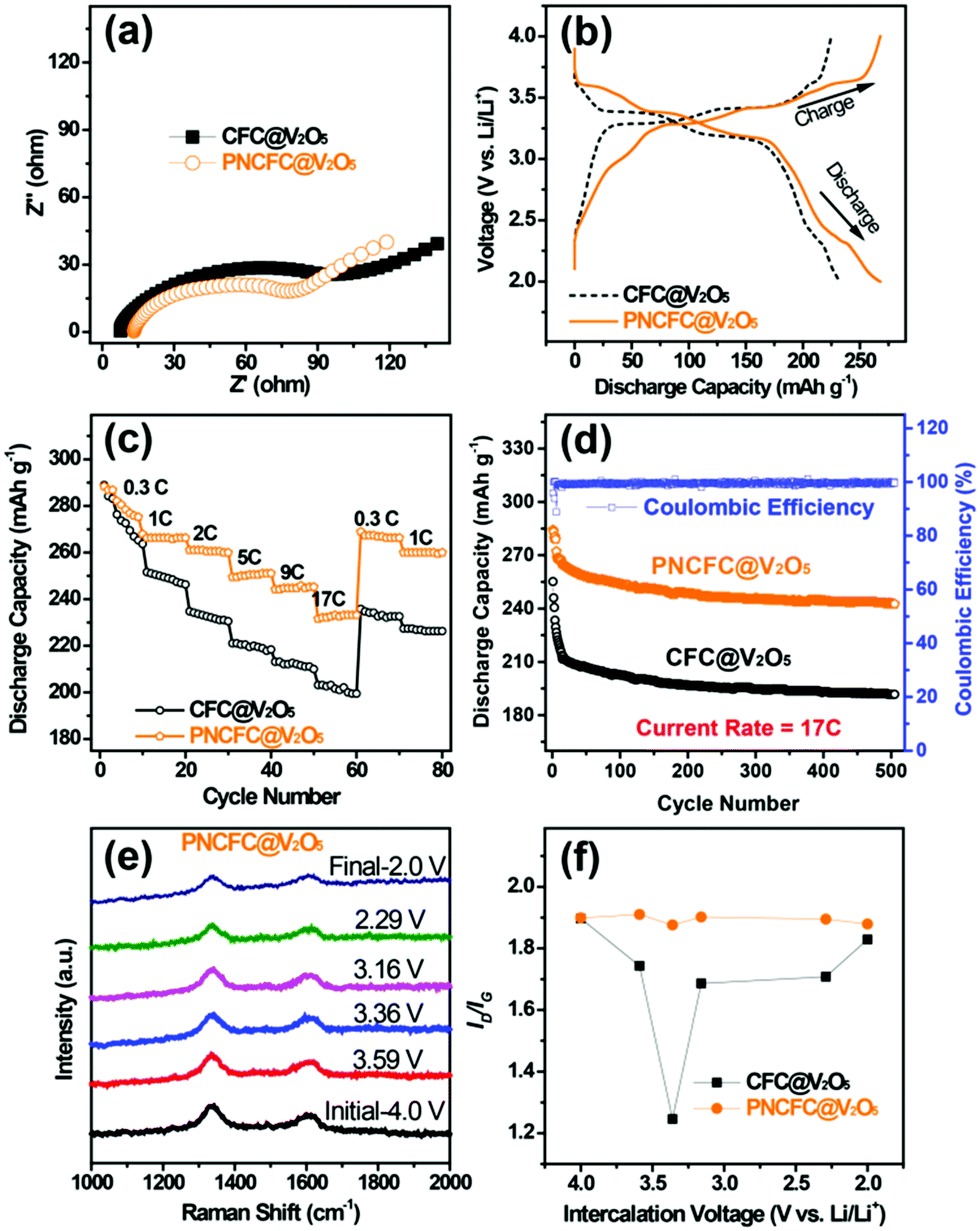 | ||
| Fig. 4 Li storage properties. (a) The Nyquist plots, (b) charge–discharge curves at a current density of 2.0C, (c) rate capability and (d) cycling stability at a current density of 2.0C of both CFC@V2O5 and PNCFC@V2O5. (e) In situ Raman spectra of PNCFC@V2O5 at different intercalated voltages. (f) The plot of the ID/IGvs. intercalated voltages of CFC@V2O5 and PNCFC@V2O5. | ||
In situ Raman analysis
Additional analysis of the intercalation of Li-ions was carried out by in situ Raman spectra for the two electrodes. Raman analysis is an effective strategy to study the effect of defect carbon and graphitic carbon in a sample.27 Detail information on the set-up can be found in Fig. S19 (ESI†). The response of the current collector D and G peaks towards Li-ion intercalation at different voltages can be effective for determining the impact of the different CFC current collectors. The Raman spectra collected for CFC@V2O5 showed a non-uniform variation in the carbon D and G bands at different Li-ion voltages (Fig. S19e, ESI†), while that of PNCFC@V2O5 showed nearly uniform variations in the D and G bands (Fig. 4e). The ID/IG values of the electrodes are plotted against the intercalation at different voltages (Fig. 4f) and the ID/IG value of the PNCFC@V2O5 cell is nearly the same at different Li-ion voltages, but varies and is non-uniform for CFC@V2O5. This result interprets that the V2O5 NWs are not strictly attached to the CFC surface leading to instability of the CFC@V2O5 electrode. However, the ID/IG values for PNCFC@V2O5 indicate that there are strong interfacial interactions between the shell layer of the PNCFC and V2O5 NWs. These interactions are highly stable regardless of the voltage. Such strict adherence between the PNCFC and V2O5 NWs allows easy diffusion of Li-ions and transportation of electrons into the entire electrode cavity. Both the theoretical analyses and experimental results are consistent and also account for the higher capacity retention and better stability of the PNCFC@V2O5 electrode over the CFC@V2O5.Discussion
Charge redistribution and adsorption energy capability
The electronic interaction between V2O5 and PNCFC demonstrates a crucial role in enhancing the Li-storage properties of PNCFC@V2O5. DFT calculations were further employed to investigate the charge distribution relationship between V2O5 and carbon scaffolds. The optimized model structures for CFC@V2O5 and PNCFC@V2O5 are displayed in Fig. S2 and S5 (ESI†), respectively. Fig. 5a distinctly displayed the natural bonding orbital (NBO) charge distribution in the optimized CFC@V2O5 and PNCFC@V2O5 obtained from DFT calculations.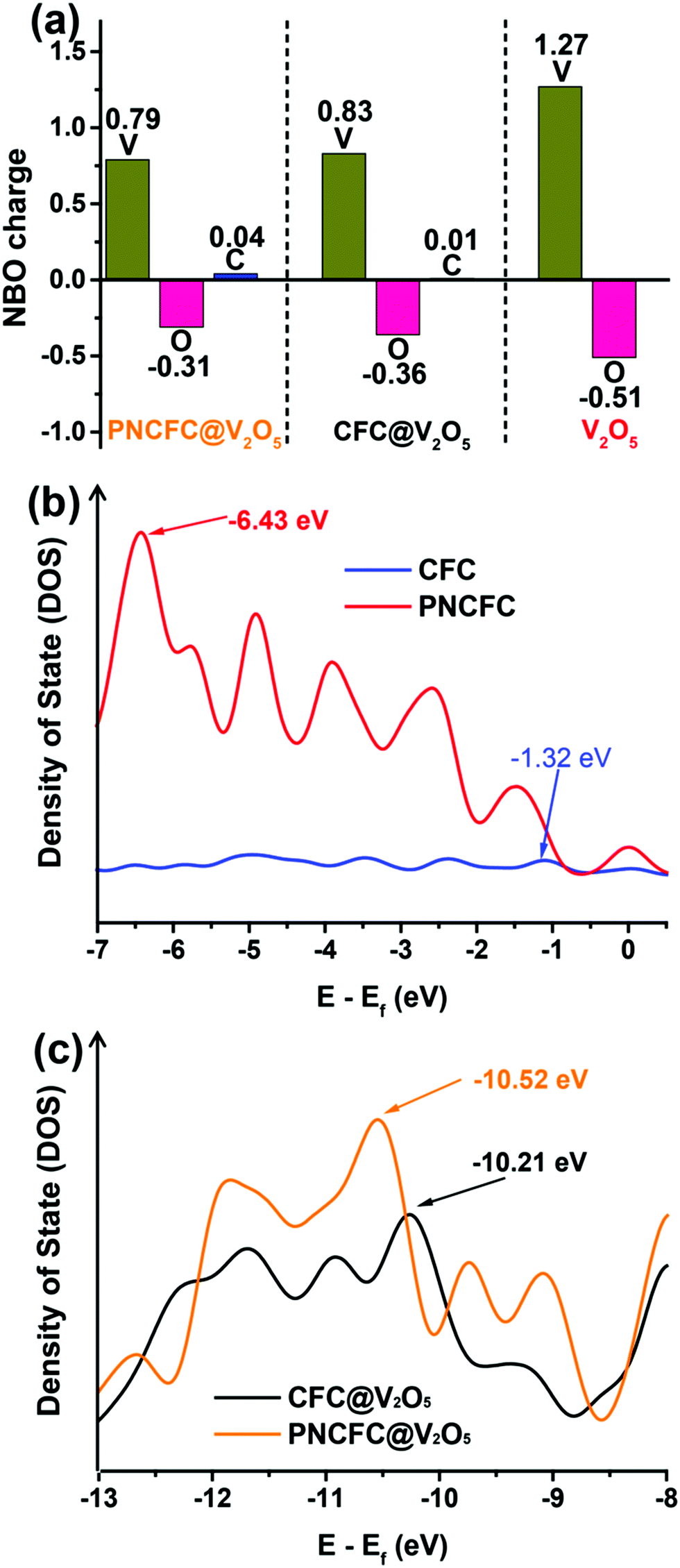 | ||
| Fig. 5 (a) NBO charge redistribution of V2O5, CFC@V2O5 and PNCFC@V2O5. (b) DOS plots of CFC and PNCFC and their corresponding p-bands. (c) DOS plots of CFC@V2O5 and PNCFC@V2O5 and their corresponding p-bands. | ||
The NBO charge distribution of pristine V2O5 was also introduced for comparison (optimized model structure in Fig. S1, ESI†). The charge distribution on PNCFC@V2O5 shows that the positive charge on V is less electropositive, while the negative charge on O is less electronegative, in the order of PNCFC@V2O5 > CFC@V2O5 > V2O5. However, the positive charge on C of PNCFC@V2O5 is more electropositive. These results indicate that the surface functionalization of PNCFC could change the electron density charges on the entire electrode, leading to excellent electronic interactions between PNCFC and V2O5 compared to that of CFC@V2O5.27 To obtain further understanding on the electronic interaction between the hybrids, the analysis of the density of states (DOS) related to the Fermi level (Ef) is studied and the corresponding s, p, d, f and sum bands DOS are shown in Fig. S20 (ESI†). Firstly, in all the samples, the p-band is more intense than the s, d and f-bands and nearly the same as that of the sum-adsorption energy, indicating that the main Li-ion intercalation process is related to the p-adsorption energy. The p-adsorption energy center (Ep) relative to Ef of CFC is −1.27 eV and that of PNCFC is −6.43 eV (Fig. 5b). The results indicate that Ep of PNCFC is farther away from the Ef, facilitating the energy band-gap to reduce and allowing the p-band to undergo a distinct shift in PNCFC than CFC. The hybridization of PNCFC with V2O5 could definitely require lower adsorption energy than that of CFC-based hybrids. Indeed, the Ep in PNCFC@V2O5 also depicts a shift towards higher Ef (−10.52 eV) than CFC@V2O5 (−10.21 eV) (Fig. 5c) due to the increased bonding strength of the PNCFC-based hybrid, which originates from the easy release and pulling of electrons from PNCFC@V2O5. Hence, the energies of the charge density and valence band electrons will increase, enabling rapid electron transfer towards promoting fast Li-ion intercalation kinetics for enhanced Li-ion storage properties. Finally, both the NBO charge distribution and DOS results indicate changes in the bonding or electronic states and also in accordance with the XPS, finally affirming that there exist stronger electronic interactions between PNCFC and V2O5 than pristine CFC and V2O5.
TiO2-based anode: electrochemical properties and mechanism studies
To obtain further understanding of the LiIR-based mechanism, we also utilized a TiO2 anode, which is also well known for its poor electrical conductivity leading to its slow kinetics and poor rate capability. Similar to V2O5, theoretical predictions on TiO2 hybridized with CFC and PNCFC were studied. DFT calculations conducted to determine the ΔGH* required for Li-ion intercalation through CFC@TiO2 and PNCFC@TiO2. The optimized CFC@TiO2 and PNCFC@TiO2 cluster structures used for DFT calculations are presented in Fig. S21 and S22 (ESI†), respectively. As a Li-ion passes across the optimized CFC@TiO2 structure (Fig. S23, ESI†), the ΔGH* required for the transportation of Li-ions throughout CFC@TiO2 is 1.11 eV (Fig. 6a). However, the maximum ΔGH* required is 0.51 eV for PNCFC@TiO2 to transport the Li-ion through the entire PNCFC@TiO2 structure (Fig. S24, ESI†), which is also lower than that of CFC@TiO2. These results also correlate with that of V2O5 in Fig. 1. NBO charge distribution studies were also performed for the TiO2-based electrodes. Pristine TiO2 was also employed for comparison (optimized structure in Fig. S25, ESI†). The enlarged snapshot of covalent bonds between O (a functional group of ECC) and Ti (from TiO2) and an enlarged snapshot of covalent bonds between C (a functional group of ECC) and O (from TiO2) are shown in Fig. S26 (ESI†).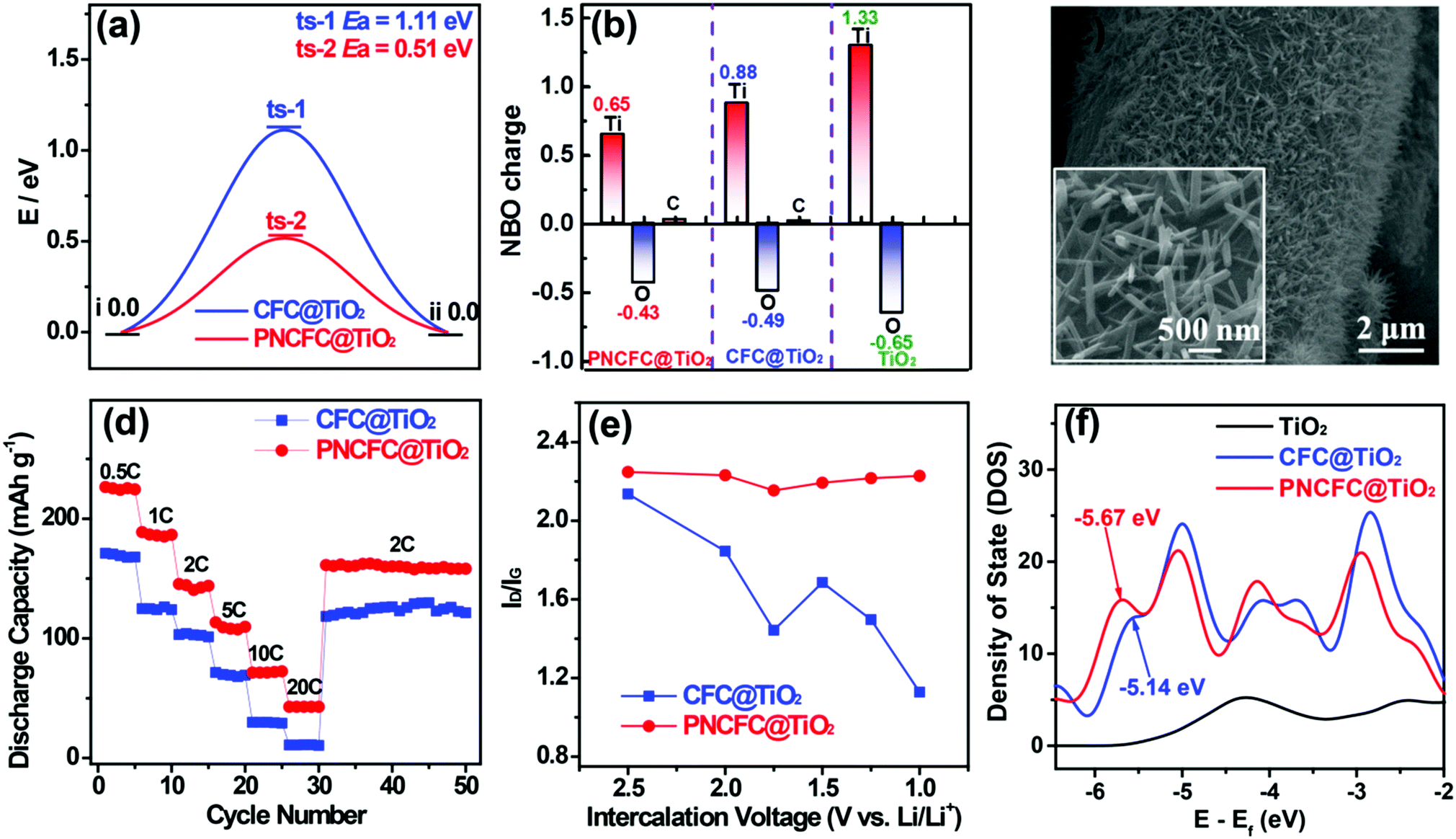 | ||
| Fig. 6 Properties of CFC@TiO2 and PNCFC@TiO2. (a) Calculated migration ΔGH* to Li-ion diffusion through CFC@TiO2 and PNCFC@TiO2. (b) NBO charge redistribution of CFC@TiO2 and PNCFC@TiO2. (c) SEM images of PNCFC@TiO2 NWs. (d) Rate performance and (e) plot of the ID/IGvs. intercalated voltages for CFC@TiO2 and PNCFC@TiO2. (f) DOS plots of TiO2, CFC@TiO2 and PNCFC@TiO2 and their corresponding p-bands. | ||
According to the NBO charge redistribution in Fig. 6b, compared to CFC@TiO2 and pristine TiO2, the charge value on the positively charged Ti decreases, the positive charge on C increases and O is negatively charged in PNCFC@TiO2, indicating stronger electronic interactions between PNCFC and TiO2 of PNCFC@TiO2. Hence, we hydrothermally coated rutile-type TiO2 nanowires (TiO2 NWs) (Fig. S27, ESI†) on CFC (CFC@TiO2 NWs) and PNCFC (PNCFC@TiO2 NWs) according to our previous work (Fig. 6c).11,16 Because TiO2 is known for its poor electrical conductivity, the rate capability performance was investigated at different current densities and in the voltage range of 1.0–3.0 V (vs. Li/Li+), where the PNCFC@TiO2 exhibited superior storage properties to CFC@TiO2 (Fig. 6d). In situ Raman spectra also show that the PNCFC@TiO2 is more stable than CFC@TiO2 (Fig. 6e). Furthermore, the plot of ID/IG values against intercalation voltages collected from the whole results obtained for the TiO2-based electrodes are in-line with those of the V2O5-based electrodes. Moreover, the Ep of PNCFC@TiO2 is −5.67 eV, which is also farther from the Ef compared to CFC@TiO2 (Fig. 6f), indicating a shift in the Ep, and could facilitate rapid Li-ion kinetics in PNCFC@TiO2 than CFC@TiO2. Hence, these results did not clearly confirm that the Li-ion storage properties of LiIR-based TMO electrodes can be effectively modulated by tuning the p-adsorption energy level but also deciphered understanding of the large variation in performance enhancement in carbon scaffold-TMO electrode hybrids.
Conclusions
In summary, we systematically demonstrated the theoretical and experimental insights into the storage mechanism and large variation in the Li-ion storage performance of different carbon scaffolds as current collectors for LiIR-based TMO electrodes. LiIR-based electrodes, such as V2O5 and TiO2 NWs, were directly synthesized on two different carbon current collectors with different surface properties (CFC and PNCFC). DFT calculations revealed that Li-ion adsorption occurred at a lower adsorption energy (ΔGH*) level of 7.75 and 4.64 eV for PNCFC@V2O5 and PNCFC@TiO2, respectively compared to those of CFC@V2O5 (18.21 eV), V2O5 (29.04 eV), CFC@TiO2 (6.67 eV) and TiO2 (11.34 eV). Electrochemical performance confirmed that the V2O5 and TiO2 NWs coated on PNCFC exhibited better Li-storage activity than those hybridized on CFC. Additionally, in situ Raman spectra analysis revealed that PNCFC could serve as a better scaffold for the electrodes in enhancing the cycling stability and rate capability of the PNCFC-based electrodes compared to CFC. XPS, NBO charge distribution and DOS analyses showed that the PNCFC-based electrodes are attributed to stronger electronic interaction between the PNCFC and TMOs due to the shift of p-adsorption energy level. This work deciphers insight into the Li-storage mechanism and performance modulation of carbon-based current collectors and LiIR-based electrodes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21875292 and 51525202), the National Key Research and Development Program of China (2016YFA0202604), the Fundamental Research Funds for the Central Universities (17lgjc36) and Science Starting Foundation of Hunan University (531118010182).References
- T. Zhou, Y. Zheng, H. Gao, S. Min, S. Li, H. K. Liu and Z. Guo, Adv. Sci., 2015, 2, 1500027 CrossRef.
- V. Aravindan, Y.-S. Lee and S. Madhavi, Adv. Energy Mater., 2015, 5, 1402225 CrossRef.
- S. Cao, Z. Xue, C. Yang, J. Qin, L. Zhang, P. Yu, S. Wang, Y. Zhao, X. Zhang and R. Liu, Nano Energy, 2018, 50, 25–34 CrossRef CAS.
- S. Joshi, Q. Wang, A. Puntambekar and V. Chakrapani, ACS Energy Lett., 2017, 2, 1257–1262 CrossRef CAS.
- Z. Yao, X. Xia, D. Xie, Y. Wang, C.-a. Zhou, S. Liu, S. Deng, X. Wang and J. Tu, Adv. Funct. Mater., 2018, 28, 1802756 CrossRef.
- X. Xia, D. Chao, C. F. Ng, J. Lin, Z. Fan, H. Zhang, Z. X. Shen and H. J. Fan, Mater. Horiz., 2015, 2, 237–244 RSC.
- X. Xia, S. Deng, S. Feng, J. Wu and J. Tu, J. Mater. Chem. A, 2017, 5, 21134–21139 RSC.
- F. Wang, X. Wang, Z. Chang, Y. Zhu, L. Fu, X. Liu and Y. Wu, Nanoscale Horiz., 2016, 1, 272–289 RSC.
- C. Wang, L. Wu, H. Wang, W. Zuo, Y. Li and J. Liu, Adv. Funct. Mater., 2015, 25, 3524–3533 CrossRef CAS.
- V. Augustyn, E. R. White, J. Ko, G. Grüner, B. C. Regan and B. Dunn, Mater. Horiz., 2014, 1, 219–223 RSC.
- M.-S. Balogun, M. Yu, C. Li, T. Zhai, Y. Liu, X. Lu and Y. Tong, J. Mater. Chem. A, 2014, 2, 10825–10829 RSC.
- Y. Dai, Q. Li, S. Tan, Q. Wei, Y. Pan, X. Tian, K. Zhao, X. Xu, Q. An, L. Mai and Q. Zhang, Nano Energy, 2017, 40, 73–81 CrossRef CAS.
- Y. Yue and H. Liang, Adv. Energy Mater., 2017, 7, 1602545 CrossRef.
- J. Yao, Y. Li, R. C. Massé, E. Uchaker and G. Cao, Energy Stor. Mater., 2018, 11, 205–259 CrossRef.
- S. Wang, K. A. Owusu, L. Mai, Y. Ke, Y. Zhou, P. Hu, S. Magdassi and Y. Long, Appl. Energy, 2018, 211, 200–217 CrossRef CAS.
- M.-S. Balogun, C. Li, Y. Zeng, M. Yu, Q. Wu, M. Wu, X. Lu and Y. Tong, J. Power Sources, 2014, 272, 946–953 CrossRef CAS.
- B. Long, M.-S. Balogun, L. Luo, Y. Luo, W. Qiu, S. Song, L. Zhang and Y. Tong, Small, 2017, 13, 1702081 CrossRef.
- L. Shen, B. Ding, P. Nie, G. Cao and X. Zhang, Adv. Energy Mater., 2013, 3, 1484–1489 CrossRef CAS.
- M.-S. Balogun, Y. Luo, F. Lyu, F. Wang, H. Yang, H. Li, C. Liang, M. Huang, Y. Huang and Y. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 9733–9744 CrossRef CAS.
- Y. Shi, L. Wen, G. Zhou, J. Chen, S. Pei, K. Huang, H. M. Cheng and F. Li, 2D Mater., 2015, 2, 024004 CrossRef.
- S. Yoon, S. Lee, S. Kim, K.-W. Park, D. Cho and Y. Jeong, J. Power Sources, 2015, 279, 495–501 CrossRef CAS.
- G. Qian, X. Liao, Y. Zhu, F. Pan, X. Chen and Y. Yang, ACS Energy Lett., 2019, 4, 690–701 CrossRef CAS.
- X. Min, B. Sun, S. Chen, M. Fang, X. Wu, Y. g. Liu, A. Abdelkader, Z. Huang, T. Liu, K. Xi and R. Vasant Kumar, Energy Stor. Mater., 2019, 16, 597–606 CrossRef.
- S. Liu, Z. Wang, C. Yu, H. B. Wu, G. Wang, Q. Dong, J. Qiu and A. Eychmüller, Adv. Mater., 2013, 25, 3462–3467 CrossRef CAS.
- Y. Deng, S. Dong, Z. Li, H. Jiang, X. Zhang and X. Ji, Small Methods, 2018, 2, 1700332 CrossRef.
- X. Huang, G. Diao, S. Li, M.-S. Balogun, N. Li, Y. Huang, Z.-Q. Liu and Y. Tong, RSC Adv., 2018, 8, 17056–17059 RSC.
- M.-S. Balogun, H. Yang, Y. Luo, W. Qiu, Y. Huang, Z.-Q. Liu and Y. Tong, Energy Environ. Sci., 2018, 11, 1859–1869 RSC.
- H.-P. Feng, L. Tang, G.-M. Zeng, J. Tang, Y.-C. Deng, M. Yan, Y.-N. Liu, Y.-Y. Zhou, X.-Y. Ren and S. Chen, J. Mater. Chem. A, 2018, 6, 7310–7337 RSC.
- M.-S. Balogun, M. Yu, Y. Huang, C. Li, P. Fang, Y. Liu, X. Lu and Y. Tong, Nano Energy, 2015, 11, 348–355 CrossRef CAS.
- G. X. Pan, X. H. Xia, F. Cao, J. Chen and Y. J. Zhang, Electrochim. Acta, 2014, 149, 349–354 CrossRef CAS.
- Y. Chu, L. Guo, B. Xi, Z. Feng, F. Wu, Y. Lin, J. Liu, D. Sun, J. Feng, Y. Qian and S. Xiong, Adv. Mater., 2018, 30, 1704244 CrossRef PubMed.
- Y. Zheng, T. Zhou, X. Zhao, W. K. Pang, H. Gao, S. Li, Z. Zhou, H. Liu and Z. Guo, Adv. Mater., 2017, 29, 1700396 CrossRef.
- J. Hu, C. Ouyang, S. A. Yang and H. Y. Yang, Nanoscale Horiz., 2019, 4, 457–463 RSC.
- S. Ullah, P. A. Denis and F. Sato, Appl. Mater. Today, 2017, 9, 333–340 CrossRef.
- M.-S. Balogun, W. Qiu, F. Lyu, Y. Luo, H. Meng, J. Li, W. Mai, L. Mai and Y. Tong, Nano Energy, 2016, 26, 446–455 CrossRef CAS.
- D. Chao, X. Xia, J. Liu, Z. Fan, C. F. Ng, J. Lin, H. Zhang, Z. X. Shen and H. J. Fan, Adv. Mater., 2014, 26, 5794–5800 CrossRef CAS.
- D. Kong, X. Li, Y. Zhang, X. Hai, B. Wang, X. Qiu, Q. Song, Q.-H. Yang and L. Zhi, Energy Environ. Sci., 2016, 9, 906–911 RSC.
- M.-S. Balogun, W. Qiu, H. Yang, W. Fan, Y. Huang, P. Fang, G. Li, H. Ji and Y. Tong, Energy Environ. Sci., 2016, 9, 3411–3416 RSC.
- X. Fang, M. Ge, J. Rong and C. Zhou, ACS Nano, 2014, 8, 4876–4882 CrossRef CAS.
- K. Wang, S. Luo, Y. Wu, X. He, F. Zhao, J. Wang, K. Jiang and S. Fan, Adv. Funct. Mater., 2013, 23, 846–853 CrossRef CAS.
- D. Chao, C. Zhu, X. Xia, J. Liu, X. Zhang, J. Wang, P. Liang, J. Lin, H. Zhang and Z. X. Shen, Nano Lett., 2014, 15, 565–573 CrossRef.
- W. Wang, W. Liu, Y. Zeng, Y. Han, M. Yu, X. Lu and Y. Tong, Adv. Mater., 2015, 27, 3572–3578 CrossRef CAS.
- H. Song, C. Liu, C. Zhang and G. Cao, Nano Energy, 2016, 22, 1–10 CrossRef CAS.
- S. D. Perera, B. Patel, N. Nijem, K. Roodenko, O. Seitz, J. P. Ferraris, Y. J. Chabal and K. J. Balkus, Adv. Energy Mater., 2011, 1, 936–945 CrossRef CAS.
- Y. Song, T.-Y. Liu, B. Yao, T.-Y. Kou, D.-Y. Feng, X.-X. Liu and Y. Li, Small, 2017, 13, 1700067 CrossRef PubMed.
- Y. Huang, Y. Lu, Y. Lin, Y. Mao, G. Ouyang, H. Liu, S. Zhang and Y. Tong, J. Mater. Chem. A, 2018, 6, 24740–24747 RSC.
- Y. Huang, L. Hu, R. Liu, Y. Hu, T. Xiong, W. Qiu, M. S. Balogun, A. Pan and Y. Tong, Appl. Catal., B, 2019, 251, 181–194 CrossRef CAS.
- Y. Sun, K. Xu, Z. Wei, H. Li, T. Zhang, X. Li, W. Cai, J. Ma, H. J. Fan and Y. Li, Adv. Mater., 2018, 30, 1802121 CrossRef.
- G. Diao, M.-S. Balogun, S.-Y. Tong, X. Guo, X. Huang, Y. Mao and Y. Tong, J. Mater. Chem. A, 2018, 6, 15274–15283 RSC.
- D. Liu, Y. Liu, A. Pan, K. P. Nagle, G. T. Seidler, Y.-H. Jeong and G. Cao, J. Phys. Chem. C, 2011, 115, 4959–4965 CrossRef CAS.
- K. Ye, K. Li, Y. Lu, Z. Guo, N. Ni, H. Liu, Y. Huang, H. Ji and P. Wang, TrAC, Trends Anal. Chem., 2019, 116, 102–108 CrossRef CAS.
- M. Gotić, S. Popović, M. Ivanda and S. Musić, Mater. Lett., 2003, 57, 3186–3192 CrossRef.
- G. Wang, H. Wang, X. Lu, Y. Ling, M. Yu, T. Zhai, Y. Tong and Y. Li, Adv. Mater., 2014, 26, 2676–2682 CrossRef CAS.
- P. Zhang, L. Zhao, Q. An, Q. Wei, L. Zhou, X. Wei, J. Sheng and L. Mai, Small, 2016, 12, 1082–1090 CrossRef CAS.
- A. Eftekhari, Sustainable Energy Fuels, 2017, 1, 2053–2060 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00327d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |